
Interaction between Antifungal Isoxazolo[3,4-b]Pyridin 3(1H)-One Derivatives and Human Serum Proteins Analyzed with Biomimetic Chromatography and QSAR Approach


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Reagents
2.2. Analytes
2.3. HSA-HPLC Analysis
2.4. QSRR Analysis
2.5. In-Silico Calculation
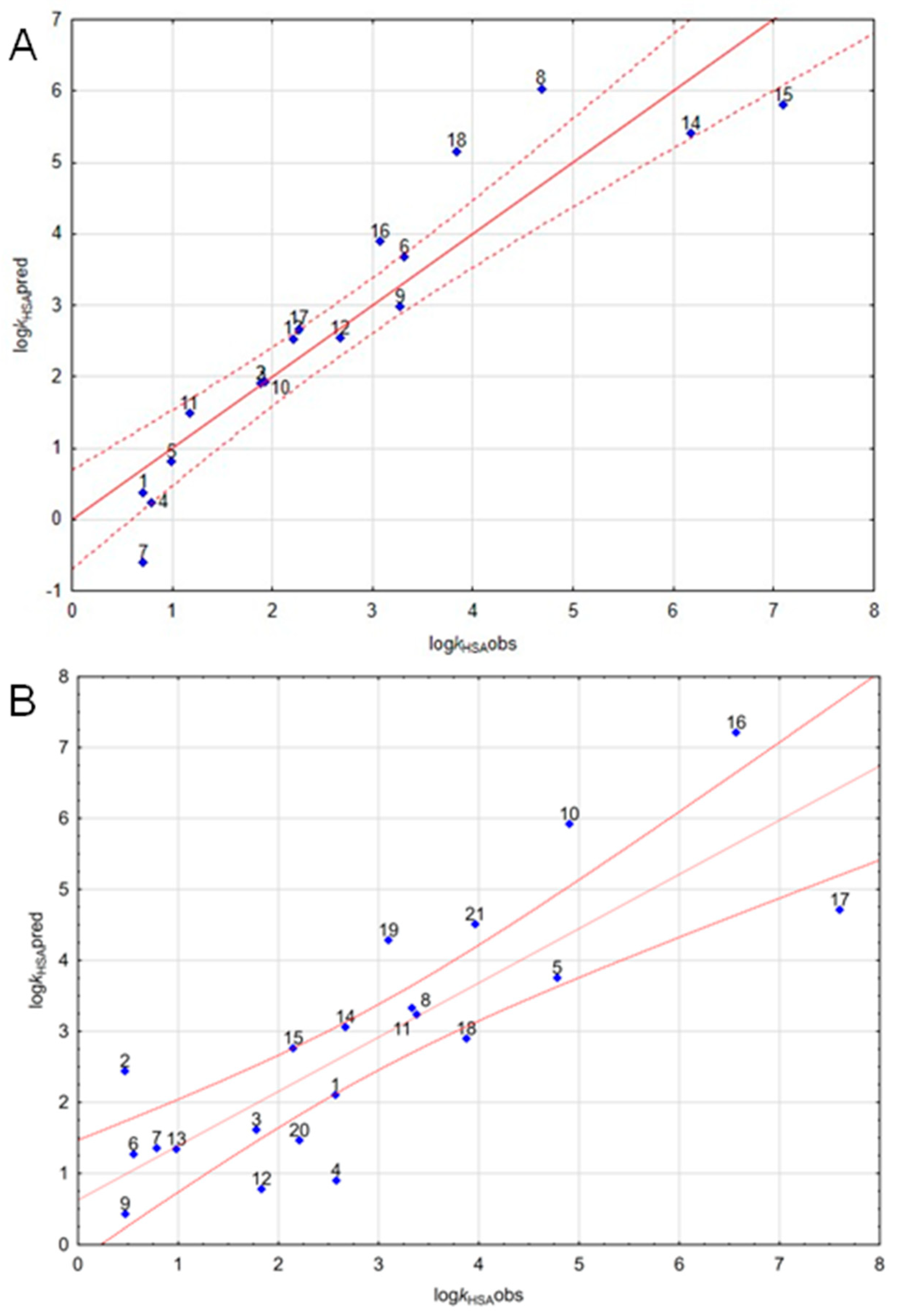
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wong, S.S.W.; Samaranayake, L.P.; Seneviratne, C.J. In pursuit of the ideal antifungal agent for Candida infections: High-throughput screening of small molecules. Drug Discov. Today 2014, 19, 1721–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallabhaneni, S.; Mody, R.K.; Walker, T.; Chiller, T. The Global Burden of Fungal Diseases. Infect. Dis. Clin. N. Am. 2016, 30, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Seneviratne, C.J.; Rosa, E.A.R. Editorial: Antifungal drug discovery: New theories and new therapies. Front. Microbiol. 2016, 7, 728. [Google Scholar] [CrossRef] [PubMed]
- Sanglard, D.; Coste, A.; Ferrari, S. Antifungal drug resistance mechanisms in fungal pathogens from the perspective of transcriptional gene regulation. FEMS Yeast Res. 2009, 9, 1029–1050. [Google Scholar] [CrossRef] [Green Version]
- Clancy, C.J.; Nguyen, M.H. At what cost echinocandin resistance? J. Infect. Dis. 2011, 204, 499–501. [Google Scholar] [CrossRef] [Green Version]
- Seneviratne, C.J.; Jin, L.J.; Samaranayake, Y.H.; Samaranayake, L.P. Cell density and cell aging as factors modulating antifungal resistance of Candida albicans biofilms. Antimicrob. Agents Chemother 2008, 52, 3259–3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, N.R.H.; Bicanic, T.; Salim, R.; Hope, W. Liposomal Amphotericin B (AmBisome®): A Review of the Pharmacokinetics, Pharmacodynamics, Clinical Experience and Future Directions. Drugs 2016, 76, 485–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanat, K.; Brzezinska, E.; Sobanska, A. Aspects of Drug-Protein Binding and Methods of Analyzing the Phenomenon. Curr. Pharm. Des. 2018, 24, 2974–2985. [Google Scholar] [CrossRef]
- Lambrinidis, G.; Vallianatou, T.; Tsantili-Kakoulidou, A. In vitro, in silico and integrated strategies for the estimation of plasma protein binding. Rev. Adv. Drug Deliv. Rev. 2015, 23, 27–45. [Google Scholar] [CrossRef]
- Zsila, F. Subdomain IB is the third major drug binding region of human serum albumin: Toward the three-sites model. Mol. Pharm. 2013, 10, 1668–1682. [Google Scholar] [CrossRef]
- Saczewski, J.; Fedorowicz, J.; Kedzia, A.; Ziolkowska-Klinkosz, M.; Jalinska, A. Synthesis and Antifungal Activity of Some 4,6-Dimethylisoxazolo[3,4- b]pyridin-3(1H)-one Derivatives. Med. Chem. 2016, 12, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Saczewski, J.; Kedzia, A.; Jalińska, A. New derivatives of 4,6-dimethylisoxazolo[3,4-b]pyridin-3(1H)-one: Synthesis, tautomerism, electronic structure and antibacterial activity. Heterocycl. Commun. 2014, 20, 215. [Google Scholar] [CrossRef]
- Saczewski, J.; Fedorowicz, J.; Korcz, M.; Saczewski, F.; Wicher, B.; Gdaniec, M.; Konopacka, A. Experimental and theoretical studies on the tautomerism and reactivity of isoxazolo[3,4-b]quinolin-3(1H)-ones. Tetrahedron 2015, 71, 8975–8984. [Google Scholar] [CrossRef]
- Ciura, K.; Fedorowicz, J.; Žuvela, P.; Lovrić, M.; Kapica, H.; Baranowski, P.; Sawicki, W.; Wong, M.W.; Sączewski, J. Affinity of Antifungal Isoxazolo[3,4-b]pyridine-3(1H)-Ones to Phospholipids in Immobilized Artificial Membrane (IAM) Chromatography. Molecules 2020, 25, 4835. [Google Scholar] [CrossRef] [PubMed]
- Ciura, K.; Fedorowicz, J.; Andrić, F.; Žuvela, P.; Greber, K.E.; Baranowski, P.; Kawczak, P.; Nowakowska, J.; Baçzek, T.; Saçzewski, J. Lipophilicity determination of antifungal isoxazolo[3,4-b]pyridin-3(1H)-ones and their N1-substituted derivatives with chromatographic and computational methods. Molecules 2019, 24, 4311. [Google Scholar] [CrossRef] [Green Version]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew-Burke-Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef] [Green Version]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Valko, K.L. Application of biomimetic HPLC to estimate in vivo behavior of early drug discovery compounds. Futur. Drug Discov. 2019, 1, FDD11. [Google Scholar] [CrossRef] [Green Version]
- Valko, K.; Nunhuck, S.; Bevan, C.; Abraham, M.H.; Reynolds, D.P. Fast Gradient HPLC Method to Determine Compounds Binding to Human Serum Albumin. Relationships with Octanol/Water and Immobilized Artificial Membrane Lipophilicity. J. Pharm. Sci. 2003, 92, 2236–2248. [Google Scholar] [CrossRef]
- Hubicka, U.; Żuromska-Witek, B.; Komsta, Ł.; Krzek, J. Lipophilicity study of fifteen fluoroquinolones by reversed-phase thin-layer chromatography. Anal. Methods 2015, 7, 3841–3848. [Google Scholar] [CrossRef]
- Dąbrowska, M.; Starek, M.; Komsta, Ł.; Szafrański, P.; Stasiewicz-Urban, A.; Opoka, W. Assessment of the chromatographic lipophilicity of eight cephalosporins on different stationary phases. Eur. J. Pharm. Sci. 2017, 101, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Tropsha, A.; Gramatica, P.; Gombar, V. The Importance of Being Earnest: Validation is the Absolute Essential for Successful Application and Interpretation of QSPR Models. QSAR Comb. Sci. 2003, 22, 69–77. [Google Scholar] [CrossRef]
- Lučić, B.; Batista, J.; Bojović, V.; Lovrić, M.; Kržić, A.S.; Bešlo, D.; Nadramija, D.; Vikić-Topić, D. Estimation of random accuracy and its use in validation of predictive quality of classification models within predictive challenges. Croat. Chem. Acta 2019, 92, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Taraji, M.; Haddad, P.R.; Amos, R.I.J.; Talebi, M.; Szucs, R.; Dolan, J.W.; Pohl, C.A. Error measures in quantitative structure-retention relationships studies. J. Chromatogr. A 2017, 1524, 298–302. [Google Scholar] [CrossRef]
- Reutlinger, M.; Koch, C.P.; Reker, D.; Todoroff, N.; Schneider, P.; Rodrigues, T.; Schneider, G. Chemically advanced template search (CATS) for scaffold-hopping and prospective target prediction for “orphan” molecules. Mol. Inform. 2013, 32, 133–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciura, K.; Ulenberg, S.; Kapica, H.; Kawczak, P.; Belka, M.; Bączek, T. Drug affinity to human serum albumin prediction by retention of cetyltrimethylammonium bromide pseudostationary phase in micellar electrokinetic chromatography and chemically advanced template search descriptors. J. Pharm. Biomed. Anal. 2020, 188, 113423. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| No | logkHSA | logkw * | CHIIAM * | PPB% ** |
|---|---|---|---|---|
| 1 | 2.57 | 2.36 | nd | 83.78 |
| 2 | 0.47 | 3.98 | 12.92 | 93.94 |
| 3 | 1.78 | 2.87 | 24.85 | 85.62 |
| 4 | 2.58 | 3.76 | nd | 92.81 |
| 5 | 4.78 | 3.68 | 34.01 | 61.59 |
| 6 | 0.56 | 2.53 | 14.91 | 42.95 |
| 7 | 0.79 | 2.90 | 22.14 | 34.49 |
| 8 | 3.38 | 2.77 | 29.99 | 75.69 |
| 9 | 0.47 | 2.58 | 16.92 | 30.01 |
| 10 | 4.90 | 3.83 | 33.98 | 64.76 |
| 11 | 3.33 | 3.69 | 34.30 | 62.85 |
| 12 | 1.83 | 3.28 | 28.65 | 42.03 |
| 13 | 0.98 | 2.81 | 21.39 | 85.85 |
| 14 | 2.67 | 3.14 | 28.10 | 11.46 |
| 15 | 2.15 | 2.97 | 26.61 | 11.89 |
| 16 | 6.57 | 3.33 | 33.40 | 49.73 |
| 17 | 7.60 | 3.50 | 33.78 | 68.75 |
| 18 | 3.88 | 2.95 | nd | 11.13 |
| 19 | 3.10 | 3.21 | 29.78 | 54.04 |
| 20 | 2.21 | 3.23 | 27.84 | 26.44 |
| 21 | 3.96 | 3.24 | 30.37 | 61.70 |
| No | CATS3D_08_AL | R8v+ | F03[C-N] | JGI5 | MATS6v |
|---|---|---|---|---|---|
| 1 | 0 | 0.004 | 6 | 0.028 | −0.012 |
| 2 | 0 | 0.012 | 9 | 0.027 | 0.007 |
| 3 | 0 | 0.009 | 7 | 0.038 | −0.147 |
| 4 | 0 | 0.012 | 9 | 0.028 | −0.055 |
| 5 | 0 | 0.013 | 7 | 0.028 | 0.008 |
| 6 | 0 | 0.003 | 5 | 0.045 | −0.120 |
| 7 | 0 | 0.003 | 5 | 0.044 | −0.330 |
| 8 | 0 | 0.012 | 7 | 0.032 | 0.097 |
| 9 | 0 | 0.001 | 5 | 0.043 | −0.384 |
| 10 | 3 | 0.011 | 7 | 0.031 | −0.193 |
| 11 | 0 | 0.011 | 6 | 0.038 | −0.263 |
| 12 | 0 | 0.006 | 6 | 0.045 | −0.327 |
| 13 | 0 | 0.007 | 6 | 0.045 | −0.111 |
| 14 | 0 | 0.012 | 7 | 0.034 | −0.115 |
| 15 | 0 | 0.011 | 7 | 0.034 | −0.058 |
| 16 | 2 | 0.019 | 7 | 0.034 | 0.051 |
| 17 | 4 | 0.010 | 7 | 0.032 | −0.172 |
| 18 | 0 | 0.010 | 6 | 0.038 | 0.029 |
| 19 | 2 | 0.010 | 7 | 0.034 | −0.339 |
| 20 | 0 | 0.011 | 7 | 0.043 | −0.080 |
| 21 | 2 | 0.011 | 7 | 0.034 | −0.006 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciura, K.; Fedorowicz, J.; Kapica, H.; Pastewska, M.; Sawicki, W.; Sączewski, J. Interaction between Antifungal Isoxazolo[3,4-b]Pyridin 3(1H)-One Derivatives and Human Serum Proteins Analyzed with Biomimetic Chromatography and QSAR Approach. Processes 2021, 9, 512. https://doi.org/10.3390/pr9030512
Ciura K, Fedorowicz J, Kapica H, Pastewska M, Sawicki W, Sączewski J. Interaction between Antifungal Isoxazolo[3,4-b]Pyridin 3(1H)-One Derivatives and Human Serum Proteins Analyzed with Biomimetic Chromatography and QSAR Approach. Processes. 2021; 9(3):512. https://doi.org/10.3390/pr9030512
Chicago/Turabian StyleCiura, Krzesimir, Joanna Fedorowicz, Hanna Kapica, Monika Pastewska, Wiesław Sawicki, and Jarosław Sączewski. 2021. "Interaction between Antifungal Isoxazolo[3,4-b]Pyridin 3(1H)-One Derivatives and Human Serum Proteins Analyzed with Biomimetic Chromatography and QSAR Approach" Processes 9, no. 3: 512. https://doi.org/10.3390/pr9030512
APA StyleCiura, K., Fedorowicz, J., Kapica, H., Pastewska, M., Sawicki, W., & Sączewski, J. (2021). Interaction between Antifungal Isoxazolo[3,4-b]Pyridin 3(1H)-One Derivatives and Human Serum Proteins Analyzed with Biomimetic Chromatography and QSAR Approach. Processes, 9(3), 512. https://doi.org/10.3390/pr9030512