
Calcium Carbonate as Functional Filler in Polyamide 12-Manipulation of the Thermal and Mechanical Properties


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Functional Filler Production
Surface Modification and Amount
2.3. Composite Manufacturing
Formulation Mixing and Compounding
2.4. Composite Analysis
2.4.1. Scanning Electron Microscopy
2.4.2. Thermal Response Analysis
Melt Flow Index
Differential Scanning Calorimetry
2.4.3. Mechanical Tensile Properties
3. Results and Discussion
3.1. Mineral Difference
3.2. Homogeneity of Filler Distribution
3.3. Melt Properties
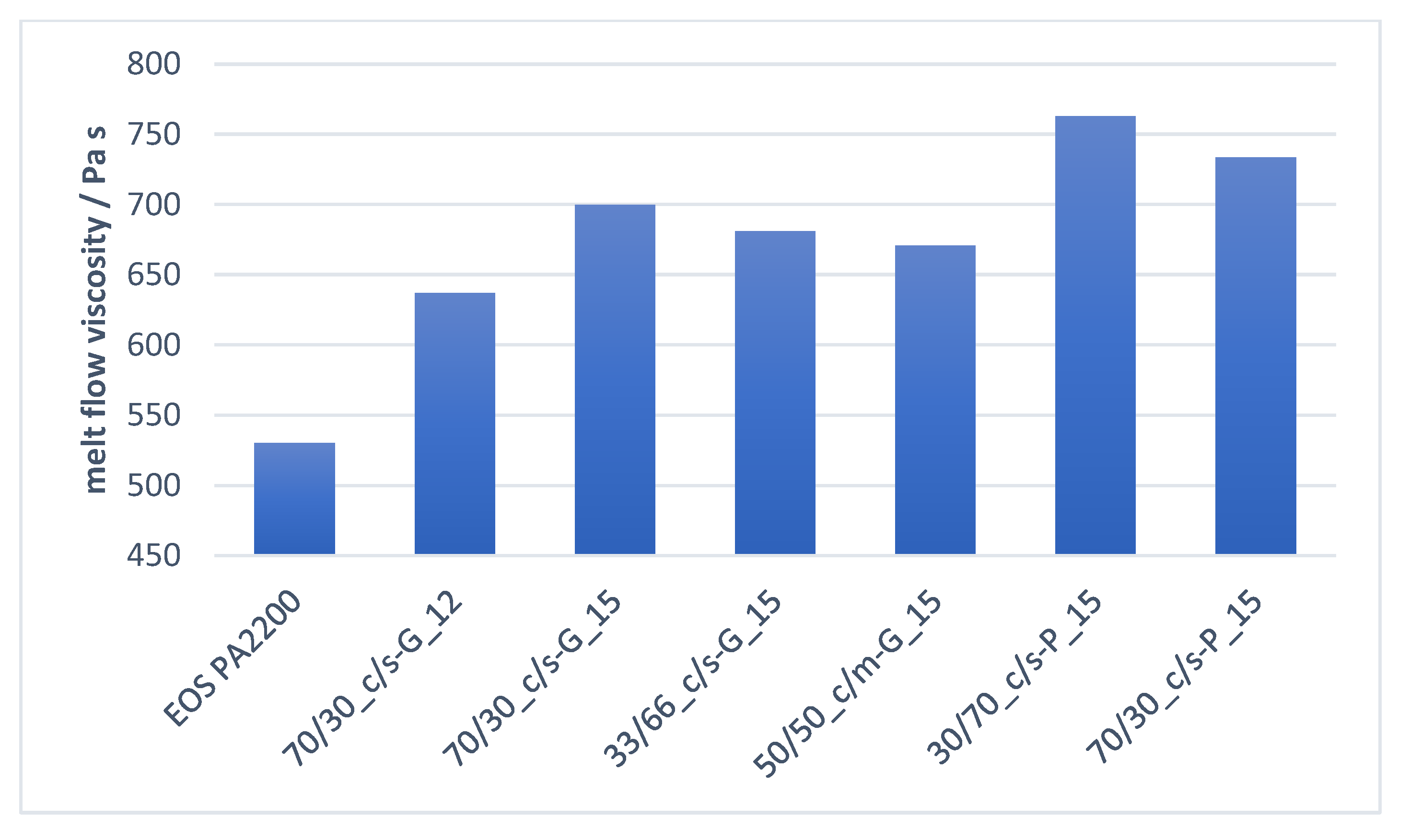
3.3.1. Influence of Untreated Functional Filler Ratio/Morphology on Melt Properties
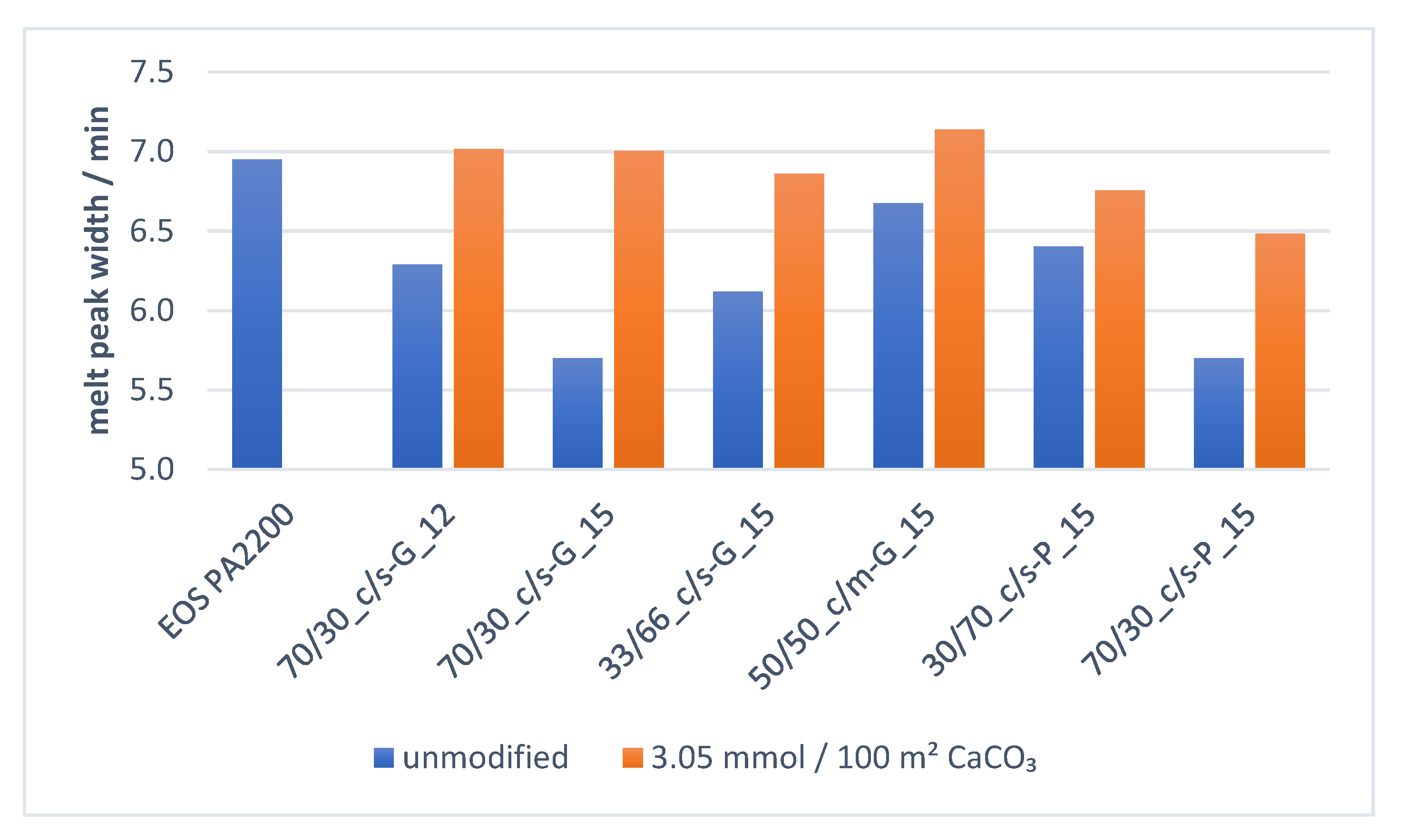
3.3.2. Effect of Surface Modifier Amount on Melt Properties
3.4. Crystallization Properties
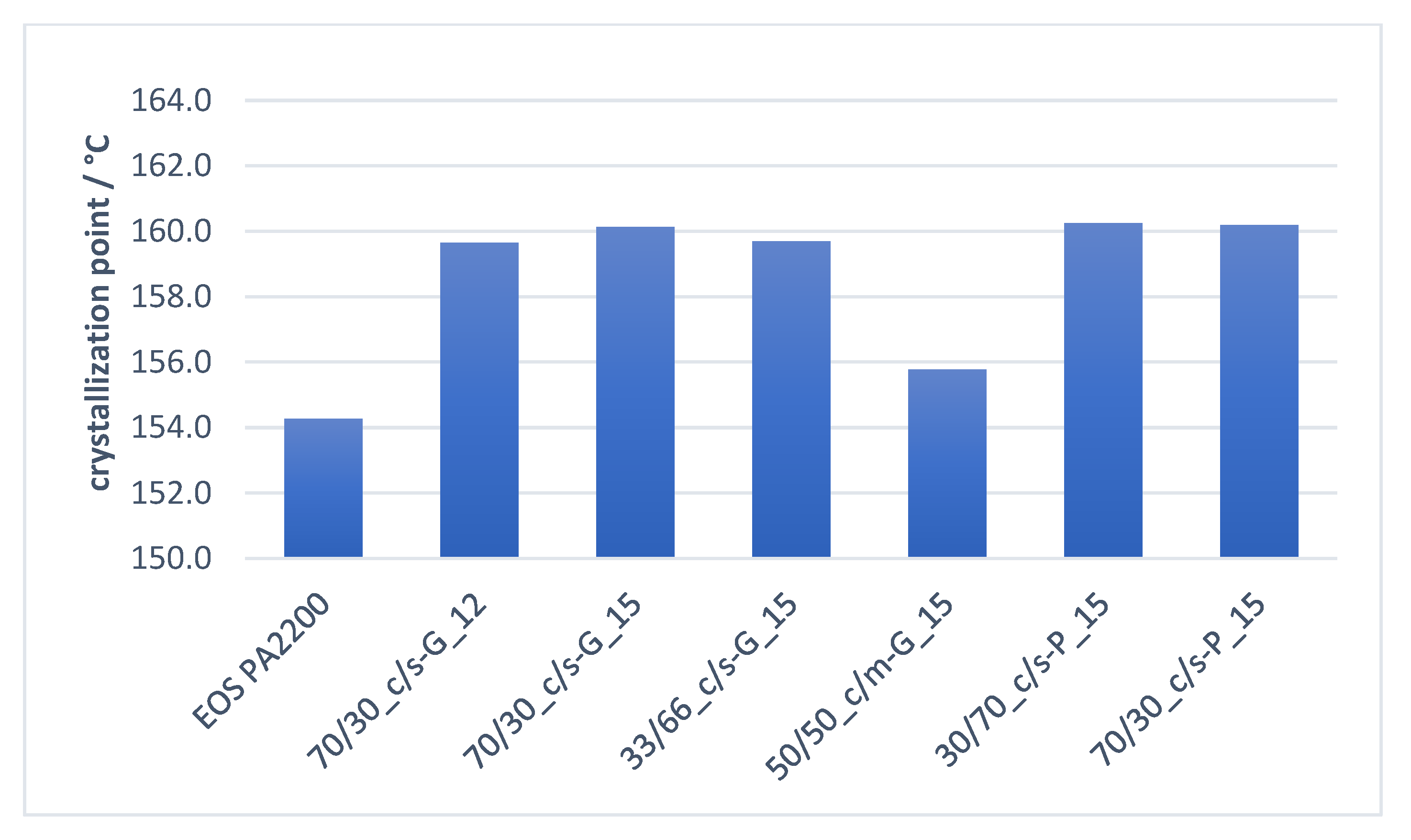
3.4.1. Influence of Untreated Functional Filler Ratio/Morphology on Crystallization Properties
3.4.2. Effect of Surface Modifier Amount on Crystallization Properties
3.5. Ductility
3.5.1. Influence of Untreated Functional Filler Ratio/Morphology on Ductility
3.5.2. Effect of Surface Modifier Amount on Ductility
4. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Schmid, M. Laser Sintering with Plastics: Technology, Processes, and Materials; Carl Hanser Verlag GmbH Co. KG: Munich, Germany, 2018. [Google Scholar]
- Mueller, B. Additive manufacturing technologies–rapid prototyping to direct digital manufacturing. Assem. Autom. 2012, 32. [Google Scholar] [CrossRef]
- Hague, R.; Mansour, S.; Saleh, N. Design opportunities with rapid manufacturing. Assem. Autom. 2003, 23, 346–356. [Google Scholar] [CrossRef]
- Gibson, I.; Rosen, D.; Stucker, B. Additive Manufacturing Technologies: 3D Printing, Rapid Prototyping, and Direct Digital Manufacturing; Springer: New York, NY, USA, 2014. [Google Scholar]
- Kumar, S. Selective Laser Sintering/Melting. In Comprehensive Materials Processing; Hashmi, S., Batalha, G.F., Van Tyne, C.J., Yilbas, B., Eds.; Elsevier: Oxford, UK, 2014; pp. 93–134. [Google Scholar]
- Liu-Lan, L.; Yu-Sheng, S.; Fan-di, Z.; Shu-huai, H. Microstructure of selective laser sintered polyamide. J. Wuhan Univ. Technol. Mater. Sci. Ed. 2003, 18, 60–63. [Google Scholar] [CrossRef]
- Childs, T.; Berzins, M.; Ryder, G.; Tontowi, A. Selective laser sintering of an amorphous polymer—simulations and experiments. Proc. Inst. Mech. Eng. Part. B J. Eng. Manuf. 1999, 213, 333–349. [Google Scholar] [CrossRef]
- Schmidt, M.; Pohle, D.; Rechtenwald, T. Selective laser sintering of PEEK. CIRP Ann. 2007, 56, 205–208. [Google Scholar] [CrossRef]
- Koo, J.; Lao, S.; Ho, W.; Ngyuen, K.; Cheng, J.; Pilato, L.; Wissler, G.; Ervin, M. Polyamide nanocomposites for selective laser sintering. In Proceedings of International Solid Freeform Fabrication Symposium; University of Texas Libraries: Austin, TX, USA, 2006; pp. 392–409. [Google Scholar]
- Schmid, M.; Amado, A.; Wegener, K. Polymer Powders for Selective Laser Sintering (SLS); ETH-Zürich: Zürich, Switzerland, 2014. [Google Scholar]
- Kim, J.; Creasy, T. Selective laser sintering characteristics of nylon 6/clay-reinforced nanocomposite. Polym. Test. 2004, 23, 629–636. [Google Scholar] [CrossRef]
- Goodridge, R.; Tuck, C.; Hague, R. Laser sintering of polyamides and other polymers. Prog. Mater. Sci. 2012, 57, 229–267. [Google Scholar] [CrossRef]
- Guo, Y.; Jiang, K.; Bourell, D.L. Preparation and laser sintering of limestone PA 12 composite. Polym. Test. 2014, 37, 210–215. [Google Scholar] [CrossRef]
- Sreenivasan, R.; Goel, A.; Bourell, D.L. Sustainability issues in laser-based additive manufacturing. Phys. Procedia 2010, 5, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Ford, S.; Despeisse, M. Additive manufacturing and sustainability: An exploratory study of the advantages and challenges. J. Clean. Prod. 2016, 137, 1573–1587. [Google Scholar] [CrossRef]
- Le Bourhis, F.; Kerbrat, O.; Hascoet, J.-Y.; Mognol, P. Sustainable manufacturing: Evaluation and modeling of environmental impacts in additive manufacturing. Int. J. Adv. Manuf. Technol. 2013, 69, 1927–1939. [Google Scholar] [CrossRef] [Green Version]
- Ippolito, F.; Rentsch, S.; Hübner, G.; Claypole, T.; Gane, P. Influence of calcium carbonate on polyamide 12 regarding melting, formability and crystallization properties. Compos. Part. B Eng. 2019, 164, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Gysau, D. Füllstoffe: Grundlagen und Anwendungen; Vincentz Network GmbH & Co. KG: Hannover, Germany, 2006. [Google Scholar]
- Shi, X.; Rosa, R.; Lazzeri, A. On the coating of precipitated calcium carbonate with stearic acid in aqueous medium. Langmuir 2010, 26, 8474–8482. [Google Scholar] [CrossRef]
- Goodman, H. Surface-Modified Fillers for Polymer Resins Compositions. Patent GB0321703A, 16 September 2003. [Google Scholar]
- Ippolito, F.; Hübner, G.; Claypole, T.; Gane, P. Influence of the Surface Modification of Calcium Carbonate on Polyamide 12 Composites. Polymers 2020, 12, 1295. [Google Scholar] [CrossRef] [PubMed]
- Azadi, M.; Ferdosi Heragh, M.; Bidi, M.A. Electrochemical Characterizations of Epoxy Coatings Embedded by Modified Calcium Carbonate Particles. Prog. Color. Colorants Coat. 2020, 13, 213–222. [Google Scholar]
- Chen, X.; Qian, X.; An, X. Using calcium carbonate whiskers as papermaking filler. BioResources 2011, 6, 2435–2447. [Google Scholar]
- Grönfors, J. Use of Fillers in Paper and Paperboard Grades. Ph.D. Thesis, Tampere University of Applied Sciences, Tampere, Finland, 2010. [Google Scholar]
- Katz, H.S.; Mileski, J.; Melewski, J.V. Handbook of Fillers for Plastics; Springer Science & Business Media: Berlin/Heidelberg, Germany, 1987. [Google Scholar]
- Ramadhani, A.; Herda, E.; Triaminingsih, S. The effect of brushing with toothpaste containing nano calcium carbonate upon nanofill composite resin surface roughness. In Proceedings of Journal of Physics: Conference Series; IOP Publishing: Bristol, UK, 2017; p. 012103. [Google Scholar]
- Thenepalli, T.; Jun, A.Y.; Han, C.; Ramakrishna, C.; Ahn, J.W. A strategy of precipitated calcium carbonate (CaCO3) fillers for enhancing the mechanical properties of polypropylene polymers. Korean J. Chem. Eng. 2015, 32, 1009–1022. [Google Scholar] [CrossRef]
- Stirnimann, T.; Di Maiuta, N.; Gerard, D.E.; Alles, R.; Huwyler, J.; Puchkov, M. Functionalized calcium carbonate as a novel pharmaceutical excipient for the preparation of orally dispersible tablets. Pharm. Res. 2013, 30, 1915–1925. [Google Scholar] [CrossRef]
- Xanthos, M. Functional Fillers for Plastics; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Bollström, R.; Gane, P. Spot Application of a Transparent Nanoclay Layer to Provide Post-print Laser Marking Functionality on High Brightness 100% Calcium Carbonate Coatings. Adv. Print. Media Technol. 2016, 43, 33. [Google Scholar]
- Erdogan, N.; Eken, H.A. Precipitated calcium carbonate production, synthesis and properties. Physicochem. Probl. Miner. Process. 2017, 53, 57–68. [Google Scholar]
- Ippolito, F.; Hübner, G.; Claypole, T.; Gane, P. Impact of Bimodal Particle Size Distribution Ratio of Functional Calcium Carbonate Filler on Thermal and Flowability Properties of Polyamide 12. Appl. Sci. 2021, 11, 641. [Google Scholar] [CrossRef]
- Kruth, J.-P.; Levy, G.; Schindel, R.; Craeghs, T.; Yasa, E. Consolidation of polymer powders by selective laser sintering. In Proceedings of the 3rd International Conference on Polymers and Moulds Innovations, Ghent, Belgium, 17–19 September 2008; pp. 15–30. [Google Scholar]
- Höhne, G.W.H.; Hemminger, W.; Flammersheim, H.-J. Theoretical fundamentals of differential scanning calorimeters. In Differential Scanning Calorimetry; Springer: Berlin/Heidelberg, Germany, 1996; pp. 21–40. [Google Scholar]
- Wunderlich, B. Thermal Analysis of Polymeric Materials; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Drummer, D.; Rietzel, D.; Kühnlein, F. Development of a characterization approach for the sintering behavior of new thermoplastics for selective laser sintering. Phys. Procedia 2010, 5, 533–542. [Google Scholar] [CrossRef] [Green Version]
- Koutsoukos, P.G.; Kontoyannis, C.G. Precipitation of calcium carbonate in aqueous solutions. J. Chem. Soc. Faraday Trans. Phys. Chem. Condens. Phases 1984, 80, 1181–1192. [Google Scholar] [CrossRef]
- Da Silva, A.; Rocha, M.; Moraes, M.; Valente, C.; Coutinho, F. Mechanical and rheological properties of composites based on polyolefin and mineral additives. Polym. Test. 2002, 21, 57–60. [Google Scholar] [CrossRef]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | PCC Calcite | PCC Aragonite | GCC | |
|---|---|---|---|---|
| Rhombohedral | Scalenohedral | Fine Ground | ||
| Solubility product (Ksp) | 3.36 × 10−9 | 6.00 × 10−9 | ||
| Density (g cm−3) | 2.71 | 2.93 | ||
| Hardness (Mohs scale) | 3.0 | 3.0 | 3.5–4.0 | 3.0 |
| Refractive index | 1.58 | 1.58 | 1.63 | 1.58 |
| Coordination number | 6 | 9 | ||
| Specific gravity | 2.71 | 2.71 | 2.92 | 2.71 |
| TAPPI brightness * /% | 99 | 99 | 99 | 95 |
| Omyacarb 10-AV | EOS PA2200 | |
|---|---|---|
| producer/supplier | Omya International AG | EOS e-Manufacturing |
| volume-based median particle size, dv50 | 9 μm | 60 μm |
| particle shape | irregular | spherical |
| approx. thermal conductivity at 298 K | 1.3 Wm⁻1 K⁻1 | 0.2 Wm⁻1 K⁻1 |
| approx. specific heat | 0.8 kJkg⁻1 K⁻1 | 1.2 kJkg⁻1 K⁻1 |
| Amino Hexanoic Acid | |
|---|---|
| producer/supplier | Sigma Aldrich |
| CAS Number | 60-32-2 |
| Linear Formula | C6H12NO2 |
| Molecular weight | 131.17 g mol⁻1 |
| Filler Nomenclature | Filler Type | dv10 /μm | dv50 /μm | dv90 /μm | SSA /m2g−1 |
|---|---|---|---|---|---|
| c-G | coarse sized GCC | 1.0 | 4.9 | 14.8 | 1.8 |
| m-G | medium sized GCC | 0.8 | 2.1 | 5.3 | 3.7 |
| s-G | small sized GCC | 0.5 | 1.1 | 2.7 | 7.7 |
| c-P | coarse sized PCC | 1.0 | 5.0 | 9.1 | 1.3 |
| s-P | small sized PCC | 0.5 | 2.1 | 3.7 | 6.4 |
| Mixed Filler Nomenclature | Single Components | Mix Proportions | Specific Surface Area /m2g−1 | ||
|---|---|---|---|---|---|
| Coarse Filler | Fine Filler | Amount Coarse Filler /w/w% | Amount Fine Filler /w/w% | ||
| 70/30_c/s-G | c-G | s-G | 70 | 30 | 3.6 |
| 33/66_c/s-G | c-G | s-G | 33 | 66 | 5.7 |
| 50/50_c/m-G | c-G | m-G | 50 | 50 | 2.7 |
| 30/70_c/s-P | c-P | s-P | 30 | 70 | 4.9 |
| 70/30_c/s-P | c-P | s-P | 70 | 30 | 2.8 |
| Treated Filler Nomenclature | Modifier Surface Concentration /mmol Per 100 m2 Filler | Modifier Amount /% by Weight |
|---|---|---|
| c-G.1 | 3.05 | 0.7 |
| m-G.1 | 3.05 | 1.5 |
| s-G.1 | 3.05 | 3.1 |
| c-P.1 | 3.05 | 0.5 |
| s-P.1 | 3.05 | 2.6 |
| Treated Filler Nomenclature | Modifier Surface Concentration /mmol Per 100 m2 Filler | Modifier Amount /% by Weight |
|---|---|---|
| c-G.2 | 2.3 | 0.5 |
| s-G.2 | 2.3 | 2.3 |
| c-P.2 | 2.3 | 0.4 |
| s-P.2 | 2.3 | 1.9 |
| Filler Nomenclature | Coarse Filler | Fine Filler | Amount Coarse Filler /w/w% | Amount Fine Filler /w/w% | Amount Untreated Coarse Filler /w/w% |
|---|---|---|---|---|---|
| 70/30_c/s-G.2 | c-G.2 | s-G.2 | 70 | 30 | 0 |
| 70/30_c/s-P.2 | c-P.2 | s-P.2 | 70 | 30 | 0 |
| 70/30_c/s-G.3 | c-G | s-G.2 | 0 | 30 | 70 |
| 70/30_c/s-P.3 | c-P | s-P.2 | 0 | 30 | 70 |
| Compound Nomenclature | Filler Used | Filler Amount /w/w% | Total Carbonate Surface Per 100 g Polyamide 12 /m2 | Coarse Filler Surface Per 100 g Polyamide 12 /m2 | Fine Filler Surface Per 100 g Polyamide 12 /m2 |
|---|---|---|---|---|---|
| 70/30_c/s-G_12 | 70/30_c/s-G | 12 | 49 | 17 | 32 |
| 70/30_c/s-G_15 | 70/30_c/s-G | 15 | 63 | 22 | 41 |
| 33/66_c/s-G_15 | 33/66_c/s-G | 15 | 101 | 11 | 90 |
| 50/50_c/m-G_15 | 50/50_c/m-G | 15 | 48 | 16 | 32 |
| 30/70_c/s-P_15 | 30/70_c/s-P | 15 | 86 | 7 | 79 |
| 70/30_c/s-P_15 | 70/30_c/s-P | 15 | 50 | 16 | 34 |
| 70/30_c/s-G.1_12 | 70/30_c/s-G.1 | 12 | 49 | 17 | 32 |
| 70/30_c/s-G.1_15 | 70/30_c/s-G.1 | 15 | 63 | 22 | 41 |
| 33/66_c/s-G.1_15 | 33/66_c/s-G.1 | 15 | 101 | 11 | 90 |
| 50/50_c/m-G.1_15 | 50/50_c/m-G.1 | 15 | 48 | 16 | 32 |
| 30/70_c/s-P.1_15 | 30/70_c/s-P.1 | 15 | 86 | 7 | 79 |
| 70/30_c/s-P.1_15 | 70/30_c/s-P.1 | 15 | 50 | 16 | 34 |
| 70/30_c/s-G.2_12 | 70/30_c/s-G.2 | 12 | 49 | 17 | 32 |
| 70/30_c/s-G.2_15 | 70/30_c/s-G.2 | 15 | 63 | 22 | 41 |
| 70/30_c/s-P.2_15 | 70/30_c/s-P.2 | 15 | 50 | 16 | 34 |
| 70/30_c/s-G.3_12 | 70/30_c/s-G.3 | 12 | 49 | 17 | 32 |
| 70/30_c/s-G.3_15 | 70/30_c/s-G.3 | 15 | 63 | 22 | 41 |
| 70/30_c/s-P.3_15 | 70/30_c/s-P.3 | 15 | 50 | 16 | 34 |
| Uncoated GCC Blend | Surface Modification | Reduced Modification | Uncoated Coarse GCC | |
| Melt flowability | Reduced | Improved | Improved | Improved |
| Melt transition | Enhanced | Effect lost | Slight regain | Slight regain |
| Crystallization point | Increased | Increased | Increased | Increased |
| Crystallization time | 20% time needed | Regain of 150% | Regain of 150% | Regain of 150% |
| Ductility | 70% lost | 60% regain | 60% regain | 60% regain |
| Uncoated PCC blend | Modified PCC | Uncoated coarse PCC | ||
| Melt flowability | Reduced | Improved | Improved | |
| Melt transition | Enhanced | Slight regain | High regained | |
| Crystallization point | Increased | Increased | Increased | |
| Crystallization time | 20% time needed | Regain of 60% | Regain of 60% | |
| ductility | 30 % lost | no effect | no effect |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ippolito, F.; Hübner, G.; Claypole, T.; Gane, P. Calcium Carbonate as Functional Filler in Polyamide 12-Manipulation of the Thermal and Mechanical Properties. Processes 2021, 9, 937. https://doi.org/10.3390/pr9060937
Ippolito F, Hübner G, Claypole T, Gane P. Calcium Carbonate as Functional Filler in Polyamide 12-Manipulation of the Thermal and Mechanical Properties. Processes. 2021; 9(6):937. https://doi.org/10.3390/pr9060937
Chicago/Turabian StyleIppolito, Fabio, Gunter Hübner, Tim Claypole, and Patrick Gane. 2021. "Calcium Carbonate as Functional Filler in Polyamide 12-Manipulation of the Thermal and Mechanical Properties" Processes 9, no. 6: 937. https://doi.org/10.3390/pr9060937
APA StyleIppolito, F., Hübner, G., Claypole, T., & Gane, P. (2021). Calcium Carbonate as Functional Filler in Polyamide 12-Manipulation of the Thermal and Mechanical Properties. Processes, 9(6), 937. https://doi.org/10.3390/pr9060937