
Protein L—More Than Just an Affinity Ligand


Abstract
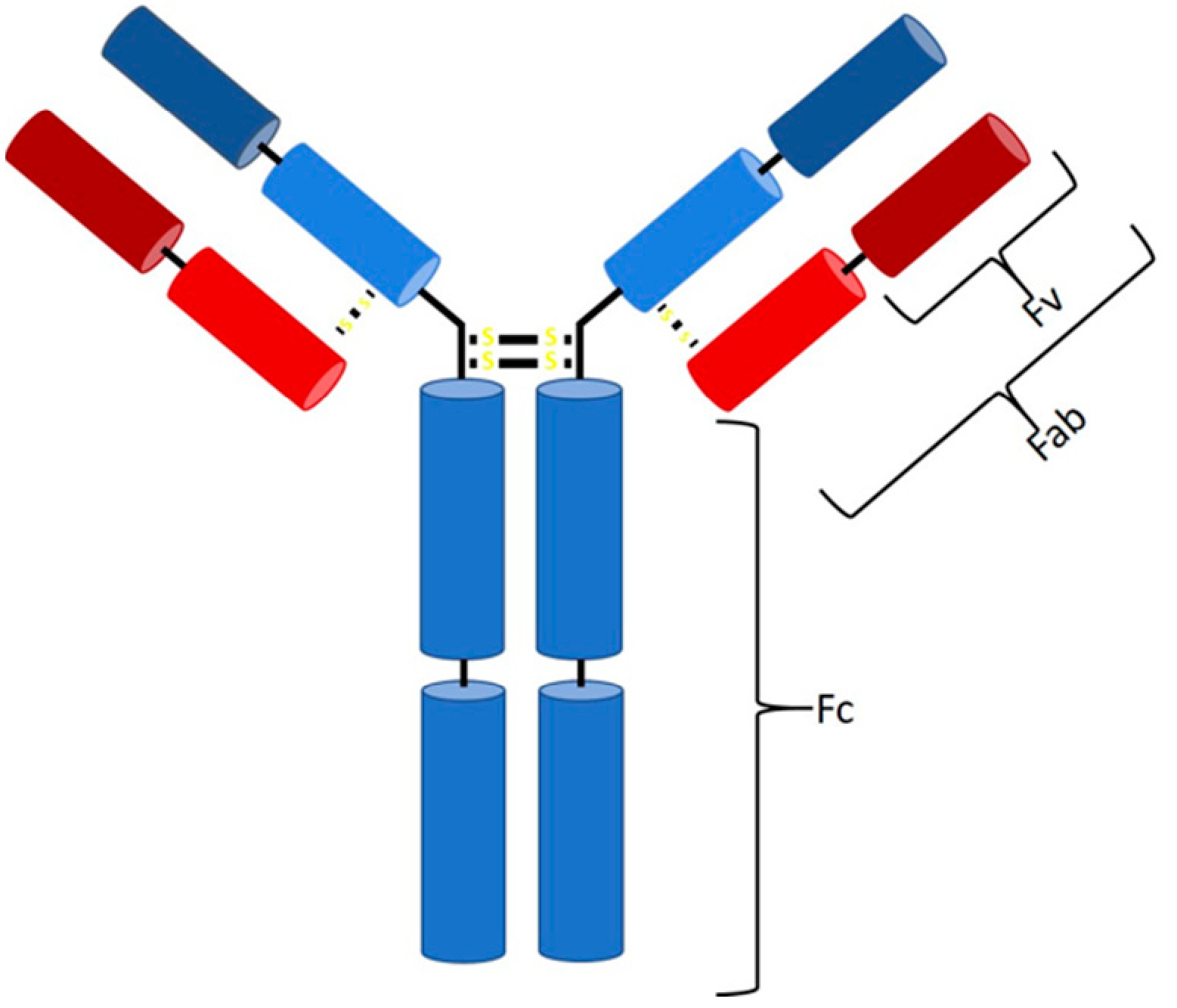
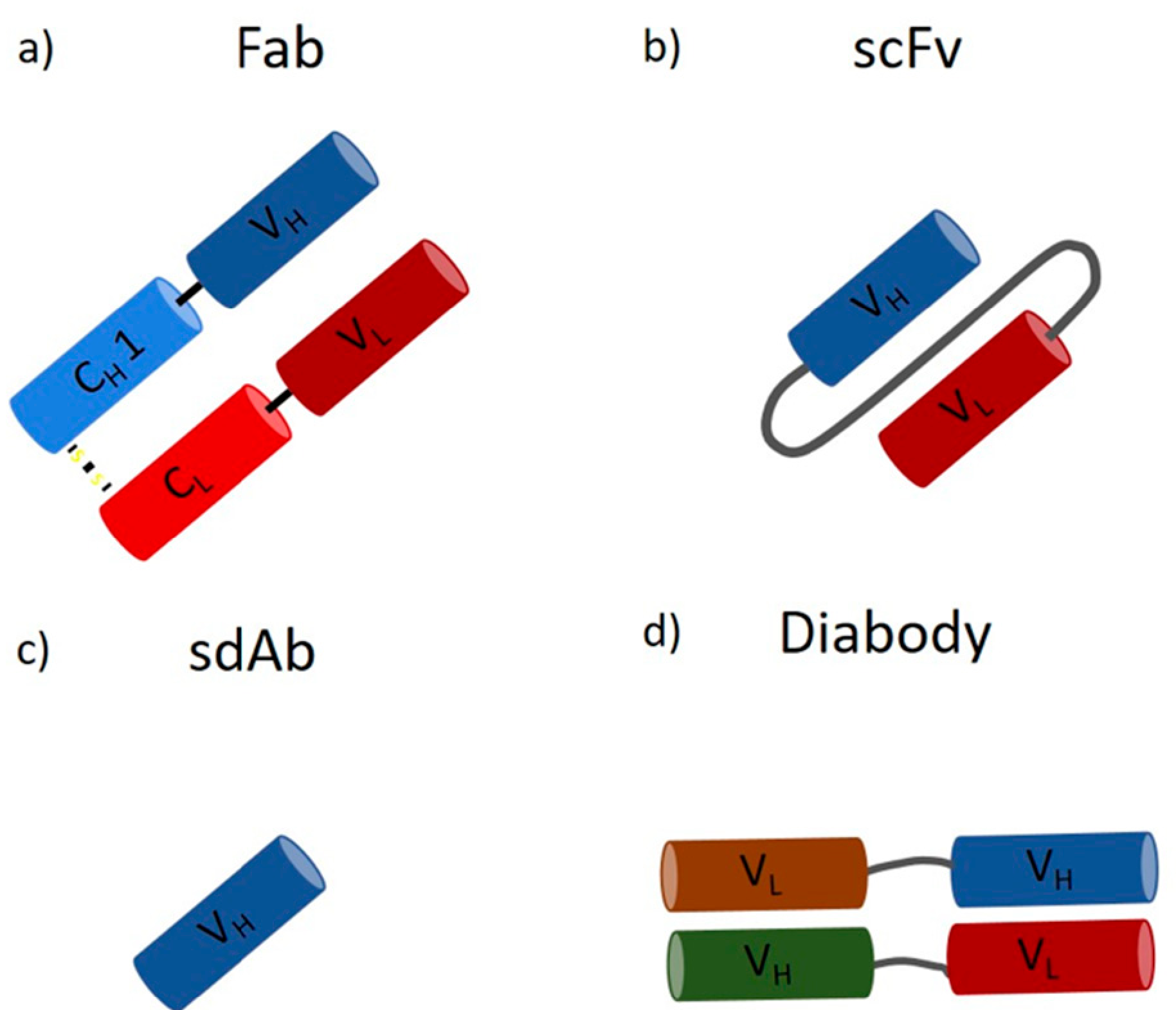
:1. Introduction
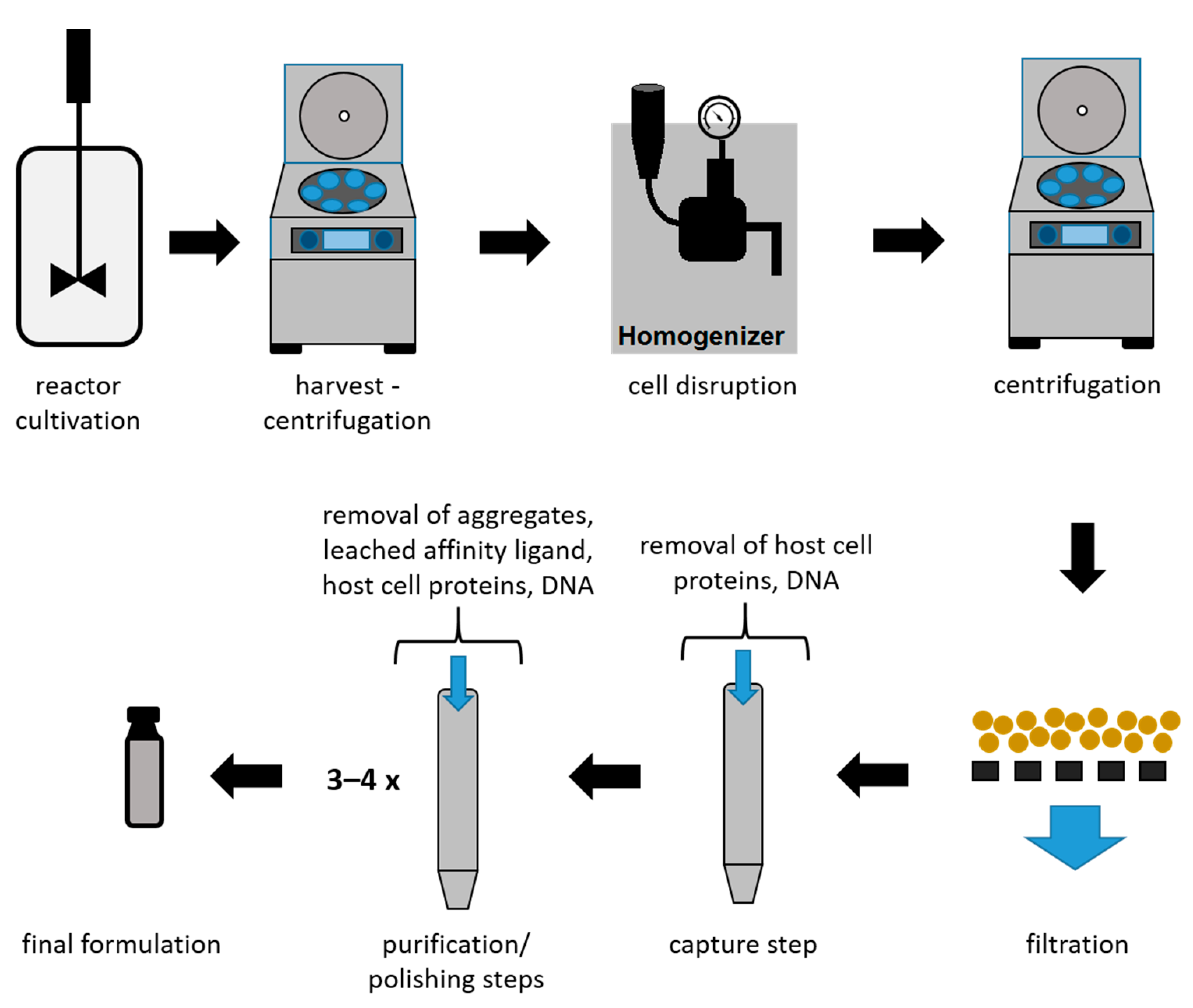
2. Downstream Processing of mAbs and Fabs
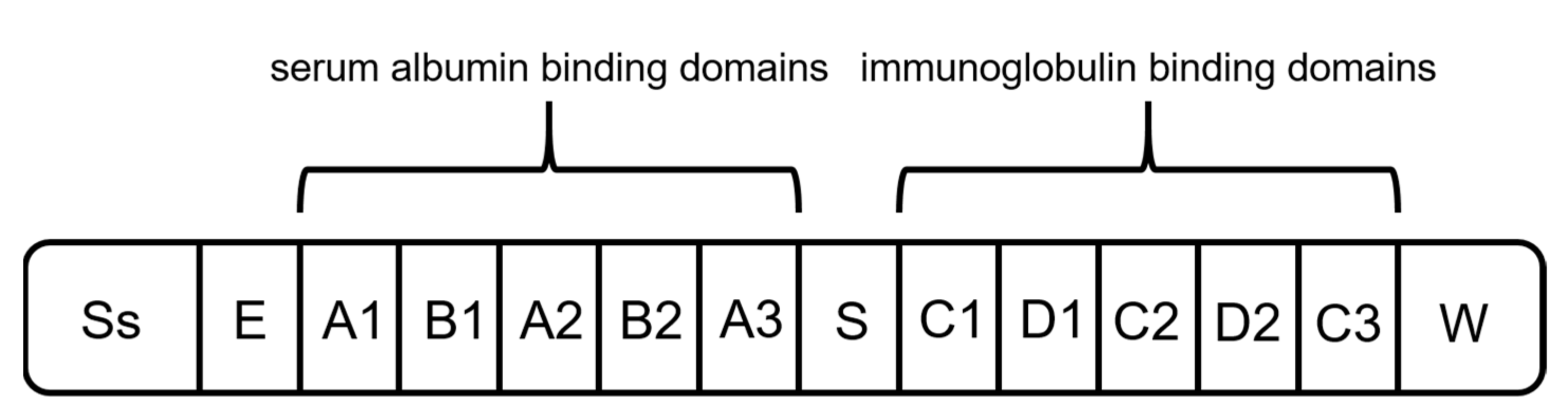
2.1. Protein A
2.2. Protein G
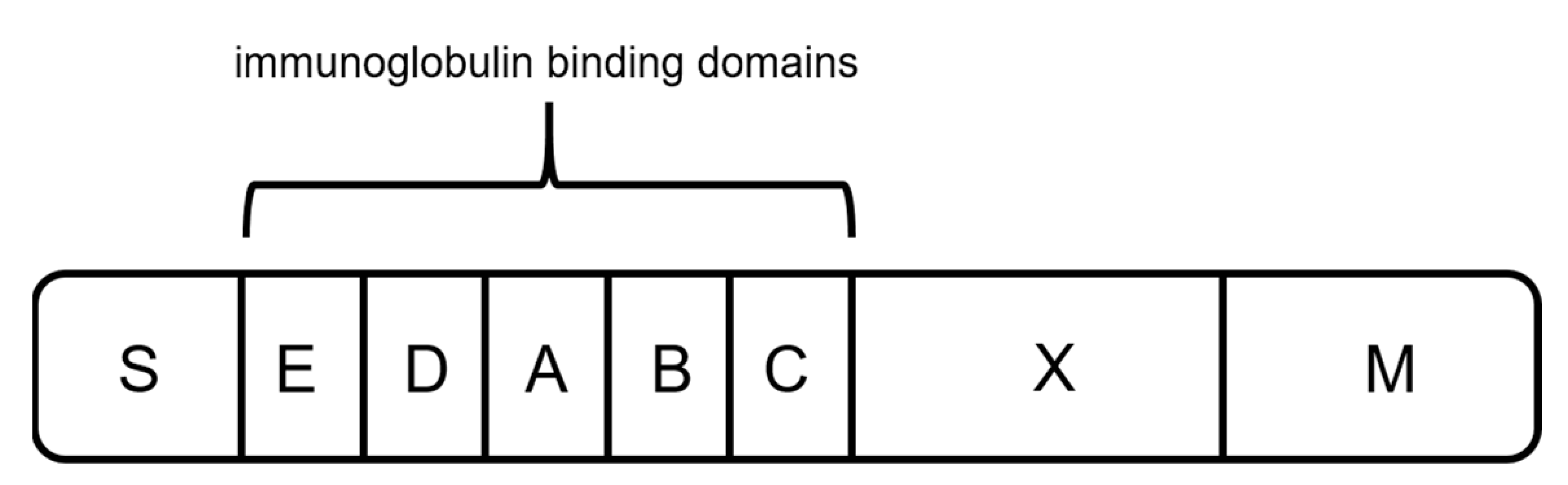
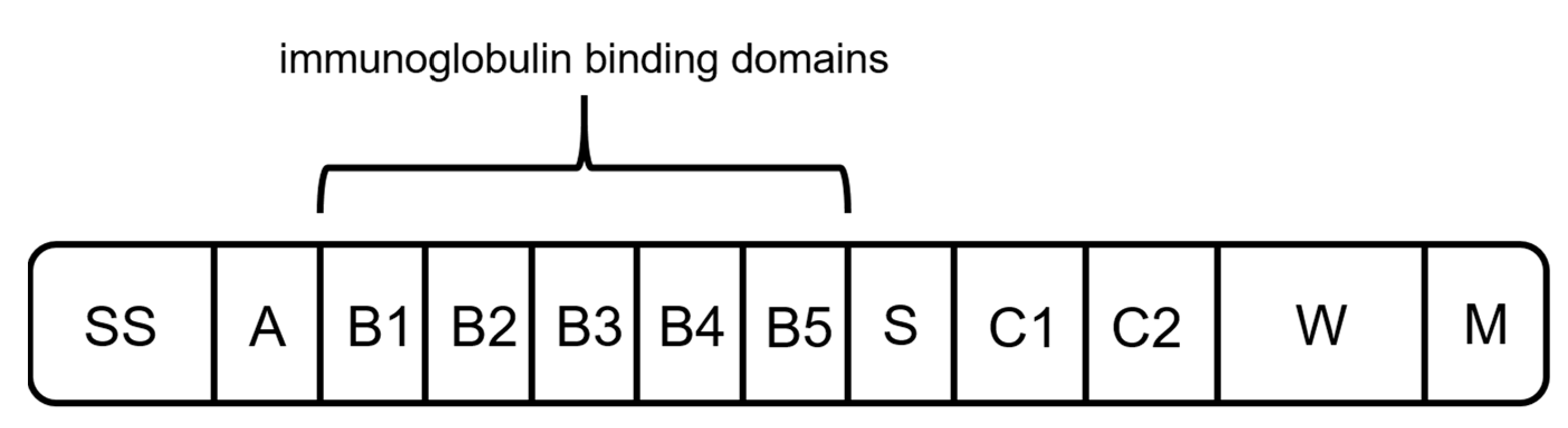
2.3. Protein L
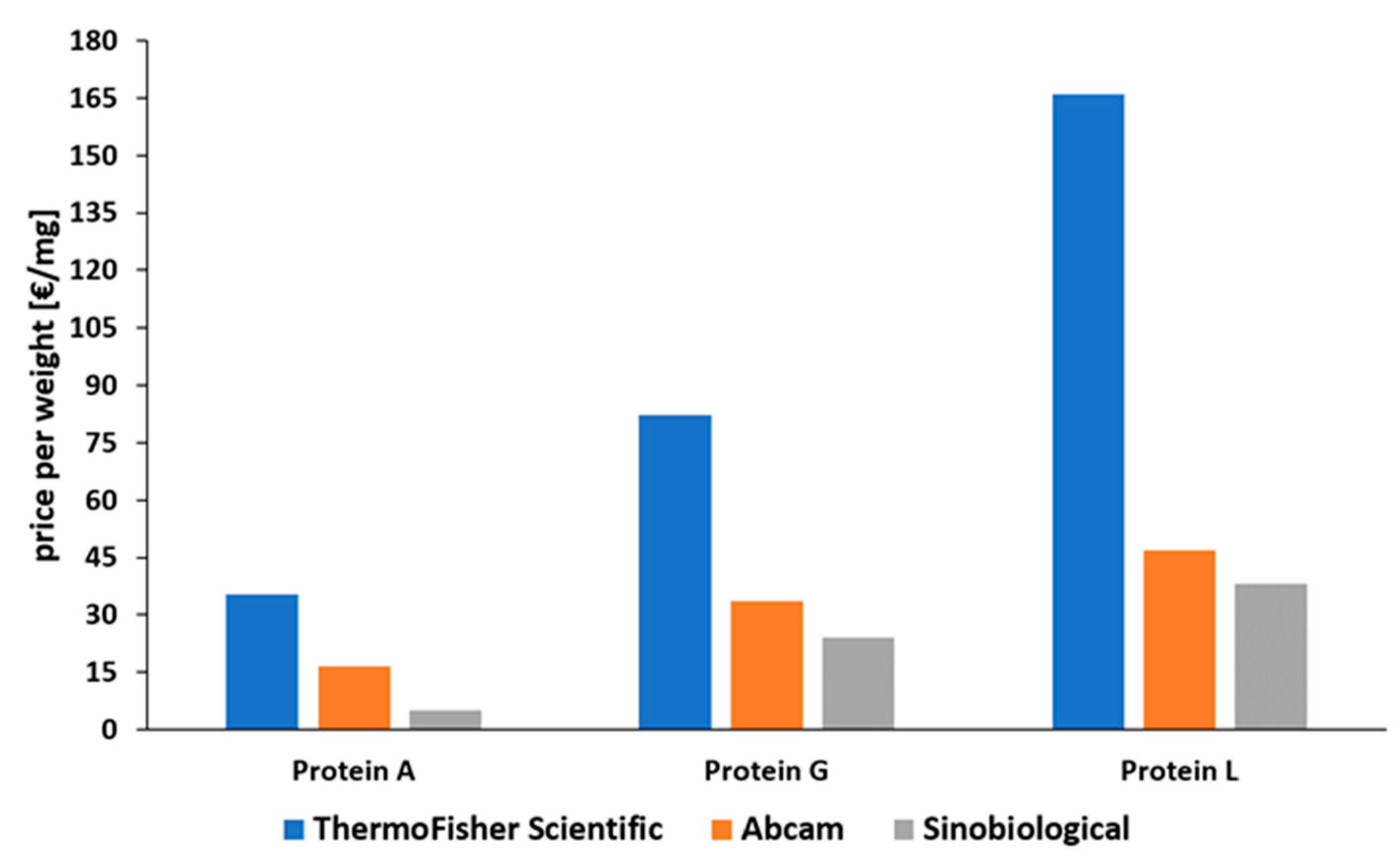
2.4. Comparison of Proteins A, G, and L
3. Is Protein L the Future?
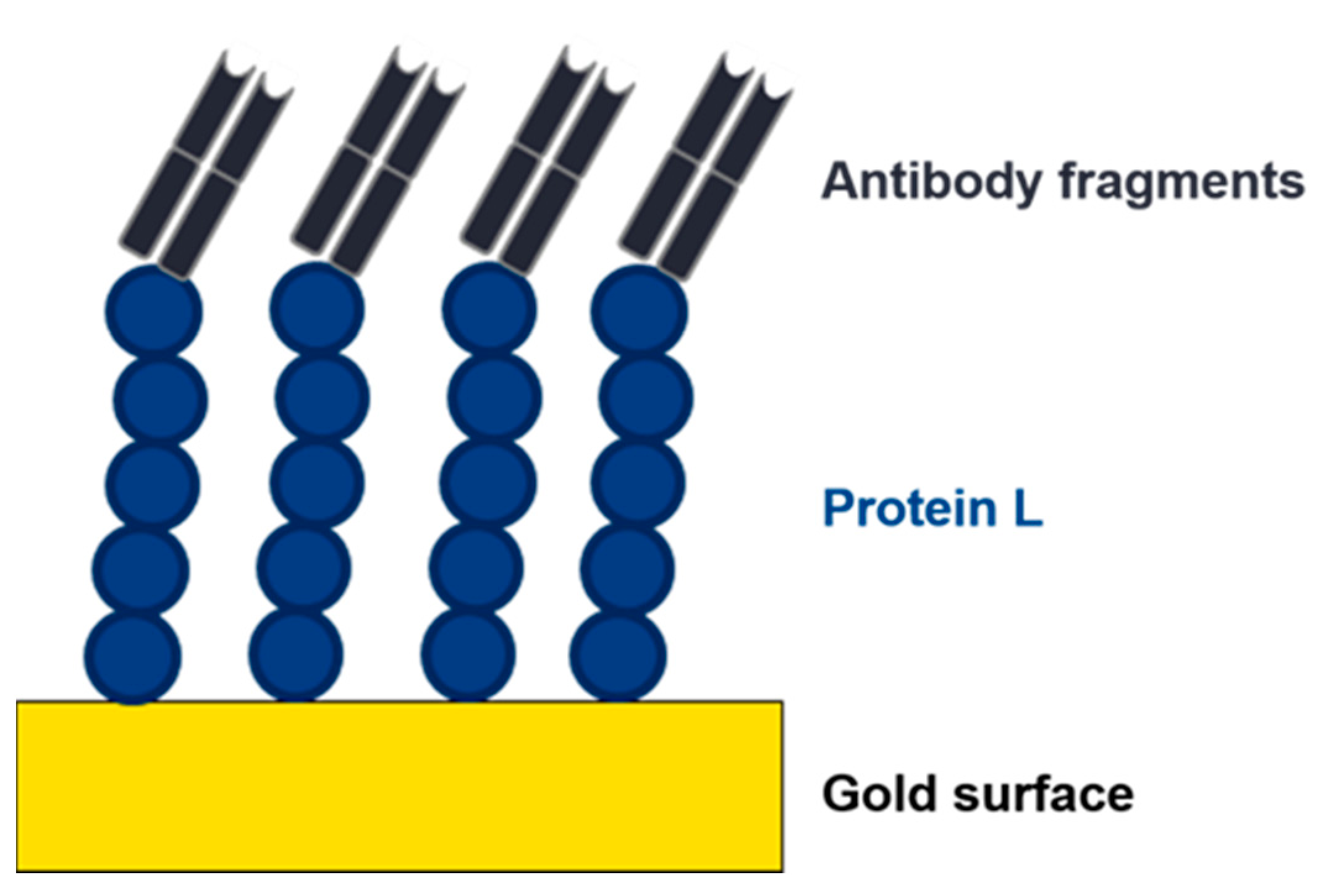
4. Further Applications of Protein L in Biomanufacturing
5. Final Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. mAbs 2015, 7, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grattendick, K.; Pross, S. Immunoglobulins. In xPharm: The Comprehensive Pharmacology Reference; Elsevier: Amsterdam, The Netherlands, 2007; pp. 1–6. [Google Scholar] [CrossRef]
- Conroy, P.J.; Hearty, S.; Leonard, P.; O’Kennedy, R.J. Antibody production, design and use for biosensor-based applications. Semin. Cell Dev. Biol. 2009, 20, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Burrell, C.J.; Howard, C.R.; Murphy, F.A. Adaptive Immune Responses to Infection. In Fenner and White’s Medical Virology; Elsevier: Amsterdam, The Netherlands, 2017; pp. 65–76. [Google Scholar] [CrossRef]
- Hnasko, R.M. The Biochemical Properties of Antibodies and Their Fragments. In Methods in Molecular Biology; Metzler, J.B., Ed.; Humana Press: New York, NY, USA, 2015; Volume 1318, pp. 1–14. [Google Scholar] [CrossRef]
- Nelson, A.L. Antibody fragments. In mAbs; Taylor & Francis: Oxfordshire, UK, 2010; Volume 2, pp. 77–83. [Google Scholar] [CrossRef] [Green Version]
- Esela-Culang, I.; Ekunik, V.; Eofran, Y. The Structural Basis of Antibody-Antigen Recognition. Front. Immunol. 2013, 4, 302. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Shen, G.; Yu, R. Aspects of recent development of immunosensors. Electrochem. Biosens. 2008, 237–260. [Google Scholar] [CrossRef]
- Schroeder, H.W.; Cavacini, L. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 2010, 125, S41–S52. [Google Scholar] [CrossRef] [Green Version]
- Gould, H.J.; Beavil, R.L. IgE. In Encyclopedia of Immunology; Elsevier: Amsterdam, The Netherlands, 1998; pp. 1202–1208. [Google Scholar] [CrossRef]
- Edholm, E.-S.; Bengten, E.; Wilson, M. Insights into the function of IgD. Dev. Comp. Immunol. 2011, 35, 1309–1316. [Google Scholar] [CrossRef]
- Woof, J.M.; Kerr, M.A. The function of immunoglobulin A in immunity. J. Pathol. 2005, 208, 270–282. [Google Scholar] [CrossRef] [Green Version]
- Sommerfeld, S.; Strube, J. Challenges in biotechnology production—generic processes and process optimization for monoclonal antibodies. Chem. Eng. Process. Process. Intensif. 2005, 44, 1123–1137. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [Green Version]
- Painter, R.H. IgG. In Encyclopedia of Immunology; Elsevier: Amsterdam, The Netherlands, 1998; pp. 1208–1211. [Google Scholar] [CrossRef]
- Rispens, T.; Vidarsson, G. Human IgG Subclasses. In Antibody Fc; Elsevier: Amsterdam, The Netherlands, 2014; pp. 159–177. [Google Scholar] [CrossRef]
- Bates, A.; Power, C.A. David vs. Goliath: The Structure, Function, and Clinical Prospects of Antibody Fragments. Antibodies 2019, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Grodzki, A.C.; Berenstein, E. Introduction to the Purification of Antibodies. In Immunocytochemical Methods and Protocols; Methods in Molecular Biology (Methods and Protocols); Humana Press: Totowa, NJ, USA, 2010; Volume 588, pp. 11–13. [Google Scholar] [CrossRef]
- Andrew, S.M.; Titus, J.A. Purification of Immunoglobulin G. In Current Protocols in Immunology; Wiley: Hoboken, NJ, USA, 2001. [Google Scholar]
- Miller, L.G.; Goldstein, G.; Murphy, M.; Ginns, L.C. Reversible Alterations in Immunoregulatory T Cells in Smoking. Chest 1982, 82, 526–529. [Google Scholar] [CrossRef]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1–30. [Google Scholar] [CrossRef]
- Tomita, M.; Tsumoto, K. Hybridoma technologies for antibody production. Immunotherapy 2011, 3, 371–380. [Google Scholar] [CrossRef]
- Spadiut, O.; Capone, S.; Krainer, F.; Glieder, A.; Herwig, C. Microbials for the production of monoclonal antibodies and antibody fragments. Trends Biotechnol. 2014, 32, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Shukla, A.A.; Thömmes, J. Recent advances in large-scale production of monoclonal antibodies and related proteins. Trends Biotechnol. 2010, 28, 253–261. [Google Scholar] [CrossRef]
- Gerngross, T.U. Advances in the production of human therapeutic proteins in yeasts and filamentous fungi. Nat. Biotechnol. 2004, 22, 1409–1414. [Google Scholar] [CrossRef]
- Gerngross, T. Production of Complex Human Glycoproteins in Yeast. Adv. Exp. Med. Biol. 2005, 564, 139. [Google Scholar] [CrossRef] [Green Version]
- Rodrigo, G.; Gruvegård, M.; Van Alstine, J.M. Antibody Fragments and Their Purification by Protein L Affinity Chromatography. Antibodies 2015, 4, 259–277. [Google Scholar] [CrossRef] [Green Version]
- Nelson, A.L.; Reichert, J.M. Development trends for therapeutic antibody fragments. Nat. Biotechnol. 2009, 27, 331–337. [Google Scholar] [CrossRef]
- Xenaki, K.T.; Oliveira, S.; Henegouwen, P.M.P.V.B.E. Antibody or Antibody Fragments: Implications for Molecular Imaging and Targeted Therapy of Solid Tumors. Front. Immunol. 2017, 8, 1287. [Google Scholar] [CrossRef]
- Ahamadi-Fesharaki, R.; Fateh, A.; Vaziri, F.; Solgi, G.; Siadat, S.D.; Mahboudi, F.; Rahimi-Jamnani, F. Single-Chain Variable Fragment-Based Bispecific Antibodies: Hitting Two Targets with One Sophisticated Arrow. Mol. Ther. Oncolytics 2019, 14, 38–56. [Google Scholar] [CrossRef] [Green Version]
- Rader, C. Overview on Concepts and Applications of Fab Antibody Fragments. Curr. Protoc. Protein Sci. 2009, 55, 6.9.1–6.9.14. [Google Scholar] [CrossRef]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef]
- Wasalathanthri, D.P.; Rehmann, M.S.; Song, Y.; Gu, Y.; Mi, L.; Shao, C.; Chemmalil, L.; Lee, J.; Ghose, S.; Borys, M.C.; et al. Technology outlook for real-time quality attribute and process parameter monitoring in biopharmaceutical development—A review. Biotechnol. Bioeng. 2020, 117, 3182–3198. [Google Scholar] [CrossRef]
- Guerra, A.; Von Stosch, M.; Glassey, J. Toward biotherapeutic product real-time quality monitoring. Crit. Rev. Biotechnol. 2019, 39, 289–305. [Google Scholar] [CrossRef]
- Arakawa, T.; Tsumoto, K.; Ejima, D. Alternative downstream processes for production of antibodies and antibody fragments. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 2032–2040. [Google Scholar] [CrossRef]
- Prasnikar, J.; Škerlj, T. New product development process and time-to-market in the generic pharmaceutical industry. Ind. Mark. Manag. 2006, 35, 690–702. [Google Scholar] [CrossRef]
- ICH Guideline Q8 (R2) on Pharmaceutical Development. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-11.pdf (accessed on 13 June 2020).
- Katsuda, T.; Sonoda, H.; Kumada, Y.; Yamaji, H. Production of Antibody Fragments in Escherichia coli. In Antibody Engineering. Methods in Molecular Biology (Methods and Protocols); Humana Press: Totowa, NJ, USA, 2012; Volume 907, pp. 305–324. [Google Scholar] [CrossRef]
- Wurm, D.J.; Slouka, C.; Bosilj, T.; Herwig, C.; Spadiut, O. How to trigger periplasmic release in recombinant Escherichia coli: A comparative analysis. Eng. Life Sci. 2016, 17, 215–222. [Google Scholar] [CrossRef]
- Ramos-De-La-Peña, A.M.; González-Valdez, J.; Aguilar, O. Protein A chromatography: Challenges and progress in the purification of monoclonal antibodies. J. Sep. Sci. 2019, 42, 1816–1827. [Google Scholar] [CrossRef]
- Shukla, A.A.; Hubbard, B.; Tressel, T.; Guhan, S.; Low, D. Downstream processing of monoclonal antibodies—Application of platform approaches. J. Chromatogr. B 2007, 848, 28–39. [Google Scholar] [CrossRef]
- Mustafaoglu, N.; Kiziltepe, T.; Bilgicer, B. Antibody purification via affinity membrane chromatography method utilizing nucleotide binding site targeting with a small molecule. Analyst 2016, 141, 6571–6582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayyar, B.V.; Arora, S.; Murphy, C.; O’Kennedy, R. Affinity chromatography as a tool for antibody purification. Methods 2012, 56, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Chahar, D.S.; Ravindran, S.; Pisal, S. Monoclonal antibody purification and its progression to commercial scale. Biologicals 2020, 63, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ghose, S.; Hubbard, B.; Cramer, S.M. Evaluation and comparison of alternatives to Protein A chromatography: Mimetic and hydrophobic charge induction chromatographic stationary phases. J. Chromatogr. A 2006, 1122, 144–152. [Google Scholar] [CrossRef]
- Fahrner, R.L.; Knudsen, H.L.; Basey, C.D.; Galan, W.; Feuerhelm, D.; Vanderlaan, M.; Blank, G.S. Industrial Purification of Pharmaceutical Antibodies: Development, Operation, and Validation of Chromatography Processes. Biotechnol. Genet. Eng. Rev. 2001, 18, 301–327. [Google Scholar] [CrossRef]
- Ishihara, T.; Yamamoto, S. Optimization of monoclonal antibody purification by ion-exchange chromatography: Application of simple methods with linear gradient elution experimental data. J. Chromatogr. A 2005, 1069, 99–106. [Google Scholar] [CrossRef]
- Grodzki, A.C.; Berenstein, E. Antibody Purification: Ion-Exchange Chromatography. In Immunocytochemical Methods and Protocols; Methods in Molecular Biology (Methods and Protocols); Humana Press: Totowa, NJ, USA, 2009; Volume 588, pp. 27–32. [Google Scholar] [CrossRef]
- Ghose, S.; Tao, Y.; Conley, L.; Cecchini, D. Purification of monoclonal antibodies by hydrophobic interaction chromatography under no-salt conditions. mAbs 2013, 5, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Gagnon, P. Technology trends in antibody purification. J. Chromatogr. A 2012, 1221, 57–70. [Google Scholar] [CrossRef]
- Ulmer, N.; Vogg, S.; Müller-Späth, T.; Morbidelli, M. Purification of Human Monoclonal Antibodies and Their Fragments. In Human Monoclonal Antibodies: Methods and Protocols; Steinitz, M., Ed.; Springer: New York, NY, USA, 2019; pp. 163–188. [Google Scholar] [CrossRef]
- Strube, J.; Grote, F.; Ditz, R. Bioprocess Design and Production Technology for the Future. In Biopharmaceutical Production Technology; Wiley: Hoboken, NJ, USA, 2012; pp. 657–705. [Google Scholar]
- Hage, D.S. Affinity Chromatography: A Review of Clinical Applications. Clin. Chem. 1999, 45, 593–615. [Google Scholar] [CrossRef]
- Ståhl, S.; Kronqvist, N.; Jonsson, A.; Löfblom, J. Affinity proteins and their generation. J. Chem. Technol. Biotechnol. 2012, 88, 25–38. [Google Scholar] [CrossRef]
- Kruljec, N.; Bratkovič, T. Alternative Affinity Ligands for Immunoglobulins. Bioconjugate Chem. 2017, 28, 2009–2030. [Google Scholar] [CrossRef]
- Hober, S.; Nord, K.; Linhult, M. Protein A chromatography for antibody purification. J. Chromatogr. B 2007, 848, 40–47. [Google Scholar] [CrossRef]
- Falugi, F.; Kim, H.K.; Missiakas, D.M.; Schneewind, O. Role of Protein A in the Evasion of Host Adaptive Immune Responses by Staphylococcus aureus. mBio 2013, 4, e00575-13. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.D.; DeLeo, F.R. Staphylococcus aureus Protein A Promotes Immune Suppression. mBio 2013, 4, e00764-13. [Google Scholar] [CrossRef]
- Palmqvist, N.; Foster, T.; Tarkowski, A.; Josefsson, E. Protein A is a virulence factor in Staphylococcus aureus arthritis and septic death. Microb. Pathog. 2002, 33, 239–249. [Google Scholar] [CrossRef]
- Choe, W.; Durgannavar, T.A.; Chung, S.J. Fc-Binding Ligands of Immunoglobulin G: An Overview of High Affinity Proteins and Peptides. Materials 2016, 9, 994. [Google Scholar] [CrossRef] [Green Version]
- Abouelkhair, M.A.; Bemis, D.A.; Kania, S.A. Characterization of recombinant wild-type and nontoxigenic protein A from Staphylococcus pseudintermedius. Virulence 2018, 9, 1050–1061. [Google Scholar] [CrossRef] [Green Version]
- Graille, M.; Stura, E.A.; Corper, A.L.; Sutton, B.J.; Taussig, M.J.; Charbonnier, J.-B.; Silverman, G.J. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: Structural basis for recognition of B-cell receptors and superantigen activity. Proc. Natl. Acad. Sci. USA 2000, 97, 5399–5404. [Google Scholar] [CrossRef] [Green Version]
- Romagnani, S.; Giudizi, M.G.; Del Prete, G.; Maggi, E.; Biagiotti, R.; Almerigogna, F.; Ricci, M. Demonstration on protein A of two distinct immunoglobulin-binding sites and their role in the mitogenic activity of Staphylococcus aureus Cowan I on human B cells. J. Immunol. 1982, 129, 596–602. [Google Scholar]
- Hao, J.; Xu, L.; He, H.; Du, X.; Jia, L. High-level expression of Staphylococcal Protein A in Pichia pastoris and purification and characterization of the recombinant protein. Protein Expr. Purif. 2013, 90, 178–185. [Google Scholar] [CrossRef]
- Sjöbring, U.; Björck, L.; Kastern, W. Streptococcal protein G. Gene structure and protein binding properties. J. Biol. Chem. 1991, 266, 399–405. [Google Scholar] [CrossRef]
- Zhang, H.-C.; Yang, J.; Yang, G.-W.; Wang, X.-J.; Fan, H.-T. Production of recombinant protein G through high-density fermentation of engineered bacteria as well as purification. Mol. Med. Rep. 2015, 12, 3132–3138. [Google Scholar] [CrossRef] [Green Version]
- Jha, R.K.; Gaiotto, T.; Bradbury, A.R.; Strauss, C.E. An improved Protein G with higher affinity for human/rabbit IgG Fc domains exploiting a computationally designed polar network. Protein Eng. Des. Sel. 2014, 27, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Derrick, J.P.; Wigley, D.B. Crystal structure of a streptococcal protein G domain bound to an Fab fragment. Nat. Cell Biol. 1992, 359, 752–754. [Google Scholar] [CrossRef] [PubMed]
- Nilson, B.H.; Lögdberg, L.; Kastern, W.; Björck, L.; Åkerström, B. Purification of antibodies using protein L-binding framework structures in the light chain variable domain. J. Immunol. Methods 1993, 164, 33–40. [Google Scholar] [CrossRef]
- Page, M.; Thorpe, R. Purification of IgG Using Protein A or Protein G. In The Protein Protocols Handbook; Humana Press: Totowa, NJ, USA, 2009; pp. 1761–1763. [Google Scholar] [CrossRef]
- Kastern, W.; Sjöbring, U.; Björck, L. Structure of peptostreptococcal protein L and identification of a repeated immunoglobulin light chain-binding domain. J. Biol. Chem. 1992, 267, 12820–12825. [Google Scholar] [CrossRef]
- Myhre, E.B.; Erntell, M. A non-immune interaction between the light chain of human immunoglobulin and a surface component of a Peptococcus magnus strain. Mol. Immunol. 1985, 22, 879–885. [Google Scholar] [CrossRef]
- Darcy, E.; Leonard, P.; Fitzgerald, J.; Danaher, M.; O’Kennedy, R. Purification of Antibodies Using Affinity Chromatography. In Protein Chromatography; Methods in Molecular Biology (Methods and Protocols); Humana Press: New York, NY, USA, 2011; pp. 369–382. [Google Scholar] [CrossRef]
- Graille, M.; Stura, E.A.; Housden, N.G.; Beckingham, J.A.; Bottomley, S.P.; Beale, D.; Taussig, M.J.; Sutton, B.J.; Gore, M.G.; Charbonnier, J.-B. Complex between Peptostreptococcus magnus Protein L and a Human Antibody Reveals Structural Convergence in the Interaction Modes of Fab Binding Proteins. Structure 2001, 9, 679–687. [Google Scholar] [CrossRef]
- Akerström, B.; Björck, L. Protein L: An immunoglobulin light chain-binding bacterial protein. Characterization of binding and physicochemical properties. J. Biol. Chem. 1989, 264, 19740–19746. [Google Scholar] [CrossRef]
- Wikstroem, M.; Drakenberg, T.; Forsen, S.; Sjoebring, U.; Bjoerck, L. Three-dimensional solution structure of an immunoglobulin light chain-binding domain of protein L. Comparison with the IgG-binding domains of protein G. Biochemistry 1994, 33, 14011–14017. [Google Scholar] [CrossRef]
- Nilson, B.H.; Solomon, A.; Björck, L.; Akerström, B. Protein L from Peptostreptococcus magnus binds to the kappa light chain variable domain. J. Biol. Chem. 1992, 267, 2234–2239. [Google Scholar] [CrossRef]
- Griep, R.; McDougall, J. Analysis and Purification of Antibody Fragments Using Protein A, Protein G, and Protein L. Adv. Struct. Saf. Stud. 2010, 301–315. [Google Scholar] [CrossRef]
- Tocaj, A.; Sjöbring, U.; Björck, L.; Holst, O. High level expression of protein L, an immunoglobulin-binding protein, in Escherichia coli. J. Ferment. Bioeng. 1995, 80, 1–5. [Google Scholar] [CrossRef]
- ThermoScientific Fisher. Available online: https://www.thermofisher.com/order/catalog/product/21189 (accessed on 10 December 2020).
- Abcam. Available online: https://www.abcam.com/recombinant-protein-l-ab155706.html (accessed on 10 December 2020).
- Sinobiological. Available online: https://www.sinobiological.com/recombinant-proteins/protein-l-11044-h07e (accessed on 10 December 2020).
- Chateau, M.; Nilson, B.H.K.; Erntell, M.; Myhre, E.; Magnusson, C.G.M.; Åkerström, B.; Björck, L. On the Interaction between Protein L and Immunoglobulins of Various Mammalian Species. Scand. J. Immunol. 1993, 37, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Paloni, M.; Cavallotti, C. Molecular Modeling of the Interaction of Protein L with Antibodies. ACS Omega 2017, 2, 6464–6472. [Google Scholar] [CrossRef] [PubMed]
- Sheng, S.; Kong, F. Separation of antigens and antibodies by immunoaffinity chromatography. Pharm. Biol. 2012, 50, 1038–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5, 172. [Google Scholar] [CrossRef] [Green Version]
- Persson, T.; Eckersten, H.; Elen, O.; Hjelkrem, A.-G.R.; Markgren, J.; Söderström, M.; Börjesson, T. Predicting deoxynivalenol in oats under conditions representing Scandinavian production regions. Food Addit. Contam. Part A 2017, 34, 1026–1038. [Google Scholar] [CrossRef]
- Su, Q.; Ganesh, S.; Moreno, M.; Bommireddy, Y.; Gonzalez, M.; Reklaitis, G.V.; Nagy, Z.K. A perspective on Quality-by-Control (QbC) in pharmaceutical continuous manufacturing. Comput. Chem. Eng. 2019, 125, 216–231. [Google Scholar] [CrossRef]
- Gundinger, T.; Pansy, A.; Spadiut, O. A sensitive and robust HPLC method to quantify recombinant antibody fragments in E. coli crude cell lysate. J. Chromatogr. B 2018, 1083, 242–248. [Google Scholar] [CrossRef]
- Nguyen, B.T.; Koh, G.; Lim, H.S.; Chua, A.J.S.; Ng, M.M.L.; Toh, C.-S. Membrane-Based Electrochemical Nanobiosensor for the Detection of Virus. Anal. Chem. 2009, 81, 7226–7234. [Google Scholar] [CrossRef]
- Yu, H.; Kim, K.; Ma, K.; Lee, W.; Choi, J.-W.; Yun, C.-O.; Kim, D. Enhanced detection of virus particles by nanoisland-based localized surface plasmon resonance. Biosens. Bioelectron. 2013, 41, 249–255. [Google Scholar] [CrossRef]
- Laghrib, F.; Saqrane, S.; El Bouabi, Y.; Farahi, A.; Bakasse, M.; Lahrich, S.; El Mhammedi, M. Current progress on COVID-19 related to biosensing technologies: New opportunity for detection and monitoring of viruses. Microchem. J. 2021, 160, 105606. [Google Scholar] [CrossRef]
- Douzi, B. Protein–Protein Interactions: Surface Plasmon Resonance. In Bacterial Protein Secretion Systems: Methods and Protocols; Journet, L., Cascales, E., Eds.; Humana Press: New York, NY, USA, 2017; pp. 257–275. [Google Scholar] [CrossRef]
- Sultana, A.; Lee, J.E. Measuring Protein-Protein and Protein-Nucleic Acid Interactions by Biolayer Interferometry. Curr. Protoc. Protein Sci. 2015, 79, 19.25.1–19.25.26. [Google Scholar] [CrossRef]
- Anspach, F.B.; Petsch, D. Membrane adsorbers for selective endotoxin removal from protein solutions. Process. Biochem. 2000, 35, 1005–1012. [Google Scholar] [CrossRef]
- Boi, C.; Malavasi, A.; Carbonell, R.G.; Gilleskie, G. A direct comparison between membrane adsorber and packed column chromatography performance. J. Chromatogr. A 2020, 1612, 460629. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Y.; Li, Y. Protein L chromatography: A useful tool for monitoring/separating homodimers during the purification of IgG-like asymmetric bispecific antibodies. Protein Expr. Purif. 2020, 175, 105711. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Advantages | Disadvantages | DSP Step | References |
|---|---|---|---|---|
| AC |
|
| capture | [42,43,44,45] |
| CEX |
|
| capture (Fc lacking mAbs) purification polishing | [17,27,44,46,47] |
| AEX |
|
| purification polishing | [13,44,46,47,48] |
| HIC |
|
| polishing | [49,50] |
| SEC |
|
| polishing | [51] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kittler, S.; Besleaga, M.; Ebner, J.; Spadiut, O. Protein L—More Than Just an Affinity Ligand. Processes 2021, 9, 874. https://doi.org/10.3390/pr9050874
Kittler S, Besleaga M, Ebner J, Spadiut O. Protein L—More Than Just an Affinity Ligand. Processes. 2021; 9(5):874. https://doi.org/10.3390/pr9050874
Chicago/Turabian StyleKittler, Stefan, Mihail Besleaga, Julian Ebner, and Oliver Spadiut. 2021. "Protein L—More Than Just an Affinity Ligand" Processes 9, no. 5: 874. https://doi.org/10.3390/pr9050874
APA StyleKittler, S., Besleaga, M., Ebner, J., & Spadiut, O. (2021). Protein L—More Than Just an Affinity Ligand. Processes, 9(5), 874. https://doi.org/10.3390/pr9050874