
Identification of Microbial Flora in Dry Aged Beef to Evaluate the Rancidity during Dry Aging

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dry Aging of Beef
2.2. Analysis of Microflora on Beef during Dry Aging
2.3. Quantitative Polymerase Chain Reaction PCR (q-PCR)
2.4. Measurement of the TBARS and VBN Values
2.5. Statistical Analysis
3. Results and Discussion
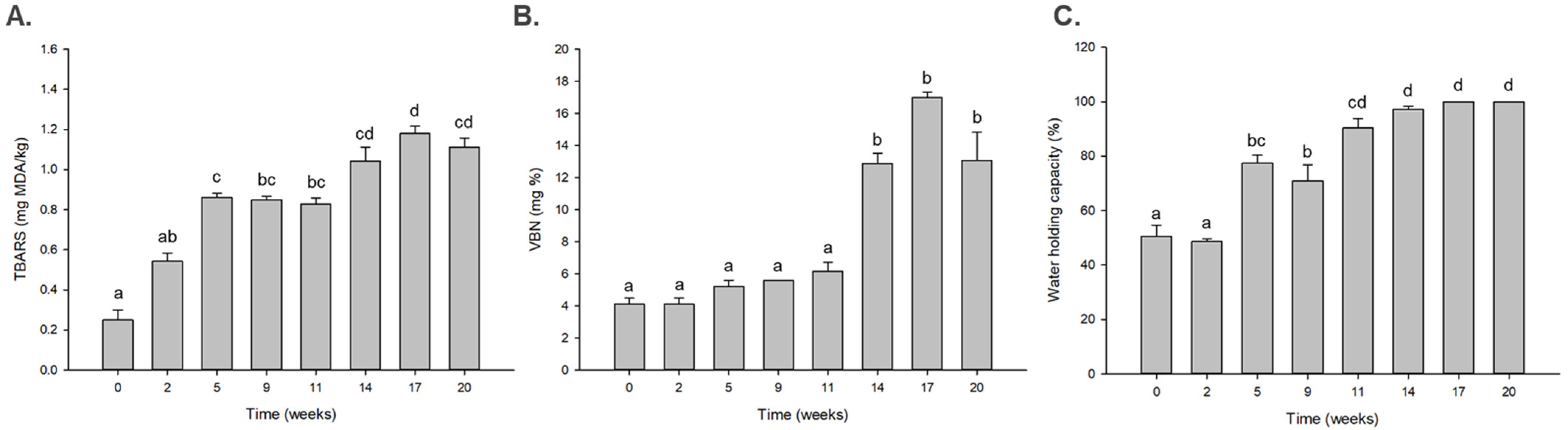
3.1. Changes in the Level of Quality Factors during Dry Aging of Beef
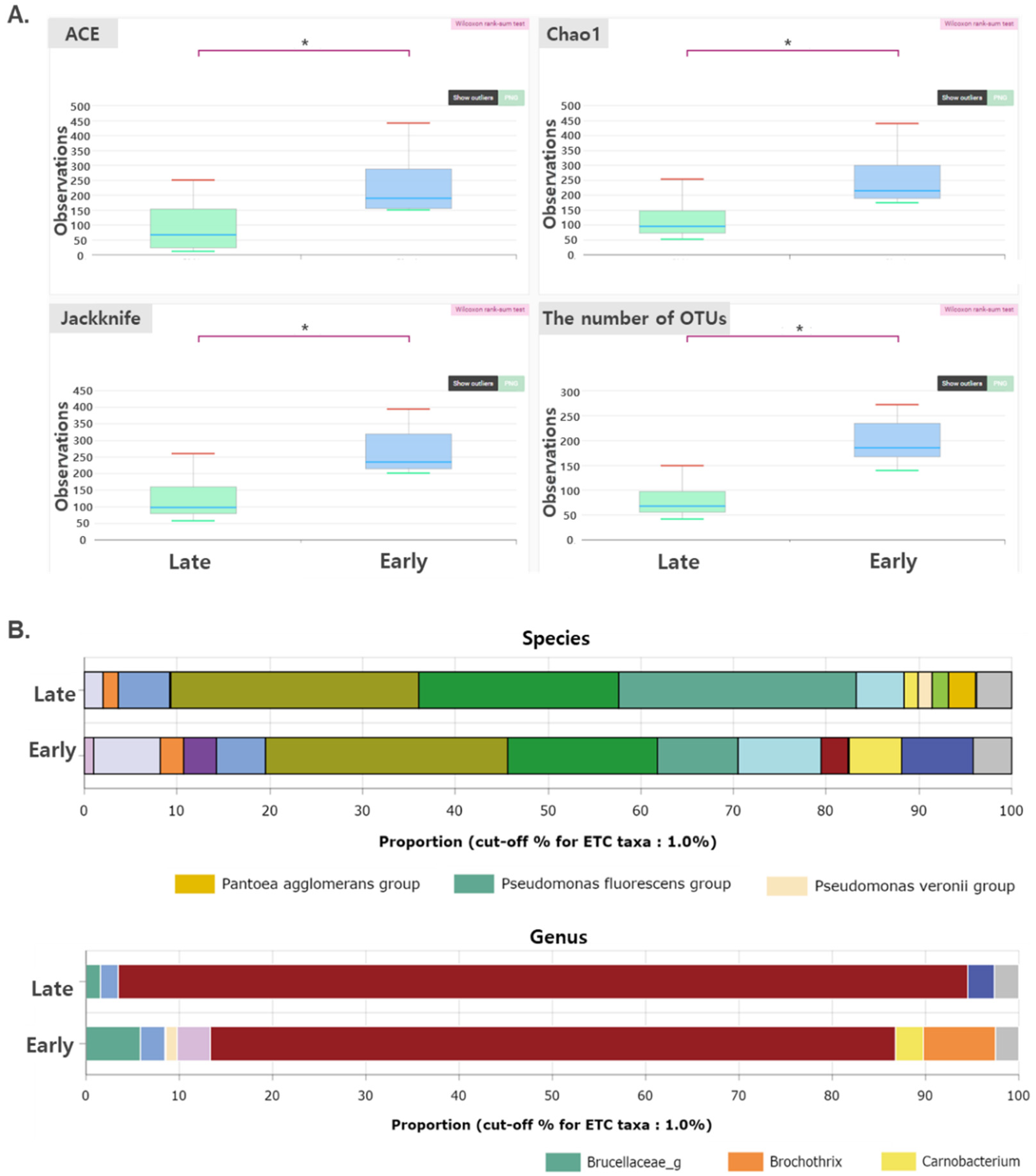
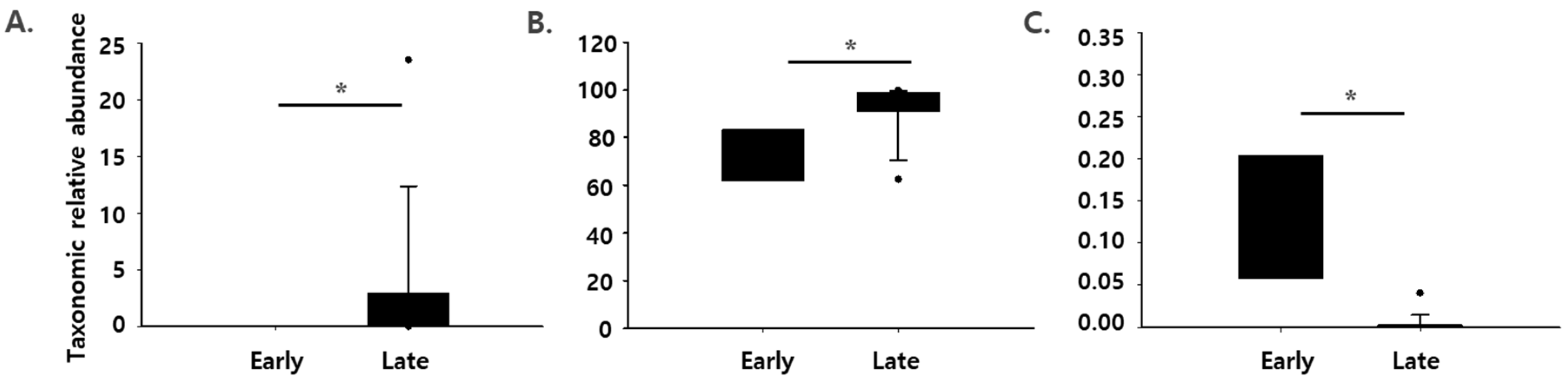
3.2. Changes in the Composition of Microflora during Dry Aging of Beef
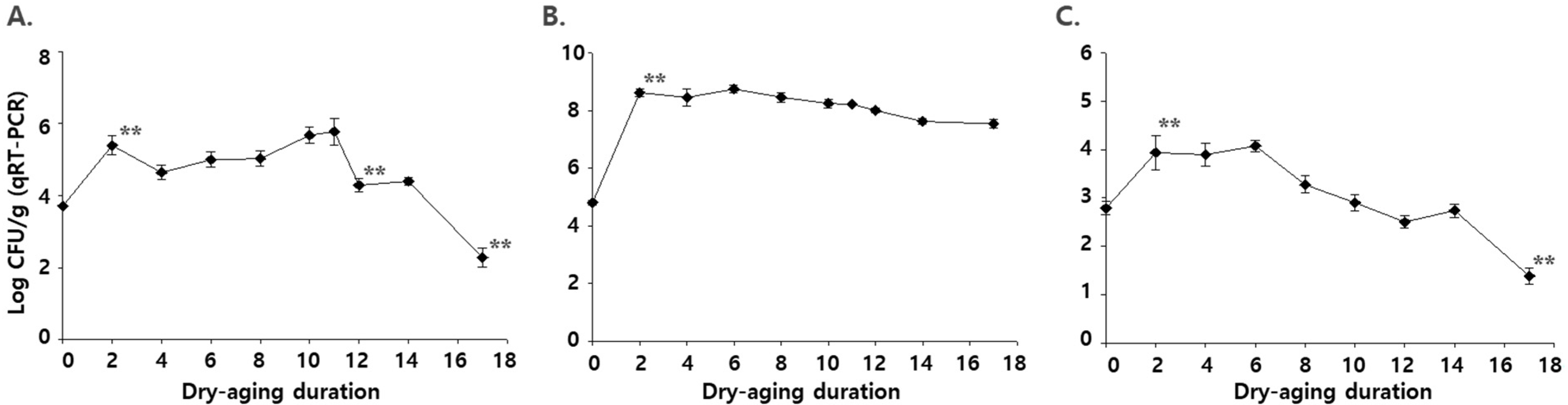
3.3. Level of the Microbes Quantified Using Real-Time q-PCR (qRT-PCR) during Dry Aging in Beef
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kim, Y.H.B.; Kemp, R.; Samuelsson, L.M. Effects of dry-aging on meat quality attributes and metabolite profiles of beef Loins. Meat Sci. 2016, 111, 168–176. [Google Scholar] [CrossRef]
- Allied Market Research. U.S. Dry Aging Beef Market—Opportunity Analysis and Industry Forecast, 2014–2020. Available online: https://www.alliedmarketresearch.com/US-dry-aging-beef-market (accessed on 5 November 2021).
- Ashaman, H.; Hastie, M.; Warner, R.; Jacob, R.; Hunyh, L. Dry-Aging—Introduction and Insights. Available online: https://www.mla.com.au/globalassets/mla-corporate/news-and-events/documents/dry-ageing-meat/introduction-and-market-insights (accessed on 5 November 2021).
- Ryu, S.; Shin, M.; Cho, S.; Hwang, I.; Kim, Y.; Oh, S. Microbial and fungal communities on dry aged beef of Hanwoo using metagenomic analysis. Foods 2020, 9, 1571. [Google Scholar] [CrossRef] [PubMed]
- Casaburi, A.; Piombino, P.; Nychas, G.J.; Villani, F.; Ercolini, D. Bacterial populations and the volatilome associated to meat spoilage. Food Microbiol. 2015, 45, 83–102. [Google Scholar] [CrossRef]
- Terkimg, N.; Witte, F.; Heinz, V. The dry aged beef paradox: Why dry aging is sometimes not better than wet aging. Meat Sci. 2021, 172, 108355. [Google Scholar]
- Raposo, A.; Perez, E.; de Faria, C.T.; Ferrus, M.A.; Carrascosa, C. Chapter 3—Food Spoilage by Pseudomonas spp. An Overview. In Foodborne Pathogens and Antibiotic Resistance; Singh, O.V., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 21–39. [Google Scholar]
- Nychas, G.-J.E.; Marshall, D.; Sofos, J. Chapter 6—Meat Poultry and Seafood. In Food Microbiology Fundamentals and Frontiers; Doyle, M.P., Beuchat, L.R., Montville, T.J., Eds.; ASM Press: New York, NY, USA, 2007. [Google Scholar]
- Gill, C.O.; Newton, K.G. The ecology of bacterial spoilage of fresh meat at chill temperatures. Meat Sci. 1978, 2, 207–217. [Google Scholar] [CrossRef]
- Lee, H.J.; Yoon, J.W.; Kim, M.; Oh, H.; Yoon, Y.; Jo, C. Changes in microbial composition on the crust by different air flow velocities and their effect on sensory properties of dry aged beef. Meat Sci. 2019, 153, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Lee, H.J.; Lee, J.; Jo, C.; Yoon, Y. Identification of microorganisms associated with the quality improvement of dry aged beef through microbiome analysis and DNA sequencing, and evaluation of their effects on beef quality. J. Food Sci. 2019, 84, 2944–2954. [Google Scholar] [CrossRef] [PubMed]
- Dave, D.; Ghaly, A.E. Meat spoilage mechanisms and preservation techniques: A critical review. Am. J. Agric. Biol. Sci. 2011, 6, 486–510. [Google Scholar]
- Byun, J.S.; Min, J.S.; Kim, I.S.; Chung, M.S.; Lee, M. Comparison of indicators of microbial quality of meat during aerobic cold storage. J. Food Prot. 2003, 66, 1733–1737. [Google Scholar] [CrossRef]
- Borch, E.; Kant-Muermans, M.L.; Blixt, Y. Bacterial spoilage of meat and cured meat products. Int. J. Food Microbiol. 1996, 33, 103–120. [Google Scholar] [CrossRef]
- Govindarajulu, S.N.; Varier, K.M.; Jayamurali, D.; Liu, W.; Chen, J.; Manoharan, N.; Li, Y.; Gajendran, B. Chapter 16—Insect Gut Microbiome and Its Applications. In Recent Advancements in Microbial Diversity; De Mandal, S., Bhatt, P., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 379–395. [Google Scholar]
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics—A guide from sampling to data analysis. Microb. Inform. Exp. 2012, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Neelakanta, G.; Sultana, H. The use of metagenomic approaches to analyze changes in microbial communities. Microbiol. Insights 2013, 6, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Gao, X.; Meng, J.; Zhang, A.; Zhou, Y.; Long, M.; Li, B.; Deng, W.; Jin, L.; Zhao, S.; et al. Metagenomic analysis of bacteria, fungi, bacteriophages, and helminths in the gut of giant pandas. Front. Microbiol. 2018, 9, 1717. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Xu, S.; Ren, Z.; Tao, L.; Jiang, J.; Zheng, S. Application of metagenomics in the human gut microbiome. World J. Gastroenterol. 2015, 21, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Bergmark, L.; Poulsen, P.H.B.; Al-Soud, W.A.; Norman, A.; Hansen, L.H.; Sørensen, S.J. Assessment of the specificity of Burkholderia and Pseudomonas qPCR assays for detection of these genera in soil using 454 pyrosequencing. FEMS Microbiol. Lett. 2012, 333, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Chumponsuk, T.; Jaroensuk, S.; Phengkhot, S.; Gentekaki, E.; Popluechai, S.; Kullawong, N. Development of genus-specific primers for quantitative PCR analysis of Streptococcus in human feces. In Proceeding of the 29th Annual Meeting of the Thai Society for Biotechnology and International Conference, Bangkok, Thailand, 23–25 November 2017. [Google Scholar]
- Dolan, A.; Burgess, C.M.; Barry, T.B.; Fanning, S.; Duffy, G. A novel quantitative reverse-transcription PCR (qRT-PCR) for the enumeration of total bacteria, using meat micro-flora as a model. J. Microbiol. Methods 2009, 77, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Witte, V.C.; Krause, G.F.; Bailey, M.E. A new extraction method for determining 2-thiobarbituric acid values of pork and beef during storage. J. Food Sci. 1970, 35, 582–585. [Google Scholar] [CrossRef]
- Conway, E.J.; Byrne, A. An absorption apparatus for the micro-determination of certain volatile substances: The micro-determination of ammonia. Biochem. J. 1933, 27, 419–429. [Google Scholar]
- Jay, J.M. Mechanism and detection of microbial spoilage in meats at low temperatures: A status report. J. Milk Food Technol. 1972, 35, 467–471. [Google Scholar] [CrossRef]
- Li, M.Y.; Zhou, G.H.; Xu, X.L.; Li, C.B.; Zhu, W.Y. Changes of bacterial diversity and main flora in chilled pork during storage using PCR-DGGE. Food Micobiol. 2006, 23, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Dainty, R.H.; Mackey, B.M. The relationship between the phenotypic properties of bacteria from chill-stored meat and spoilage processes. J. Appl. Microbiol. 1992, 73, 103S–114S. [Google Scholar] [CrossRef] [PubMed]
- Ercolini, D.; Russo, F.; Torrieri, E.; Masi, P.; Villani, F. Changes in the spoilage-related microbiota of beef during refrigerated storage under different packaging conditions. Appl. Environ. Microbiol. 2006, 72, 4663–4671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Cai, L.; Gao, T.; Li, C.; Zhou, G.; Ye, K. Comparing the quality characteristics and bacterial communities in meatballs with or without blown pack spoilage. LWT 2020, 130, 109529. [Google Scholar] [CrossRef]
- Oh, H.; Kim, S.; Lee, S.; Lee, H.; Ha, J.; Lee, J.; Choi, Y.; Choi, K.H.; Yoon, Y. Prevalence, serotype diversity, genotype and antibiotic resistance of Listeria monocytogenes isolated from carcasses and human in Korea. Food Sci. Anim. Resour. 2018, 38, 851–865. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Primer (3′–5′) | Target Gene | Reference |
|---|---|---|---|
| Pantoea spp. | F: CACTGGAAACGGTGGCTAAT | 16S rRNA | This study |
| R: CTGGGTTCATCCGATAGTGAG | |||
| Pseudomonas spp. | F: ACTTTAAGTTGGGAGGAAGGG R: ACACAGGAAATTCCACCACCC | 16S rRNA | [20] |
| Streptococcus spp. | F: CGATACATAGCCGACCTGAGA R: CCACTCTCCCCTYYTGCAC | 16S rRNA | [21] |
| Universal bacteria | |||
| Gram-positive | F: GAAAGTCCGGGCTCCATA R: ATAAGCCGGGTTCTGT | mp(G–) | [22] |
| Gram-negative | F: GAGGAAATCCRKGCTCGCAC R: AGGGGTTTACCGCGTTCC | mp(G+) | [22] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Kim, J.-C.; Park, S.; Kim, J.; Yoon, Y.; Lee, H. Identification of Microbial Flora in Dry Aged Beef to Evaluate the Rancidity during Dry Aging. Processes 2021, 9, 2049. https://doi.org/10.3390/pr9112049
Kim S, Kim J-C, Park S, Kim J, Yoon Y, Lee H. Identification of Microbial Flora in Dry Aged Beef to Evaluate the Rancidity during Dry Aging. Processes. 2021; 9(11):2049. https://doi.org/10.3390/pr9112049
Chicago/Turabian StyleKim, Sejeong, Jong-Chan Kim, Sunhyun Park, Jinkwi Kim, Yohan Yoon, and Heeyoung Lee. 2021. "Identification of Microbial Flora in Dry Aged Beef to Evaluate the Rancidity during Dry Aging" Processes 9, no. 11: 2049. https://doi.org/10.3390/pr9112049
APA StyleKim, S., Kim, J. -C., Park, S., Kim, J., Yoon, Y., & Lee, H. (2021). Identification of Microbial Flora in Dry Aged Beef to Evaluate the Rancidity during Dry Aging. Processes, 9(11), 2049. https://doi.org/10.3390/pr9112049