
Multi-Enzyme Systems in Flow Chemistry



Abstract
:1. Introduction
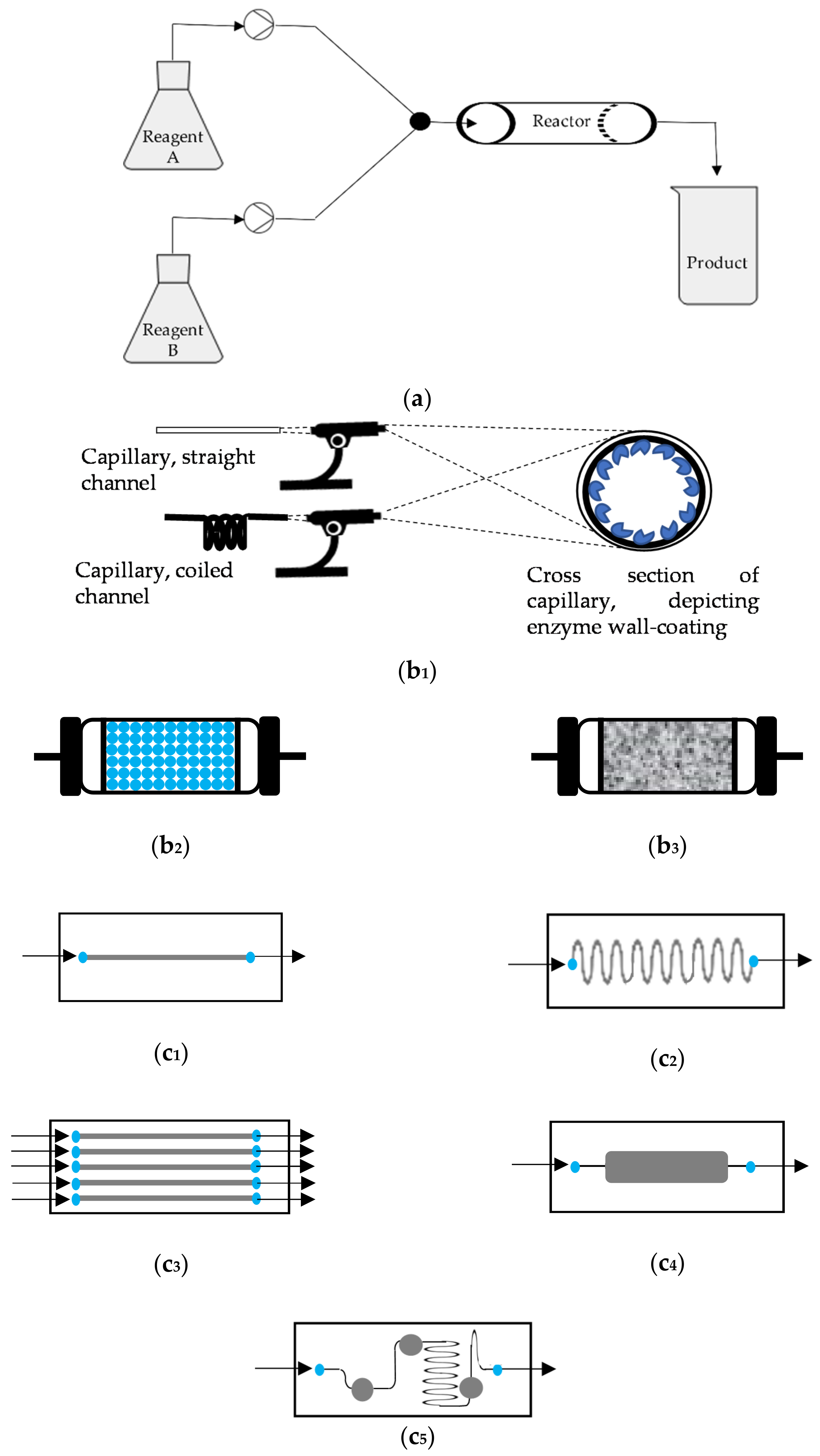
2. Flow Bioreactors
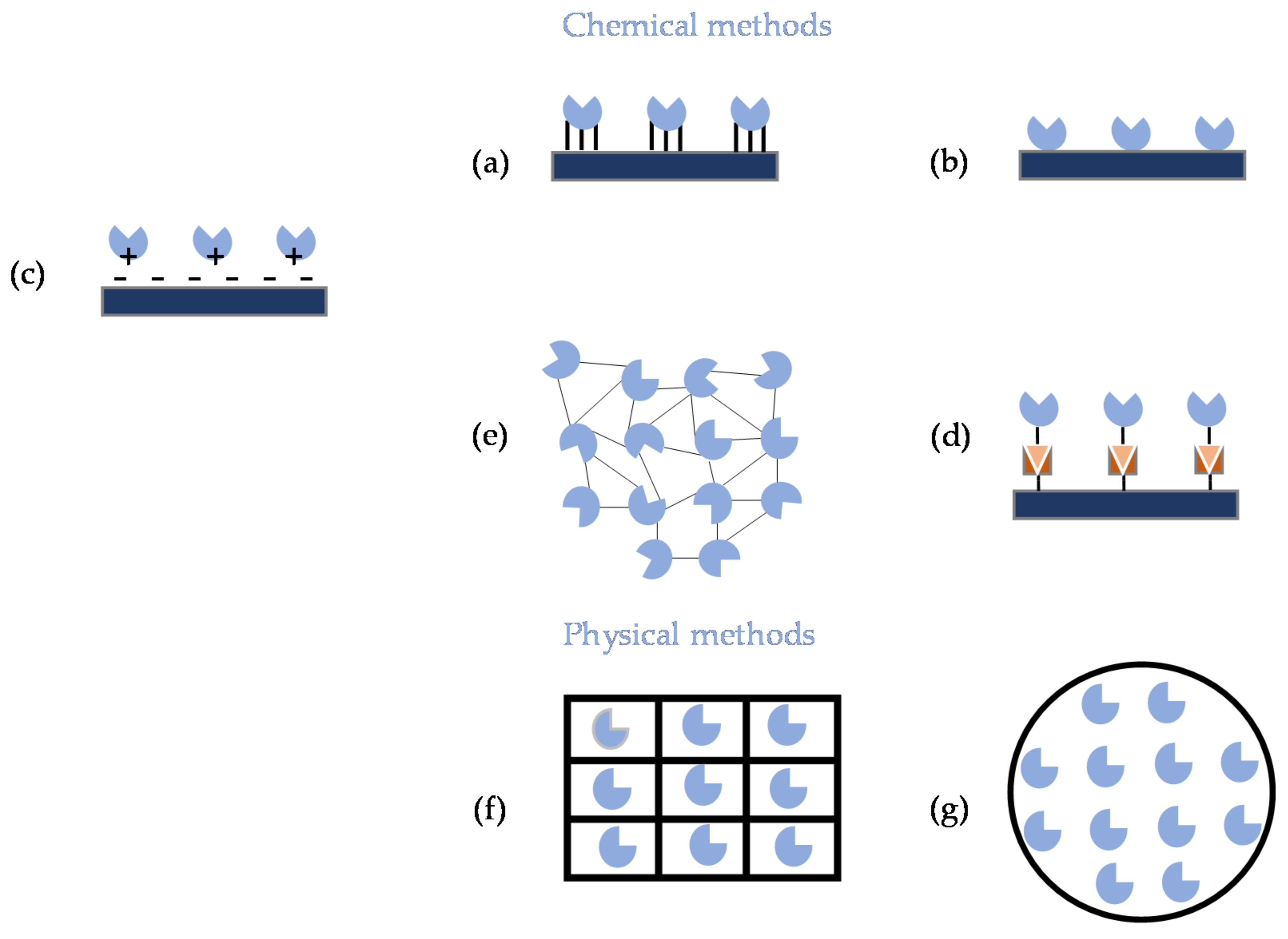
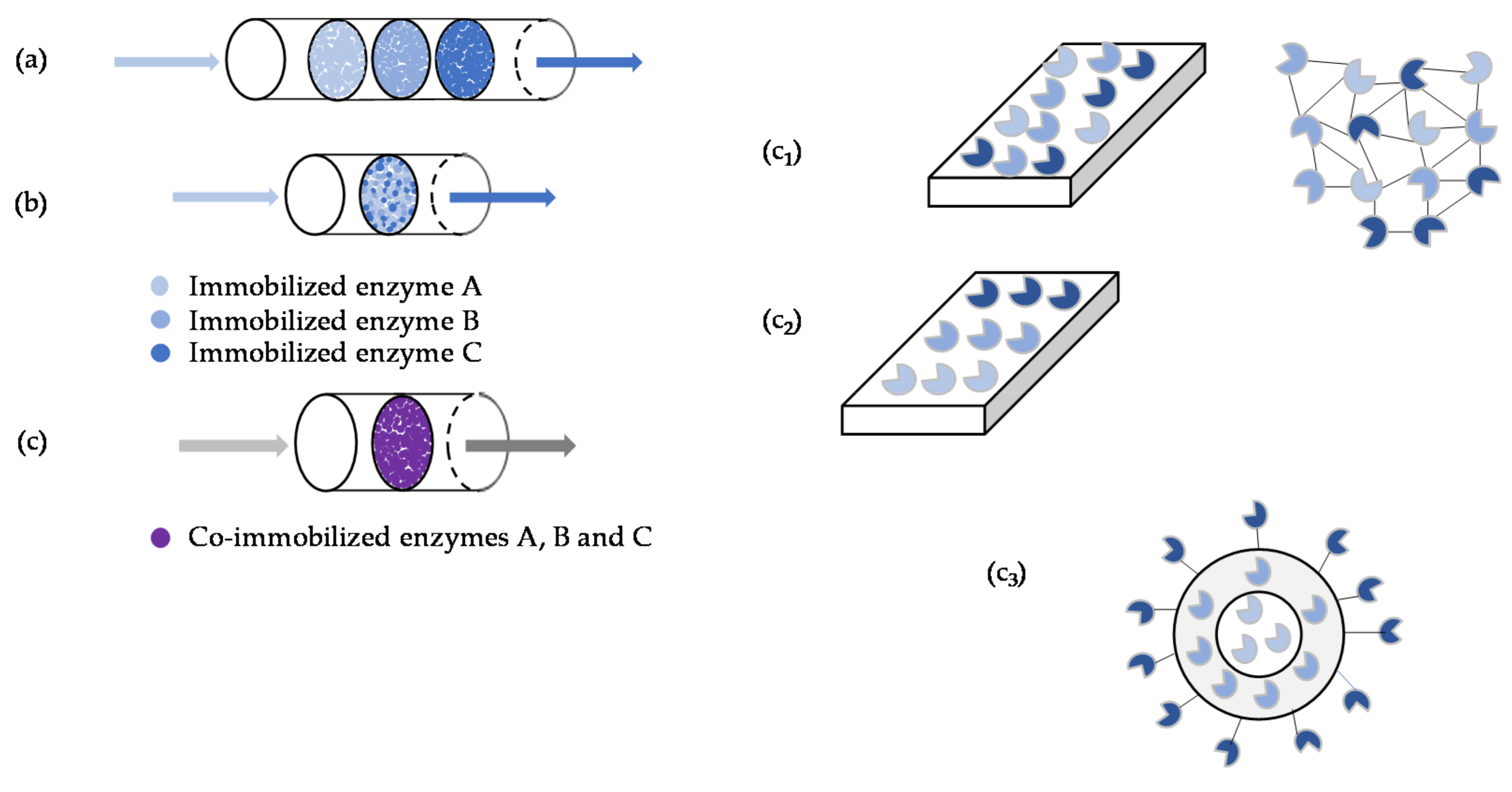
2.1. Immobilization
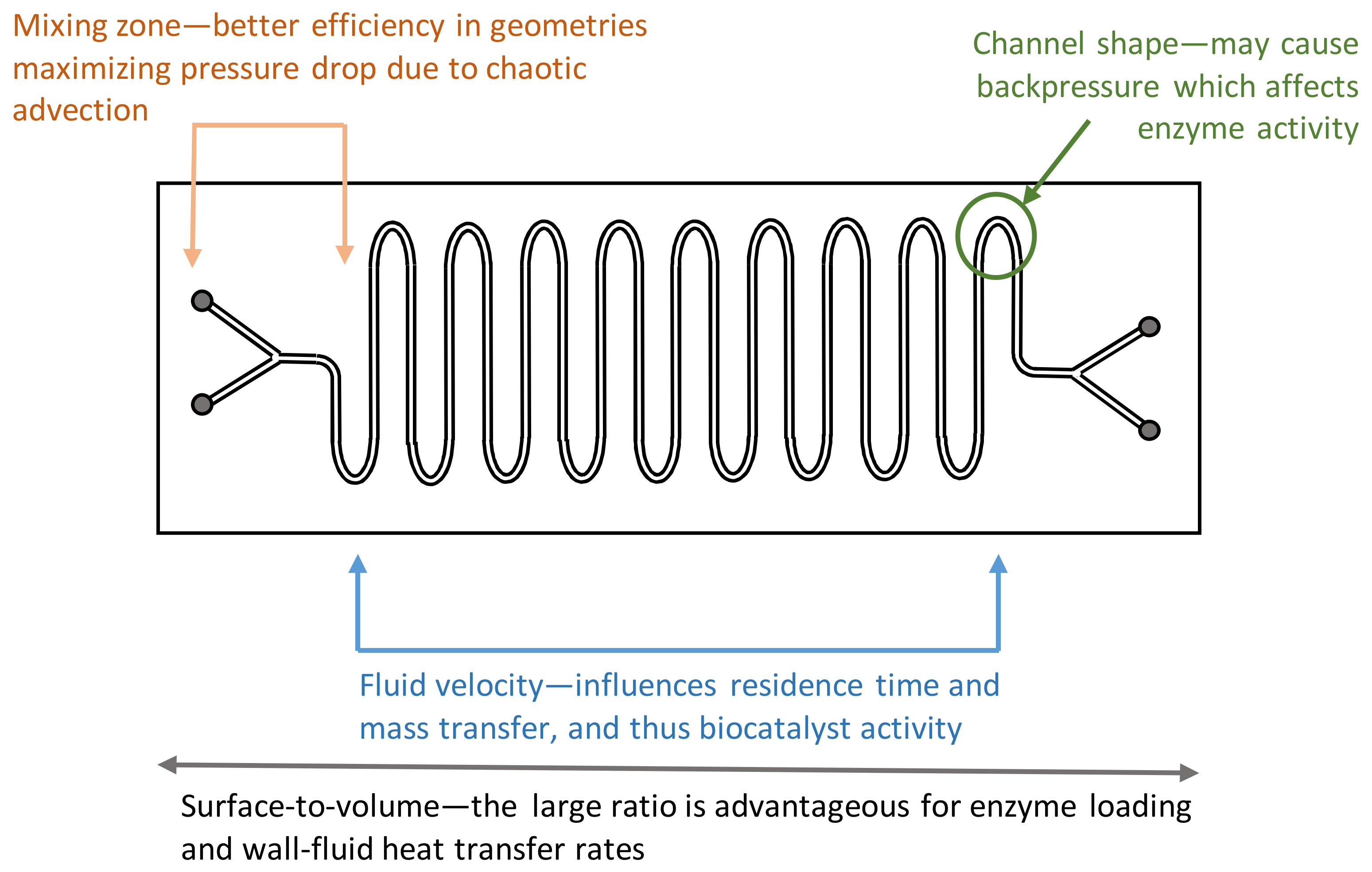
2.2. Engineering Aspects
3. Reactions with Whole Cells
4. Chemoenzymatic Reactions
5. Implementation Challenges at Processing Scale
6. Final Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Velasco-Lozano, S.; López-Gallego, F. Wiring step-wise reactions with immobilized multi-enzyme systems. Biocatal. Biotransform. 2018, 36, 184–194. [Google Scholar] [CrossRef]
- Schoffelen, S.; van Hest, J.C.M. Multi-enzyme systems: Bringing enzymes together in vitro. Soft Matter 2012, 8, 1736–1746. [Google Scholar] [CrossRef]
- Gruber, P.; Marques, M.P.C.; O’Sullivan, B.; Baganz, F.; Wohlgemuth, R.; Szita, N. Conscious coupling: The challenges and opportunities of cascading enzymatic microreactors. Biotechnol. J. 2017, 12, 1700030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basso, A.; Serban, S. Industrial applications of immobilized enzymes—A review. Mol. Catal. 2019, 479, 110607. [Google Scholar] [CrossRef]
- de Carvalho, C.C.C.R. Whole cell biocatalysts: Essential workers from Nature to the industry. Microb. Biotechnol. 2017, 10, 250–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polakovič, M.; Švitel, J.; Bučko, M.; Filip, J.; Neděla, V.; Ansorge-Schumacher, M.B.; Gemeiner, P. Progress in biocatalysis with immobilized viable whole cells: Systems development, reaction engineering and applications. Biotechnol. Lett. 2017, 39, 667–683. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, C.C.C.R.; da Fonseca, M.M.R. Biotransformations. In Comprehensive Biotechnology, 3rd ed.; Moo-Young, M., Ed.; Pergamon: Oxford, UK, 2017; pp. 574–585. [Google Scholar]
- Gross, R.; Buehler, K.; Schmid, A. Engineered catalytic biofilms for continuous large scale production of n-octanol and (S)-styrene oxide. Biotechnol. Bioeng. 2013, 110, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Todhanakasem, T.; Salangsing, O.-L.; Koomphongse, P.; Kaewket, S.; Kanokratana, P.; Champreda, V. Zymomonas mobilis Biofilm Reactor for Ethanol Production Using Rice Straw Hydrolysate Under Continuous and Repeated Batch Processes. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, B.; Tao, Y. Whole-cell biocatalysts by design. Microb. Cell Factories 2017, 16, 106. [Google Scholar] [CrossRef] [Green Version]
- Ro, D.-K.; Paradise, E.M.; Ouellet, M.; Fisher, K.J.; Newman, K.L.; Ndungu, J.M.; Ho, K.A.; Eachus, R.A.; Ham, T.S.; Kirby, J.; et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 2006, 440, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Tamborini, L.; Fernandes, P.; Paradisi, F.; Molinari, F. Flow Bioreactors as Complementary Tools for Biocatalytic Process Intensification. Trends Biotechnol. 2018, 36, 73–88. [Google Scholar] [CrossRef] [PubMed]
- De Vitis, V.; Dall’Oglio, F.; Tentori, F.; Contente, M.L.; Romano, D.; Brenna, E.; Tamborini, L.; Molinari, F. Bioprocess Intensification Using Flow Reactors: Stereoselective Oxidation of Achiral 1,3-diols with Immobilized Acetobacter aceti. Catalysts 2019, 9, 208. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Chen, Q.; Shao, L.; Jia, Y.; Zhang, X. Microfluidic immobilized enzyme reactors for continuous biocatalysis. React. Chem. Eng. 2020, 5, 9–32. [Google Scholar] [CrossRef]
- Britton, J.; Majumdar, S.; Weiss, G.A. Continuous flow biocatalysis. Chem. Soc. Rev. 2018, 47, 5891–5918. [Google Scholar] [CrossRef]
- De Santis, P.; Meyer, L.-E.; Kara, S. The rise of continuous flow biocatalysis—Fundamentals, very recent developments and future perspectives. React. Chem. Eng. 2020, 5, 2155–2184. [Google Scholar] [CrossRef]
- Wohlgemuth, R.; Plazl, I.; Žnidaršič-Plazl, P.; Gernaey, K.V.; Woodley, J.M. Microscale technology and biocatalytic processes: Opportunities and challenges for synthesis. Trends Biotechnol. 2015, 33, 302–314. [Google Scholar] [CrossRef]
- Meller, K.; Szumski, M.; Buszewski, B. Microfluidic reactors with immobilized enzymes—Characterization, dividing, perspectives. Sens. Actuators B Chem. 2017, 244, 84–106. [Google Scholar] [CrossRef]
- Arshi, S.; Nozari-Asbemarz, M.; Magner, E. Enzymatic Bioreactors: An Electrochemical Perspective. Catalysts 2020, 10, 1232. [Google Scholar] [CrossRef]
- Britton, J.; Dyer, R.P.; Majumdar, S.; Raston, C.L.; Weiss, G.A. Ten-Minute Protein Purification and Surface Tethering for Continuous-Flow Biocatalysis. Angew. Chem. Int. Ed. 2017, 56, 2296–2301. [Google Scholar] [CrossRef]
- Boehm, C.R.; Freemont, P.S.; Ces, O. Design of a prototype flow microreactor for synthetic biology in vitro. Lab A Chip 2013, 13, 3426–3432. [Google Scholar] [CrossRef] [PubMed]
- Döbber, J.; Gerlach, T.; Offermann, H.; Rother, D.; Pohl, M. Closing the gap for efficient immobilization of biocatalysts in continuous processes: HaloTag™ fusion enzymes for a continuous enzymatic cascade towards a vicinal chiral diol. Green Chem. 2018, 20, 544–552. [Google Scholar] [CrossRef]
- Roura Padrosa, D.; Benítez-Mateos, A.I.; Calvey, L.; Paradisi, F. Cell-free biocatalytic syntheses of L-pipecolic acid: A dual strategy approach and process intensification in flow. Green Chem. 2020, 22, 5310–5316. [Google Scholar] [CrossRef]
- Poppe, J.K.; Matte, C.R.; de Freitas, V.O.; Fernandez-Lafuente, R.; Rodrigues, R.C.; Záchia Ayub, M.A. Enzymatic synthesis of ethyl esters from waste oil using mixtures of lipases in a plug-flow packed-bed continuous reactor. Biotechnol. Prog. 2018, 34, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Brás, E.J.S.; Domingues, C.; Chu, V.; Fernandes, P.; Conde, J.P. Microfluidic bioreactors for enzymatic synthesis in packed-bed reactors—Multi-step reactions and upscaling. J. Biotechnol. 2020, 323, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Logan, T.C.; Clark, D.S.; Stachowiak, T.B.; Svec, F.; Fréchet, J.M.J. Photopatterning Enzymes on Polymer Monoliths in Microfluidic Devices for Steady-State Kinetic Analysis and Spatially Separated Multi-Enzyme Reactions. Anal. Chem. 2007, 79, 6592–6598. [Google Scholar] [CrossRef]
- Yin, J.; Xu, T.; Zhang, N.; Wang, H. Three-Enzyme Cascade Bioreactor for Rapid Digestion of Genomic DNA into Single Nucleosides. Anal. Chem. 2016, 88, 7730–7737. [Google Scholar] [CrossRef]
- Fornera, S.; Kuhn, P.; Lombardi, D.; Schlüter, A.D.; Dittrich, P.S.; Walde, P. Sequential Immobilization of Enzymes in Microfluidic Channels for Cascade Reactions. ChemPlusChem 2012, 77, 98–101. [Google Scholar] [CrossRef]
- Rodrigues, R.C.; Ortiz, C.; Berenguer-Murcia, Á.; Torres, R.; Fernández-Lafuente, R. Modifying enzyme activity and selectivity by immobilization. Chem. Soc. Rev. 2013, 42, 6290–6307. [Google Scholar] [CrossRef]
- Romero-Fernández, M.; Paradisi, F. Protein immobilization technology for flow biocatalysis. Curr. Opin. Chem. Biol. 2020, 55, 1–8. [Google Scholar] [CrossRef]
- Bommarius, A.S.; Paye, M.F. Stabilizing biocatalysts. Chem. Soc. Rev. 2013, 42, 6534–6565. [Google Scholar] [CrossRef]
- Chen, R.; Wei, Q.; Wei, X.; Liu, Y.; Zhang, X.; Chen, X.; Yin, X.; Xie, T. Stable and efficient immobilization of bi-enzymatic NADPH cofactor recycling system under consecutive microwave irradiation. PLoS ONE 2020, 15, e0242564. [Google Scholar] [CrossRef] [PubMed]
- Peschke, T.; Bitterwolf, P.; Hansen, S.; Gasmi, J.; Rabe, K.S.; Niemeyer, C.M. Self-Immobilizing Biocatalysts Maximize Space–Time Yields in Flow Reactors. Catalysts 2019, 9, 164. [Google Scholar] [CrossRef] [Green Version]
- Sánta-Bell, E.; Molnár, Z.; Varga, A.; Nagy, F.; Hornyánszky, G.; Paizs, C.; Balogh-Weiser, D.; Poppe, L. “Fishing and Hunting”—Selective Immobilization of a Recombinant Phenylalanine Ammonia-Lyase from Fermentation Media. Molecules 2019, 24, 4146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamad, N.R.; Marzuki, N.H.C.; Buang, N.A.; Huyop, F.; Wahab, R.A. An overview of technologies for immobilization of enzymes and surface analysis techniques for immobilized enzymes. Biotechnol. Biotechnol. Equip. 2015, 29, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Küchler, A.; Yoshimoto, M.; Luginbühl, S.; Mavelli, F.; Walde, P. Enzymatic reactions in confined environments. Nat. Nanotechnol. 2016, 11, 409–420. [Google Scholar] [CrossRef]
- Xu, K.; Chen, X.; Zheng, R.; Zheng, Y. Immobilization of Multi-Enzymes on Support Materials for Efficient Biocatalysis. Front. Bioeng. Biotechnol. 2020, 8, 660. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.-Q.; Wang, S.-S.; Li, L.-N.; Gao, J.; Zhang, Y.-W. Combined Cross-Linked Enzyme Aggregates as Biocatalysts. Catalysts 2018, 8, 460. [Google Scholar] [CrossRef] [Green Version]
- Sheldon, R.A. CLEAs, Combi-CLEAs and ‘Smart’ Magnetic CLEAs: Biocatalysis in a Bio-Based Economy. Catalysts 2019, 9, 261. [Google Scholar] [CrossRef] [Green Version]
- Homaei, A.A.; Sariri, R.; Vianello, F.; Stevanato, R. Enzyme immobilization: An update. J. Chem. Biol. 2013, 6, 185–205. [Google Scholar] [CrossRef] [Green Version]
- Sirisha, V.L.; Jain, A.; Jain, A. Chapter Nine—Enzyme Immobilization: An Overview on Methods, Support Material, and Applications of Immobilized Enzymes. In Advances in Food and Nutrition Research; Kim, S.-K., Toldrá, F., Eds.; Academic Press: Cambridge, MA, USA, 2016; Volume 79, pp. 179–211. [Google Scholar]
- Zdarta, J.; Meyer, A.S.; Jesionowski, T.; Pinelo, M. A General Overview of Support Materials for Enzyme Immobilization: Characteristics, Properties, Practical Utility. Catalysts 2018, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Taheri-Kafrani, A.; Kharazmi, S.; Nasrollahzadeh, M.; Soozanipour, A.; Ejeian, F.; Etedali, P.; Mansouri-Tehrani, H.-A.; Razmjou, A.; Yek, S.M.-G.; Varma, R.S. Recent developments in enzyme immobilization technology for high-throughput processing in food industries. Crit. Rev. Food Sci. Nutr. 2020, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Kazenwadel, F.; Franzreb, M.; Rapp, B.E. Synthetic enzyme supercomplexes: Co-immobilization of enzyme cascades. Anal. Methods 2015, 7, 4030–4037. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Wu, Y.; Zhang, S.; Tian, Y.; Yang, D.; Jiang, Z. Bioinspired construction of multi-enzyme catalytic systems. Chem. Soc. Rev. 2018, 47, 4295–4313. [Google Scholar] [CrossRef] [PubMed]
- Hwang, E.T.; Lee, S. Multienzymatic Cascade Reactions via Enzyme Complex by Immobilization. ACS Catal. 2019, 9, 4402–4425. [Google Scholar] [CrossRef]
- Ren, S.; Li, C.; Jiao, X.; Jia, S.; Jiang, Y.; Bilal, M.; Cui, J. Recent progress in multienzymes co-immobilization and multienzyme system applications. Chem. Eng. J. 2019, 373, 1254–1278. [Google Scholar] [CrossRef]
- Giannakopoulou, A.; Gkantzou, E.; Polydera, A.; Stamatis, H. Multienzymatic Nanoassemblies: Recent Progress and Applications. Trends Biotechnol. 2020, 38, 202–216. [Google Scholar] [CrossRef]
- Arana-Peña, S.; Carballares, D.; Morellon-Sterlling, R.; Berenguer-Murcia, Á.; Alcántara, A.R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Enzyme co-immobilization: Always the biocatalyst designers’ choice…or not? Biotechnol. Adv. 2020, 107584. [Google Scholar] [CrossRef]
- Ji, Q.; Wang, B.; Tan, J.; Zhu, L.; Li, L. Immobilized multienzymatic systems for catalysis of cascade reactions. Process Biochem. 2016, 51, 1193–1203. [Google Scholar] [CrossRef]
- Brena, B.; González-Pombo, P.; Batista-Viera, F. Immobilization of Enzymes: A Literature Survey. In Immobilization of Enzymes and Cells, 3rd ed.; Guisan, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 15–31. [Google Scholar]
- Wahab, R.A.; Elias, N.; Abdullah, F.; Ghoshal, S.K. On the taught new tricks of enzymes immobilization: An all-inclusive overview. React. Funct. Polym. 2020, 152, 104613. [Google Scholar] [CrossRef]
- Mateo, C.; Fernandez-Lorente, G.; Rocha-Martin, J.; Bolivar, J.M.; Guisan, J.M. Oriented Covalent Immobilization of Enzymes on Heterofunctional-Glyoxyl Supports. In Immobilization of Enzymes and Cells, 3rd ed.; Guisan, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 73–88. [Google Scholar]
- Rodriguez-Abetxuko, A.; Sánchez-deAlcázar, D.; Muñumer, P.; Beloqui, A. Tunable Polymeric Scaffolds for Enzyme Immobilization. Front. Bioeng. Biotechnol. 2020, 8, 830. [Google Scholar] [CrossRef]
- Guisan, J.M.; López-Gallego, F.; Bolivar, J.M.; Rocha-Martín, J.; Fernandez-Lorente, G. The Science of Enzyme Immobilization. In Immobilization of Enzymes and Cells: Methods and Protocols; Guisan, J.M., Bolivar, J.M., López-Gallego, F., Rocha-Martín, J., Eds.; Springer: New York, NY, USA, 2020; pp. 1–26. [Google Scholar]
- Wu, J.C.Y.; Hutchings, C.H.; Lindsay, M.J.; Werner, C.J.; Bundy, B.C. Enhanced Enzyme Stability through Site-Directed Covalent Immobilization. J. Biotechnol. 2015, 193, 83–90. [Google Scholar] [CrossRef]
- Li, Y.; Ogorzalek, T.L.; Wei, S.; Zhang, X.; Yang, P.; Jasensky, J.; Brooks, C.L.; Marsh, E.N.G.; Chen, Z. Effect of immobilization site on the orientation and activity of surface-tethered enzymes. Phys. Chem. Chem. Phys. 2018, 20, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Meyer, A.S.; Mateiu, R.V.; Pinelo, M. Cascade catalysis in membranes with enzyme immobilization for multi-enzymatic conversion of CO2 to methanol. New Biotechnol. 2015, 32, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Küchler, A.; Adamcik, J.; Mezzenga, R.; Schlüter, A.D.; Walde, P. Enzyme immobilization on silicate glass through simple adsorption of dendronized polymer–enzyme conjugates for localized enzymatic cascade reactions. RSC Adv. 2015, 5, 44530–44544. [Google Scholar] [CrossRef]
- Torres, R.; Pessela, B.C.C.; Mateo, C.; Ortiz, C.; Fuentes, M.; Guisan, J.M.; Fernandez-Lafuente, R. Reversible Immobilization of Glucoamylase by Ionic Adsorption on Sepabeads Coated with Polyethyleneimine. Biotechnol. Prog. 2004, 20, 1297–1300. [Google Scholar] [CrossRef] [PubMed]
- Hanefeld, U.; Gardossi, L.; Magner, E. Understanding enzyme immobilisation. Chem. Soc. Rev. 2009, 38, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Ngo, T.T.; Lenhoff, H.M.; Ivy, J. Biotinyl-Glucose-6-Phosphate Dehydrogenase Preparation, Kinetics, and Modulation by Avidin. Appl. Biochem. Biotechnol. 1982, 7, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Peschke, T.; Bitterwolf, P.; Rabe, K.S.; Niemeyer, C.M. Self-Immobilizing Oxidoreductases for Flow Biocatalysis in Miniaturized Packed-Bed Reactors. Chem. Eng. Technol. 2019, 42, 2009–2017. [Google Scholar] [CrossRef]
- Plž, M.; Petrovičová, T.; Rebroš, M. Semi-Continuous Flow Biocatalysis with Affinity Co-Immobilized Ketoreductase and Glucose Dehydrogenase. Molecules 2020, 25, 4278. [Google Scholar] [CrossRef]
- Fornera, S.; Bauer, T.; Schlüter, A.D.; Walde, P. Simple enzyme immobilization inside glass tubes for enzymatic cascade reactions. J. Mater. Chem. 2012, 22, 502–511. [Google Scholar] [CrossRef]
- Vong, T.; Schoffelen, S.; van Dongen, S.F.M.; van Beek, T.A.; Zuilhof, H.; van Hest, J.C.M. A DNA-based strategy for dynamic positional enzyme immobilization inside fused silica microchannels. Chem. Sci. 2011, 2, 1278–1285. [Google Scholar] [CrossRef]
- Zhang, G.; Johnston, T.; Quin, M.B.; Schmidt-Dannert, C. Developing a Protein Scaffolding System for Rapid Enzyme Immobilization and Optimization of Enzyme Functions for Biocatalysis. ACS Synth. Biol. 2019, 8, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Valikhani, D.; Bolivar, J.M.; Viefhues, M.; McIlroy, D.N.; Vrouwe, E.X.; Nidetzky, B. A Spring in Performance: Silica Nanosprings Boost Enzyme Immobilization in Microfluidic Channels. ACS Appl. Mater. Interfaces 2017, 9, 34641–34649. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Han, P.; You, C. Facilitation of cascade biocatalysis by artificial multi-enzyme complexes—A review. Chin. J. Chem. Eng. 2020, 28, 2799–2809. [Google Scholar] [CrossRef]
- Sahutoglu, A.S.; Akgul, C. Immobilisation of Aspergillus oryzae α-amylase and Aspergillus niger glucoamylase enzymes as cross-linked enzyme aggregates. Chem. Pap. 2015, 69, 433–439. [Google Scholar] [CrossRef]
- De Martino, M.T.; Tonin, F.; Yewdall, N.A.; Abdelghani, M.; Williams, D.S.; Hanefeld, U.; Rutjes, F.P.J.T.; Abdelmohsen, L.K.E.A.; van Hest, J.C.M. Compartmentalized cross-linked enzymatic nano-aggregates (c-CLEnA) for efficient in-flow biocatalysis. Chem. Sci. 2020, 11, 2765–2769. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Gui, X.; Wang, G.; Yan, Y. Improving Stability and Activity of Cross-linked Enzyme Aggregates Based on Polyethylenimine in Hydrolysis of Fish Oil for Enrichment of Polyunsaturated Fatty Acids. Appl. Biochem. Biotechnol. 2012, 166, 925–932. [Google Scholar] [CrossRef]
- Wilson, L.; Illanes, A.; Pessela, B.C.C.; Abian, O.; Fernández-Lafuente, R.; Guisán, J.M. Encapsulation of crosslinked penicillin G acylase aggregates in lentikats: Evaluation of a novel biocatalyst in organic media. Biotechnol. Bioeng. 2004, 86, 558–562. [Google Scholar] [CrossRef]
- Noori, R.; Perwez, M.; Sardar, M. Cross-linked Enzyme Aggregates: Current Developments and Applications. In Biocatalysis: Enzymatic Basics and Applications; Husain, Q., Ullah, M.F., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 83–112. [Google Scholar]
- Talekar, S.; Pandharbale, A.; Ladole, M.; Nadar, S.; Mulla, M.; Japhalekar, K.; Pattankude, K.; Arage, D. Carrier free co-immobilization of alpha amylase, glucoamylase and pullulanase as combined cross-linked enzyme aggregates (combi-CLEAs): A tri-enzyme biocatalyst with one pot starch hydrolytic activity. Bioresour. Technol. 2013, 147, 269–275. [Google Scholar] [CrossRef]
- Perwez, M.; Ahmed Mazumder, J.; Sardar, M. Preparation and characterization of reusable magnetic combi-CLEA of cellulase and hemicellulase. Enzym. Microb. Technol. 2019, 131, 109389. [Google Scholar] [CrossRef]
- Chen, S.; Wen, L.; Svec, F.; Tan, T.; Lv, Y. Magnetic metal–organic frameworks as scaffolds for spatial co-location and positional assembly of multi-enzyme systems enabling enhanced cascade biocatalysis. RSC Adv. 2017, 7, 21205–21213. [Google Scholar] [CrossRef] [Green Version]
- Talekar, S.; Joshi, A.; Kambale, S.; Jadhav, S.; Nadar, S.; Ladole, M. A tri-enzyme magnetic nanobiocatalyst with one pot starch hydrolytic activity. Chem. Eng. J. 2017, 325, 80–90. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, D.; Wu, C.; Yao, K.; Li, Z.; Shi, N.; Wen, F.; Gates, I.D. Co-immobilization of cellulase and lysozyme on amino-functionalized magnetic nanoparticles: An activity-tunable biocatalyst for extraction of lipids from microalgae. Bioresour. Technol. 2018, 263, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulou, A.; Patila, M.; Spyrou, K.; Chalmpes, N.; Zarafeta, D.; Skretas, G.; Gournis, D.; Stamatis, H. Development of a Four-Enzyme Magnetic Nanobiocatalyst for Multi-Step Cascade Reactions. Catalysts 2019, 9, 995. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; He, Z.; Wang, X.; Zhang, Q.; Wei, Q.; Ma, S.; Ma, C.; Li, J.; Wang, Q. Cascade enzymes within self-assembled hybrid nanogel mimicked neutrophil lysosomes for singlet oxygen elevated cancer therapy. Nat. Commun. 2019, 10, 240. [Google Scholar] [CrossRef]
- Del Arco, J.; Alcántara, A.R.; Fernández-Lafuente, R.; Fernández-Lucas, J. Magnetic micro-macro biocatalysts applied to industrial bioprocesses. Bioresour. Technol. 2021, 322, 124547. [Google Scholar] [CrossRef]
- Zhong, X.; Xia, H.; Huang, W.; Li, Z.; Jiang, Y. Biomimetic metal-organic frameworks mediated hybrid multi-enzyme mimic for tandem catalysis. Chem. Eng. J. 2020, 381, 122758. [Google Scholar] [CrossRef]
- Ye, N.; Kou, X.; Shen, J.; Huang, S.; Chen, G.; Ouyang, G. Metal-Organic Frameworks: A New Platform for Enzyme Immobilization. Chembiochem 2020, 21, 2585–2590. [Google Scholar] [CrossRef]
- Cen, Y.-K.; Liu, Y.-X.; Xue, Y.-P.; Zheng, Y.-G. Immobilization of Enzymes in/on Membranes and their Applications. Adv. Synth. Catal. 2019, 361, 5500–5515. [Google Scholar] [CrossRef]
- Zhao, Z.; Fu, J.; Dhakal, S.; Johnson-Buck, A.; Liu, M.; Zhang, T.; Woodbury, N.W.; Liu, Y.; Walter, N.G.; Yan, H. Nanocaged enzymes with enhanced catalytic activity and increased stability against protease digestion. Nat. Commun. 2016, 7, 10619. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.-L.; Zhu, J.; Wheeldon, I. Synthetic Protein Scaffolds for Biosynthetic Pathway Colocalization on Lipid Droplet Membranes. ACS Synth. Biol. 2017, 6, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Eş, I.; Vieira, J.D.G.; Amaral, A.C. Principles, techniques, and applications of biocatalyst immobilization for industrial application. Appl. Microbiol. Biotechnol. 2015, 99, 2065–2082. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, F.; Fernández-Lucas, J.; de la Fuente, D.; Zheng, C.; Bavaro, T.; Peters, B.; Massolini, G.; Annunziata, F.; Conti, P.; de la Mata, I.; et al. Immobilized enzyme reactors based on nucleoside phosphorylases and 2′-deoxyribosyltransferase for the in-flow synthesis of pharmaceutically relevant nucleoside analogues. Bioresour. Technol. 2020, 307, 123258. [Google Scholar] [CrossRef]
- Robescu, M.S.; Serra, I.; Terreni, M.; Ubiali, D.; Bavaro, T. A Multi-Enzymatic Cascade Reaction for the Synthesis of Vidarabine 5′-Monophosphate. Catalysts 2020, 10, 60. [Google Scholar] [CrossRef] [Green Version]
- Tamborini, L.; Previtali, C.; Annunziata, F.; Bavaro, T.; Terreni, M.; Calleri, E.; Rinaldi, F.; Pinto, A.; Speranza, G.; Ubiali, D.; et al. An Enzymatic Flow-Based Preparative Route to Vidarabine. Molecules 2020, 25, 1223. [Google Scholar] [CrossRef] [Green Version]
- Hartley, C.J.; Williams, C.C.; Scoble, J.A.; Churches, Q.I.; North, A.; French, N.G.; Nebl, T.; Coia, G.; Warden, A.C.; Simpson, G.; et al. Engineered enzymes that retain and regenerate their cofactors enable continuous-flow biocatalysis. Nat. Catal. 2019, 2, 1006–1015. [Google Scholar] [CrossRef]
- Peschke, T.; Skoupi, M.; Burgahn, T.; Gallus, S.; Ahmed, I.; Rabe, K.S.; Niemeyer, C.M. Self-Immobilizing Fusion Enzymes for Compartmentalized Biocatalysis. ACS Catal. 2017, 7, 7866–7872. [Google Scholar] [CrossRef]
- Zhong, C.; Duić, B.; Bolivar, J.M.; Nidetzky, B. Three-Enzyme Phosphorylase Cascade Immobilized on Solid Support for Biocatalytic Synthesis of Cello−oligosaccharides. ChemCatChem 2020, 12, 1350–1358. [Google Scholar] [CrossRef]
- Bitterwolf, P.; Ott, F.; Rabe, K.S.; Niemeyer, C.M. Imine Reductase Based All-Enzyme Hydrogel with Intrinsic Cofactor Regeneration for Flow Biocatalysis. Micromachines 2019, 10, 783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peschke, T.; Bitterwolf, P.; Gallus, S.; Hu, Y.; Oelschlaeger, C.; Willenbacher, N.; Rabe, K.S.; Niemeyer, C.M. Self-Assembling All-Enzyme Hydrogels for Flow Biocatalysis. Angew. Chem. Int. Ed. 2018, 57, 17028–17032. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Obst, F.; Haefner, S.; Heroldt, T.; Peiter, M.; Simon, F.; Richter, A.; Voit, B.; Appelhans, D. Hydrogel/enzyme dots as adaptable tool for non-compartmentalized multi-enzymatic reactions in microfluidic devices. React. Chem. Eng. 2019, 4, 67–77. [Google Scholar] [CrossRef]
- Lefrançois, P.; Goudeau, B.; Arbault, S. Dynamic monitoring of a bi-enzymatic reaction at a single biomimetic giant vesicle. Analyst 2020. [Google Scholar] [CrossRef] [PubMed]
- Hakala, T.A.; Bialas, F.; Toprakcioglu, Z.; Bräuer, B.; Baumann, K.N.; Levin, A.; Bernardes, G.J.L.; Becker, C.F.W.; Knowles, T.P.J. Continuous Flow Reactors from Microfluidic Compartmentalization of Enzymes within Inorganic Microparticles. ACS Appl. Mater. Interfaces 2020, 12, 32951–32960. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhao, Z.; Liu, Y.; Shao, C.; Bian, F.; Zhao, Y. Biomimetic enzyme cascade reaction system in microfluidic electrospray microcapsules. Sci. Adv. 2018, 4, eaat2816. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Kim, S.B.; Yoo, H.Y.; Lee, J.H.; Park, C.; Han, S.O.; Kim, S.W. Kinetic modeling of biodiesel production by mixed immobilized and co-immobilized lipase systems under two pressure conditions. Korean J. Chem. Eng. 2013, 30, 1272–1276. [Google Scholar] [CrossRef]
- Ji, Q.; Tan, J.; Zhu, L.; Lou, D.; Wang, B. Preparing tauroursodeoxycholic acid (TUDCA) using a double-enzyme-coupled system. Biochem. Eng. J. 2016, 105, 1–9. [Google Scholar] [CrossRef]
- Huang, X.; Li, M.; Mann, S. Membrane-mediated cascade reactions by enzyme–polymer proteinosomes. Chem. Commun. 2014, 50, 6278–6280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubey, N.C.; Tripathi, B.P.; Müller, M.; Stamm, M.; Ionov, L. Bienzymatic Sequential Reaction on Microgel Particles and Their Cofactor Dependent Applications. Biomacromolecules 2016, 17, 1610–1620. [Google Scholar] [CrossRef]
- Sun, J.; Ge, J.; Liu, W.; Lan, M.; Zhang, H.; Wang, P.; Wang, Y.; Niu, Z. Multi-enzyme co-embedded organic–inorganic hybrid nanoflowers: Synthesis and application as a colorimetric sensor. Nanoscale 2014, 6, 255–262. [Google Scholar] [CrossRef]
- Cui, C.; Chen, H.; Chen, B.; Tan, T. Genipin Cross-Linked Glucose Oxidase and Catalase Multi-enzyme for Gluconic Acid Synthesis. Appl. Biochem. Biotechnol. 2017, 181, 526–535. [Google Scholar] [CrossRef]
- Mafra, A.C.; Ulrich, L.G.; Kornecki, J.F.; Fernandez-Lafuente, R.; Tardioli, P.W.; Ribeiro, M.P. Combi-CLEAs of Glucose Oxidase and Catalase for Conversion of Glucose to Gluconic Acid Eliminating the Hydrogen Peroxide to Maintain Enzyme Activity in a Bubble Column Reactor. Catalysts 2019, 9, 657. [Google Scholar] [CrossRef] [Green Version]
- Hwang, E.T.; Seo, B.-K.; Gu, M.B.; Zeng, A.-P. Successful bi-enzyme stabilization for the biomimetic cascade transformation of carbon dioxide. Catal. Sci. Technol. 2016, 6, 7267–7272. [Google Scholar] [CrossRef]
- Rabe, K.S.; Müller, J.; Skoupi, M.; Niemeyer, C.M. Cascades in Compartments: En Route to Machine-Assisted Biotechnology. Angew. Chem. Int. Ed. 2017, 56, 13574–13589. [Google Scholar] [CrossRef] [PubMed]
- Holvey, C.P.; Roberge, D.M.; Gottsponer, M.; Kockmann, N.; Macchi, A. Pressure drop and mixing in single phase microreactors: Simplified designs of micromixers. Chem. Eng. Process. Process Intensif. 2011, 50, 1069–1075. [Google Scholar] [CrossRef]
- Nakagawa, K.; Tamura, A.; Chaiya, C. Preparation of proteolytic microreactors by freeze-drying immobilization. Chem. Eng. Sci. 2014, 119, 22–29. [Google Scholar] [CrossRef]
- Illg, T.; Hessel, V.; Löb, P.; Schouten, J.C. Novel process window for the safe and continuous synthesis of tert.-butyl peroxy pivalate in a micro-reactor. Chem. Eng. J. 2011, 167, 504–509. [Google Scholar] [CrossRef]
- Wei, H.-C.; Huang, S.-H.; Jiang, J.-A.; Lee, Y.-C. A Multichannel Calorimetric Simultaneous Assay Platform Using a Microampere Constant-Current Looped Enthalpy Sensor Array. Sensors 2017, 17, 292. [Google Scholar] [CrossRef] [Green Version]
- Van Schie, M.M.C.H.; Ebrahimi, K.H.; Hagen, W.R.; Hagedoorn, P.-L. Fast and accurate enzyme activity measurements using a chip-based microfluidic calorimeter. Anal. Biochem. 2018, 544, 57–63. [Google Scholar] [CrossRef]
- Abis, G.; Pacheco-Gómez, R.; Bui, T.T.T.; Conte, M.R. Isothermal Titration Calorimetry Enables Rapid Characterization of Enzyme Kinetics and Inhibition for the Human Soluble Epoxide Hydrolase. Anal. Chem. 2019, 91, 14865–14872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luckarift, H.R.; Ku, B.S.; Dordick, J.S.; Spain, J.C. Silica-immobilized enzymes for multi-step synthesis in microfluidic devices. Biotechnol. Bioeng. 2007, 98, 701–705. [Google Scholar] [CrossRef]
- Heinzler, R.; Fischöder, T.; Elling, L.; Franzreb, M. Toward Automated Enzymatic Glycan Synthesis in a Compartmented Flow Microreactor System. Adv. Synth. Catal. 2019, 361, 4506–4516. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.-X.; Qian, Z.-G.; Ki, C.S.; Park, Y.H.; Kaplan, D.L.; Lee, S.Y. Native-sized recombinant spider silk protein produced in metabolically engineered Escherichia coli results in a strong fiber. Proc. Natl. Acad. Sci. USA 2010, 107, 14059–14063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Saba, T.; Yiu, H.H.P.; Howe, R.F.; Anderson, J.A.; Shi, J. Cofactor NAD(P)H Regeneration Inspired by Heterogeneous Pathways. Chem 2017, 2, 621–654. [Google Scholar] [CrossRef] [Green Version]
- Kisukuri, C.M.; Andrade, L.H. Production of chiral compounds using immobilized cells as a source of biocatalysts. Org. Biomol. Chem. 2015, 13, 10086–10107. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.W.I.; Fischbach, M.; Huebner, H.; Buchholz, R.; Hummel, W.; Daussmann, T.; Wandrey, C.; Liese, A. Synthesis of enantiopure (5R)-hydroxyhexane-2-one with immobilised whole cells of Lactobacillus kefiri. Appl. Microbiol. Biotechnol. 2006, 71, 289–293. [Google Scholar] [CrossRef]
- Nagy-Gyor, L.; Abahazi, E.; Bodai, V.; Satorhelyi, P.; Erdelyi, B.; Balogh-Weiser, D.; Paizs, C.; Hornyanszky, G.; Poppe, L. Co-immobilized Whole Cells with omega-Transaminase and Ketoreductase Activities for Continuous-Flow Cascade Reactions. Chembiochem 2018, 19, 1845–1848. [Google Scholar] [CrossRef] [PubMed]
- Grabner, B.; Schweiger, A.K.; Gavric, K.; Kourist, R.; Gruber-Woelfler, H. A chemo-enzymatic tandem reaction in a mixture of deep eutectic solvent and water in continuous flow. React. Chem. Eng. 2020, 5, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Annunziata, F.; Letizia Contente, M.; Betti, D.; Pinna, C.; Molinari, F.; Tamborini, L.; Pinto, A. Efficient Chemo-Enzymatic Flow Synthesis of High Value Amides and Esters. Catalysts 2020, 10, 939. [Google Scholar] [CrossRef]
- Wigneswaran, V.; Nielsen, K.F.; Sternberg, C.; Jensen, P.R.; Folkesson, A.; Jelsbak, L. Biofilm as a production platform for heterologous production of rhamnolipids by the non-pathogenic strain Pseudomonas putida KT2440. Microb. Cell Factories 2016, 15, 181. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, N.; Schripsema, J.; Lienhardt, J.; Blaschek, H.P. Continuous solvent production by Clostridium beijerinckii BA101 immobilized by adsorption onto brick. World J. Microbiol. Biotechnol. 2000, 16, 377–382. [Google Scholar] [CrossRef]
- Kunduru, M.R.; Pometto, A.L. Continuous ethanol production by Zymomonas mobilis and Saccharomyces cerevisiae in biofilm reactors. J. Ind. Microbiol. 1996, 16, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Kurade, M.B.; Waghmode, T.R.; Patil, S.M.; Jeon, B.-H.; Govindwar, S.P. Monitoring the gradual biodegradation of dyes in a simulated textile effluent and development of a novel triple layered fixed bed reactor using a bacterium-yeast consortium. Chem. Eng. J. 2017, 307, 1026–1036. [Google Scholar] [CrossRef]
- Stojkovic, G.; Znidarsic-Plazl, P. Continuous synthesis of L-malic acid using whole-cell microreactor. Process Biochem. 2012, 47, 1102–1107. [Google Scholar] [CrossRef]
- Kim, H.J.; Huh, D.; Hamilton, G.; Ingber, D.E. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab A Chip 2012, 12, 2165–2174. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Moore, B.S. Enzymatic Cascade Reactions in Biosynthesis. Angew. Chem. Int. Ed. 2019, 58, 6846–6879. [Google Scholar] [CrossRef] [PubMed]
- Rudroff, F.; Mihovilovic, M.D.; Gröger, H.; Snajdrova, R.; Iding, H.; Bornscheuer, U.T. Opportunities and challenges for combining chemo- and biocatalysis. Nat. Catal. 2018, 1, 12–22. [Google Scholar] [CrossRef]
- Schmidt, S.; Castiglione, K.; Kourist, R. Overcoming the Incompatibility Challenge in Chemoenzymatic and Multi-Catalytic Cascade Reactions. Chem. A Eur. J. 2018, 24, 1755–1768. [Google Scholar] [CrossRef]
- Clayton, A.D.; Labes, R.; Blacker, A.J. Combination of chemocatalysis and biocatalysis in flow. Curr. Opin. Green Sustain. Chem. 2020, 26, 100378. [Google Scholar] [CrossRef]
- Suresh, A.; Shravan Ramgopal, D.; Panchamoorthy Gopinath, K.; Arun, J.; SundarRajan, P.; Bhatnagar, A. Recent advancements in the synthesis of novel thermostable biocatalysts and their applications in commercially important chemoenzymatic conversion processes. Bioresour. Technol. 2021, 323, 124558. [Google Scholar] [CrossRef]
- Losada-Garcia, N.; Cabrera, Z.; Urrutia, P.; Garcia-Sanz, C.; Andreu, A.; Palomo, J.M. Recent Advances in Enzymatic and Chemoenzymatic Cascade Processes. Catalysts 2020, 10, 1258. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, P.; Gao, S.; Wang, Z.; Luan, P.; González-Sabín, J.; Jiang, Y. Construction of chemoenzymatic cascade reactions for bridging chemocatalysis and Biocatalysis: Principles, strategies and prospective. Chem. Eng. J. 2020, 127659. [Google Scholar] [CrossRef]
- Falus, P.; Cerioli, L.; Bajnóczi, G.; Boros, Z.; Weiser, D.; Nagy, J.; Tessaro, D.; Servi, S.; Poppe, L. A Continuous-Flow Cascade Reactor System for Subtilisin A- Catalyzed Dynamic Kinetic Resolution of N-tert-Butyloxycarbonylphenylalanine Ethyl Thioester with Benzylamine. Adv. Synth. Catal. 2016, 358, 1608–1617. [Google Scholar] [CrossRef]
- Farkas, E.; Oláh, M.; Földi, A.; Kóti, J.; Éles, J.; Nagy, J.; Gal, C.A.; Paizs, C.; Hornyánszky, G.; Poppe, L. Chemoenzymatic Dynamic Kinetic Resolution of Amines in Fully Continuous-Flow Mode. Org. Lett. 2018, 20, 8052–8056. [Google Scholar] [CrossRef] [PubMed]
- Lackner, F.; Hiebler, K.; Grabner, B.; Gruber-Woelfler, H. Optimization of a Catalytic Chemoenzymatic Tandem Reaction for the Synthesis of Natural Stilbenes in Continuous Flow. Catalysts 2020, 10, 1404. [Google Scholar] [CrossRef]
- Szelwicka, A.; Zawadzki, P.; Sitko, M.; Boncel, S.; Czardybon, W.; Chrobok, A. Continuous Flow Chemo-Enzymatic Baeyer–Villiger Oxidation with Superactive and Extra-Stable Enzyme/Carbon Nanotube Catalyst: An Efficient Upgrade from Batch to Flow. Org. Process Res. Dev. 2019, 23, 1386–1395. [Google Scholar] [CrossRef]
- Blaser, H.-U.; Pugin, B.; Studer, M. Enantioselective Heterogeneous Catalysis: Academic and Industrial Challenges. In Chiral Catalyst Immobilization and Recycling; Vos, D.E.D., Vankelecom, I.F.J., Jacobs, P.A., Eds.; Wiley: Weinheim, Germany, 2000; pp. 1–17. [Google Scholar]
- Kragl, U.; Dwars, T. The development of new methods for the recycling of chiral catalysts. Trends Biotechnol. 2001, 19, 442–449. [Google Scholar] [CrossRef]
- Jones, E.; McClean, K.; Housden, S.; Gasparini, G.; Archer, I. Biocatalytic oxidase: Batch to continuous. Chem. Eng. Res. Des. 2012, 90, 726–731. [Google Scholar] [CrossRef]
- Gasparini, G.; Archer, I.; Jones, E.; Ashe, R. Scaling Up Biocatalysis Reactions in Flow Reactors. Org. Process Res. Dev. 2012, 16, 1013–1016. [Google Scholar] [CrossRef]
- Toftgaard Pedersen, A.; de Carvalho, T.M.; Sutherland, E.; Rehn, G.; Ashe, R.; Woodley, J.M. Characterization of a continuous agitated cell reactor for oxygen dependent biocatalysis. Biotechnol. Bioeng. 2017, 114, 1222–1230. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Microreactors | Mesoreactors | Macroreactors |
|---|---|---|
| At least one dimension between 10 µm and 500 µm | At least one dimension between 500 µm and a few mm | Dimensions above mm scale |
| Specific area 1 up to 50,000 m2/m3 | Specific area up to 50,000 m2/m3 | Specific area around 100 and 1000 m2/m3 |
| µL range | mL range | L to kL range |
| mg to g scale | multi-g to kg scale | kg to ton scale |
| Immobilization Method | System | Comments | Reference |
|---|---|---|---|
| Covalent | Two-step: keto reductase (KRE) and glucose dehydrogenase (GDH) | Production of (R)-4-chloro-3-hydroxybutanoate. The enzymes were co-immobilized in mesocellular siliceous foams through microwave irradiation with p-benzoquinone as crosslinking agent. The formulation retained more than 90% residual activity upon 30 days of storage at 4 °C. The formulation retained over 50% residual activity after 6 repeated batch conversion cycles, whereas upon co-immobilization by entrapment in calcium alginate the residual activity dropped to ~20%. | [32] |
| Two-step: uridine phosphorylase (UP) and purine nucleoside phosphorylase (pNP) phosphorylase | Synthesis of arabinosyladenine, an antiviral nucleoside. Enzymes were co-immobilized on glutaraldehyde activated monolithic aminopropyl silica carrier. Recirculation through the column at a flow rate of 0.5 mL.min−1 of adenine (nucleobase, 1 mM) and arabinosyluracil (sugar donor, 2 mM) resulted in 60% conversion after 24 h. | [89] | |
| Covalent and ionic | Three-step: UP, pNP and deoxyadenosine kinase (dAK) | Synthesis of vidarabine 5′-monophosphate (antiviral drug). UP and pNP were covalently bound individually to glyoxyl-agarose and dAK was bound by ionic interaction ionic interaction to functionalized Sepabeads® EC-EP. Close to full conversion of the substrate (adenine, 25 mM) was reported. | [90] |
| Covalent and affinity | Two-step: UP and pNP, each fused with His-tag binding peptide | Synthesis of vidarabine The enzymes were co-immobilized either covalently on glyoxyl-agarose or by metal-ion affinity on a hydrophylic polymer-coated controlled porosity glass beads, EziG1 (Opal). Each one of the resulting formulations was packed in a glass column that was fed with arabinofuranosyluracil (16 mM) as sugar donor and adenine (8 mM) as sugar acceptor. Eighty percent conversion was reached after either 4 h of residence time (covalent-based formulation) or 80 min (affinity-based formulation). The later displayed poor operational stability, hence the former was used for continuous production of vidarabine. Under a residence time 2 h (67% conversion) operation proceeded for 8 days, after which the product was recovered (55% yield, over 99% purity). | [91] |
| Three-step: glycerol kinase (GK)/acetate kinase (AK) + glycerol-3-phosphate dehydrogenase (GPD)/NADH oxidase (NOX) + fructose aldolase (FA), each harboring a harboring a maleimide–thiol conjugation module | Conversion of glycerol to a chiral d-fagomine precursor. GK/AK and GPD/NOX were produced as modular biocatalysts that retain and recycle their cofactors as fusion proteins, to which cofactors were covalently tethered. GK/AK, GPD/NOX and FA were covalently bound through the conjugation module to chemically trifluoroketone activated agarose beads and each of the three formulations added in packed-bed reactors disposed sequentially. Space–time yields of 70 g L−1 h−1 g−1 and total turnover numbers above 10,000 were reported | [92] | |
| Affinity | Two-step: (R)-selective alcohol dehydrogenase (RADH), (S)-selective methylglyoxal reductase and GDH, each fused with streptavidin binding peptide | Selective reduction of 5-nitrononane-2,8-dione. Each enzyme was bound to magnetic microbeads coated with streptavidin, which were introduced in a compartmentalized microfluidic packed-bed reactor. Under selected flow conditions and ratio of immobilized enzymes load, an initial conversion of 73.6% stereoselectivity exceeding 99:1 and product space–time yield of 106 g L−1day−1 were reported. | [93] |
| Two-step: RADH, and GDH, each fused with His-tag binding peptide | Selective reduction of 5-nitrononane-2,8-dione. Each enzyme was bound to Co2+ functionalized magnetic microbeads, which were introduced in a compartmentalized microfluidic packed-bed reactor. Despite noticeable decrease inactivity upon immobilization, namely for GDH, under selected immobilized enzyme load and flow conditions, 98% substrate conversion of 98% and product space–time yield of 131 g L−1day−1 was reported. The outcome compared favorably with that of a previous work [93], albeit at the cost of a decrease (~60%) in specific productivity. | [63] | |
| Two-step: KRE and GDH, each fused with His-tag binding peptide | Reduction of keto-ester (ethyl-2-methylacetoacetate) and bulky ketones (4-phenyl-2-butanone; 3′-hydroxyacetophenone) to secondary alcohols. The enzymes from crude extracts were co-immobilized under optimized ratios in a column Ni2+ functionalized crosslinked agarose, which was packed in a flow reactor. Co-immobilization reduced cofactor requirements and immobilization enhanced tolerance to high substrate concentrations (130 mM and above) as compared to the free enzymes. The immobilized enzymes were used in 20 (keto-ester) and 13 (bulky ketones) repeated batch conversion cycles with 95% substrate conversion with substrate concentration of 130 mM. | [64] | |
| Affinity (cont.) | Three-step: β-galactosidase (bGAL), glucose oxidase GOx and horseradish peroxidase (HRP) | Conversion of lactose to resorufin. Streptavidin-coated microbeads functionalized with either individual or a mixture of chemically biotinylated enzymes. The formulations were introduced in microfluidic channel, where the former immobilization approach outperformed the later. Operational parameters, e.g., flow rate, relative amount of enzymes, initial substrate concentration and total amount of biocatalyst, were optimized. | [21] |
| Three-step: sucrose phosphorylase, cellobiose phosphorylase and cellodextrin phosphorylase, each fused with Zbasic2 binding module | Synthesis of soluble cello-oligosaccharides with degree of polymerization ≤6. The enzymes were co-immobilized according to previously established ratio of activities on macroporous polymethacrylate particles coated with sulfonate groups harboring the negative charges to interact with the Zbasic2 module. The formulation was used through five repeated batch conversion cycles allowing the synthesis of 10 to 12.5 g L−1 of the intended cello-oligosaccharides from ~68 g L−1 sucrose and 12 g L−1 glucose, and retaining ~85% of the overall initial activity. Some leakage of cellobiose phosphorylase was observed, the trend ascribed to the excess of negative surface charges of the fused enzyme. | [94] | |
| Two-step: Imine reductase and GDH fused with SpyTag and SpyCatcher domain, respectively, to generate two complementary building blocks | Conversion of cyclic imines to the corresponding secondary amines. Upon incubation in magnesium-supplemented potassium phosphate buffer the two fused enzymes self-assembled to a porous hydrogel through the formation of a covalent isopeptide bond between the activated lysin residue of the SpyCatcher and the aspartic acid residue on the SpyTag domains. The catalytic hydrogel exhibited a stereoselectivity over 99%. The gel was packed in a microfluidic (150 µL volume) channel. After 40 h days of operation (10 µL min−1 feeding rate, 5 mM 3,4-dihydroisoquinoline solution) ~90% conversion was observed, whereas after 5 h of operation with unbound GDH roughly no conversion was observed, due to GDH leakage. Space–time yield of 150 g L−1.day−1 was observed at a flow rate of 100 µL.min−1. | [95] | |
| Entrapment (cont.) | Two-step: ADH and GDH fused with SpyCatcher and SpyTag domain, respectively, to generate two complementary homo-tetrameric building blocks | Selective reduction of 5-nitrononane-2,8-dione, acetophenone, 4′-chloroacetophenone and trans-4-phenyl-3-buten-2-one to the corresponding R alcohols. The two fused enzymes self-assembled to a porous hydrogel containing 77% of enzyme. The gel was packed in a microfluidic (150 µL volume) channel. After 7 days of operation (10 µL min−1 feeding rate, 5 mM substrate solution) ~70% conversion was observed with no enzyme leakage, whereas after 2 h of operation with the free enzymes roughly full leakage was observed. Spacetime yield 4.5-fold higher than previously reported [93] was observed. Mass transfer limitations were advantageously used: co-entrapment of NADP+ (cofactor) allowed for 30 h of continuous conversion with no cofactor in the feed. Stereoselectivity over 99% was observed in all reactions after 10 h of continuous operation. The gel could be stored for 30 days at 4 °C with no loss in activity. | [96] |
| Entrapment (cont.) | Two cascades, each three-step: bGAL GOx and HRP (cascade 1); and phospholipase D, choline oxidase and HRP (cascade 2) | Detection of lactose and/or glucose (cascade 1) and of phosphatidylcholine (cascade 2). The enzymes of each cascade were co-immobilized in a noncompartmentalized manner in a hydrogel matrix composed of poly(ethylene glycol) diacrylate, 2-(dimethylamino)ethyl methacrylate, and 2-hydroxyethyl methacrylate, either as bulky hydrogels or as dots (350 μm diameter) integrated into polydimethylsiloxane (PDMS)-on-glass microfluidic reactors to perform the reaction under continuous flow. Overall, immobilization increased the catalytic activity of the cascades as compared to the free form. | [97] |
| Encapsulation | Two-step: GOx and HRP | Conversion of glucose to resorufin. A mixture of GOx and HRP was encapsulated in giant unilamellar vesicles (GUV) sized from 10 to 200 μm, produced out of a liposome suspension prepared from phospholipids present in the soybean polar extract. | [98] |
| Two-step: GOx and HRP | Conversion of glucose to resorufin. The two enzymes were encapsulated inside silica microparticles. The formulation was packed in a microfluidic chamber and assessed for monitoring glucose concentration. The device operated within the range of glucose concentration found in saliva and sweat. | [99] | |
| Encapsulation and entrapment | Two-step: Alcohol oxidase and catalase | Alcohol oxidase and catalase were individually entrapped in inverse opal particles and the whole embedded in calcium alginate microcapsules to mimic hepatocytes for elementary alcohol detoxification. | [100] |
| Combi-CLEAs and covalent binding | Multi-step: cellulase, pectinase and xylanase | Saccharification of cellulose and hemicellulose. The enzymes were individually immobilized in amino-functionalized magnetic particles which were afterwards crosslinked with glutaraldehyde to yield magnetic combi-CLEAs. Immobilization improved the thermal stability of the enzymes and the formulation was used through 12 repeated batch conversion cycles with minor loss of activity. Moreover, when integrated in simultaneous saccharification and fermentation of wheat straw the formulation allowed a 1.82-fold increase in bioethanol concentration as compared to use of free enzymes. | [76] |
| Combi-CLEAs and encapsulation | Two-step: GOx and HRP | Conversion of glucose to resorufin. A mixture of GOx and HRP was engulfed inside the bowl-shaped polymersomes and the enzyme molecules were crosslinked with either genipin or glutaraldehyde to produce crosslinked enzymatic nanoaggregates inside the submicron-sized vesicles (c-CLEnA). | [71] |
| Biocatalyst | Bioreactor | Reaction | Reference |
|---|---|---|---|
| Lactobacillus kefiri | Plug flow reactor |  | [121] |
| Escherichia coli with Chromobacterium violaceum ωws-transaminase activity and Lodderomyces elongisporus with ketoreductase activity | Continuous flow reactor |  | [122] |
| Immobilized 7α- and 7β-hydroxysteroid dehydrogenases | Two column bioreactors with each enzyme or single column with both immobilized enzymes |  | [102] |
| Phenolic acid decarboxylase and a chemical Pd-catalyst | Packed-bed reactor |  | [123] |
| Acyltransferase from Mycobacterium smegmatis, in-line purification with SO2Cl, and hydrogenation step | Continuous flow reactor |  | [124] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, P.; de Carvalho, C.C.C.R. Multi-Enzyme Systems in Flow Chemistry. Processes 2021, 9, 225. https://doi.org/10.3390/pr9020225
Fernandes P, de Carvalho CCCR. Multi-Enzyme Systems in Flow Chemistry. Processes. 2021; 9(2):225. https://doi.org/10.3390/pr9020225
Chicago/Turabian StyleFernandes, Pedro, and Carla C. C. R. de Carvalho. 2021. "Multi-Enzyme Systems in Flow Chemistry" Processes 9, no. 2: 225. https://doi.org/10.3390/pr9020225
APA StyleFernandes, P., & de Carvalho, C. C. C. R. (2021). Multi-Enzyme Systems in Flow Chemistry. Processes, 9(2), 225. https://doi.org/10.3390/pr9020225