
How to Deal with Skin Biopsy in an Infant with Blisters?


Abstract
:1. Introduction
1.1. How Can the Clinical Context Provide Information Concerning the Etiology?
- -
- distribution of the lesions: localized blisters (traumatic/suction blister, local infection, solitary mastocytosis, etc.)
- -
- general symptoms: fever, systemic disorders (infection, toxic epidermal necrosis, metabolic disorder, etc.)
- -
- tense or flaccid blister/erosion (giving indications on the level of splitting)
1.2. What Is the Appropriate Technique for Skin Biopsy in This Context?
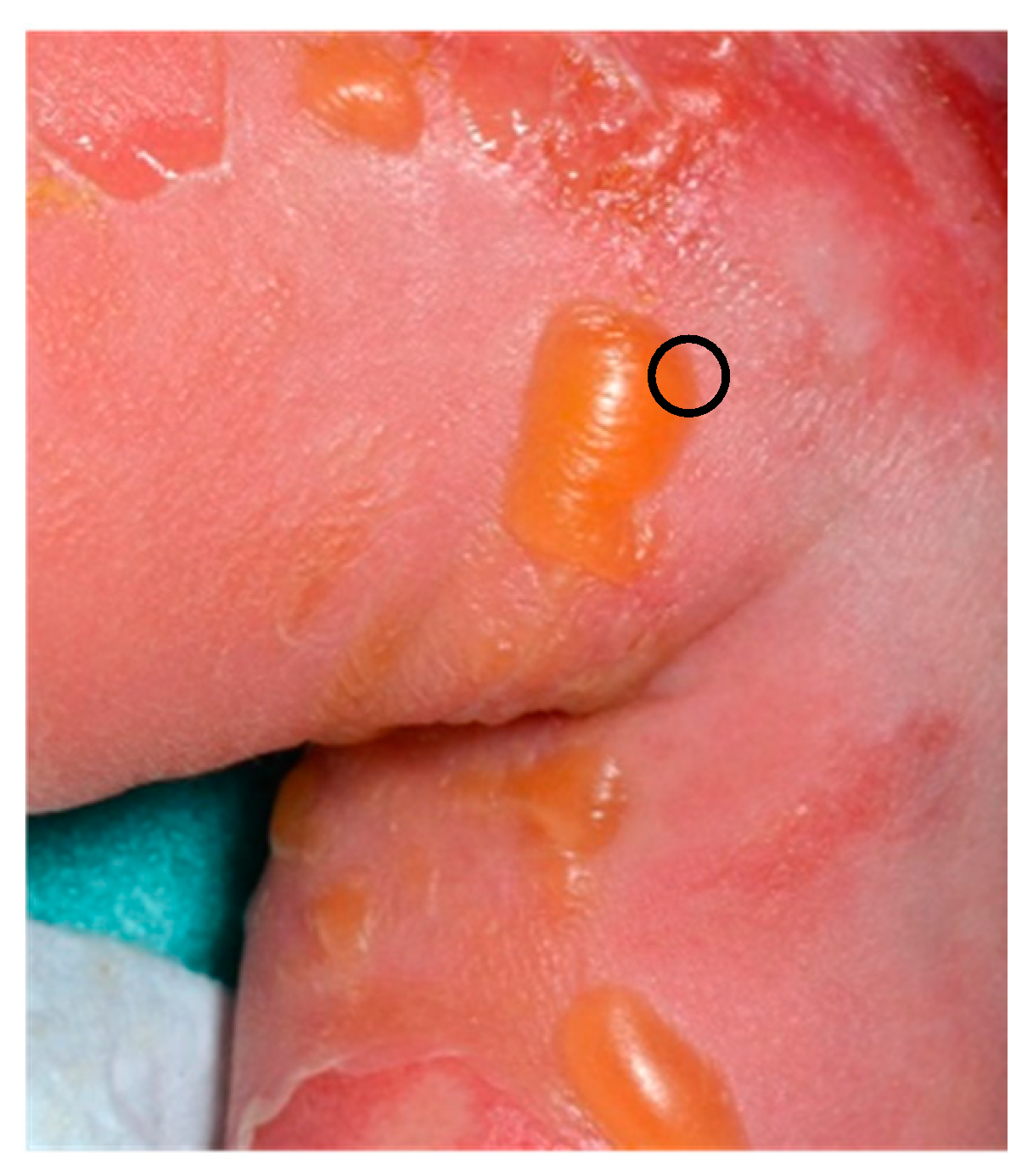
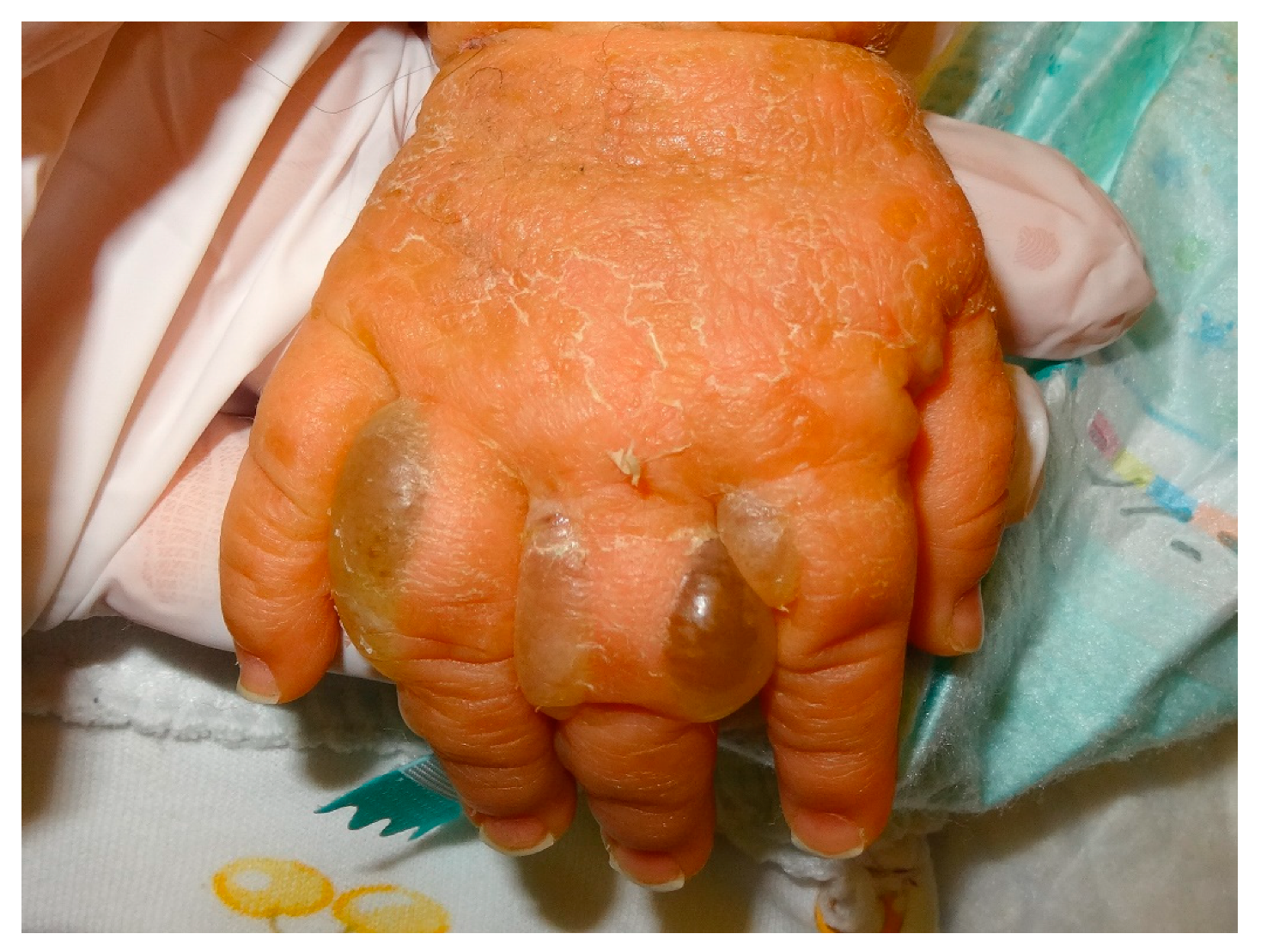
- for routine histology, it must be performed between the normal skin and the edge of the blister so that the epidermis does not detach as in Figure 1, fixed in formalin, and processed routinely (paraffin embedded, haematoxylin and eosin (HE) staining) in order to determine the anatomic level of the blister
- for immunohistochemistry, it will be frozen or can be put in Michel media
- ○
- if an autoimmune blistering disease (AIBD) is suspected, the biopsy will be performed on healthy perilesional skin and processed for direct immunofluorescence (DIF)
- ○
- if a genodermatosis is suspected, the biopsy will also be taken between the normal skin and the edge of the blister and processed for immunomapping with specialized antibodies (DIF or immunoperoxidase)
2. Etiologies
2.1. Infections
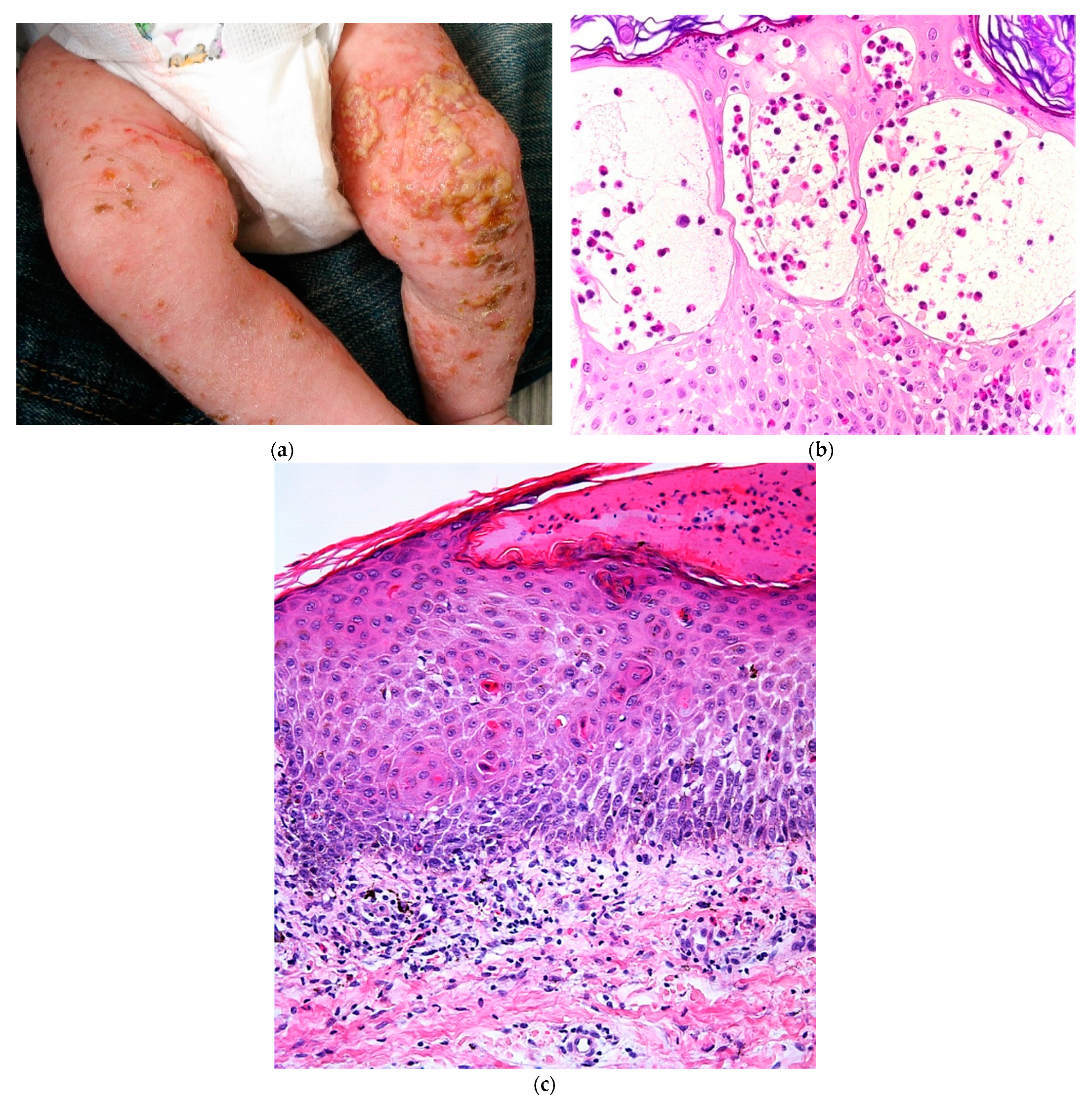
2.1.1. Staphylococcal Scalded Skin Syndrome (SSSS)
2.1.2. Histology
2.1.3. Impetigo
2.1.4. Histology
2.1.5. Scabies
2.1.6. Histology
2.2. Genodermatosis
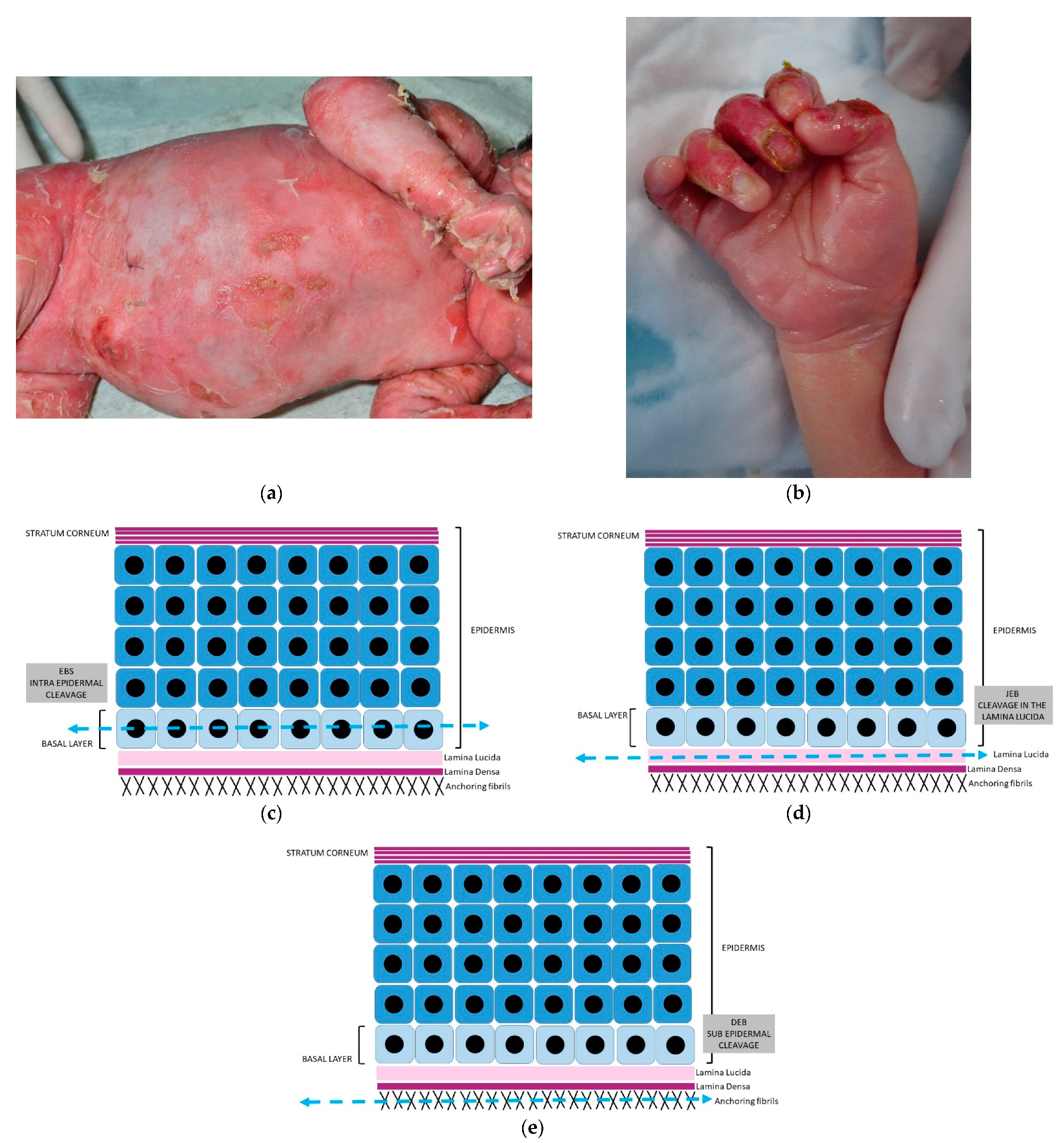
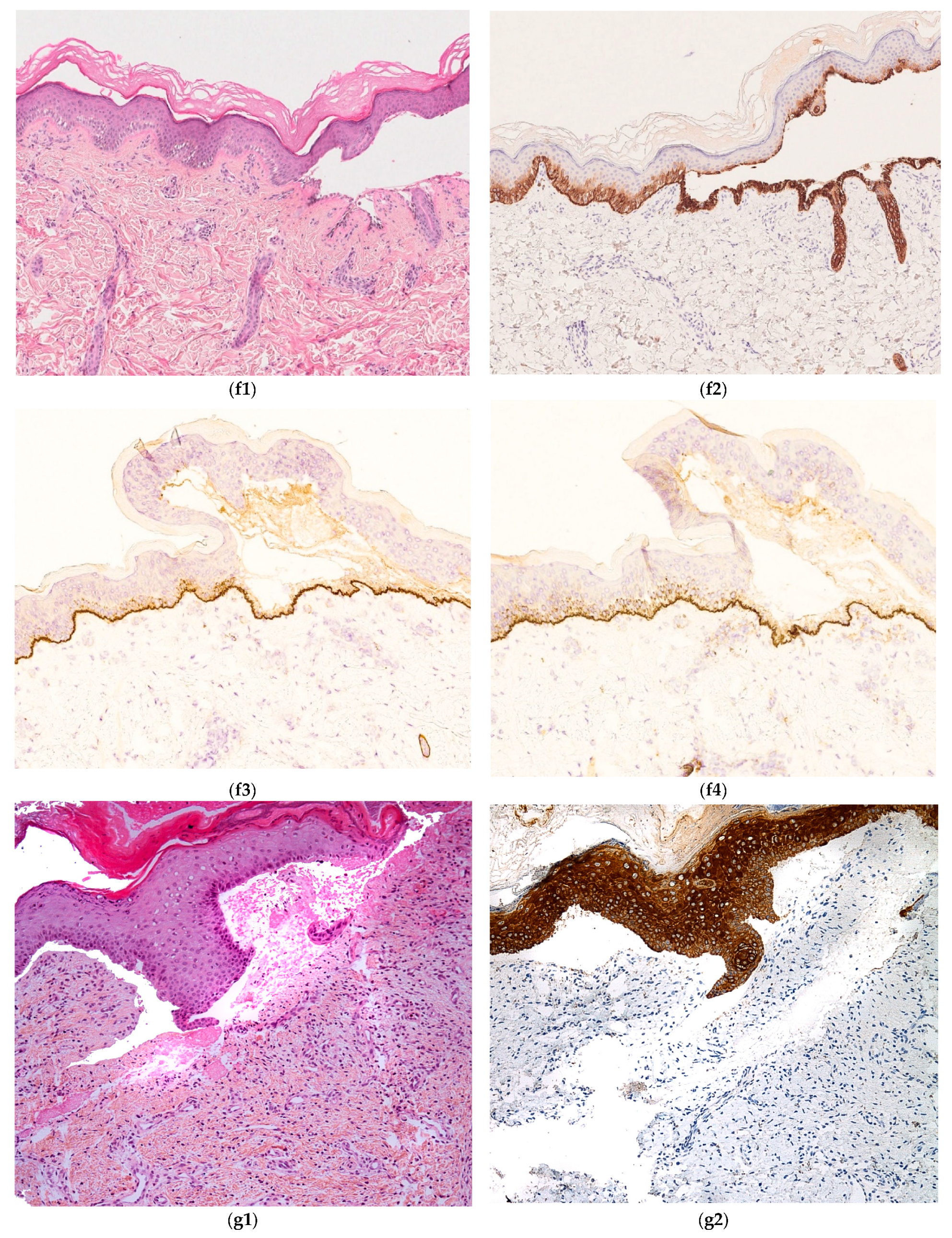
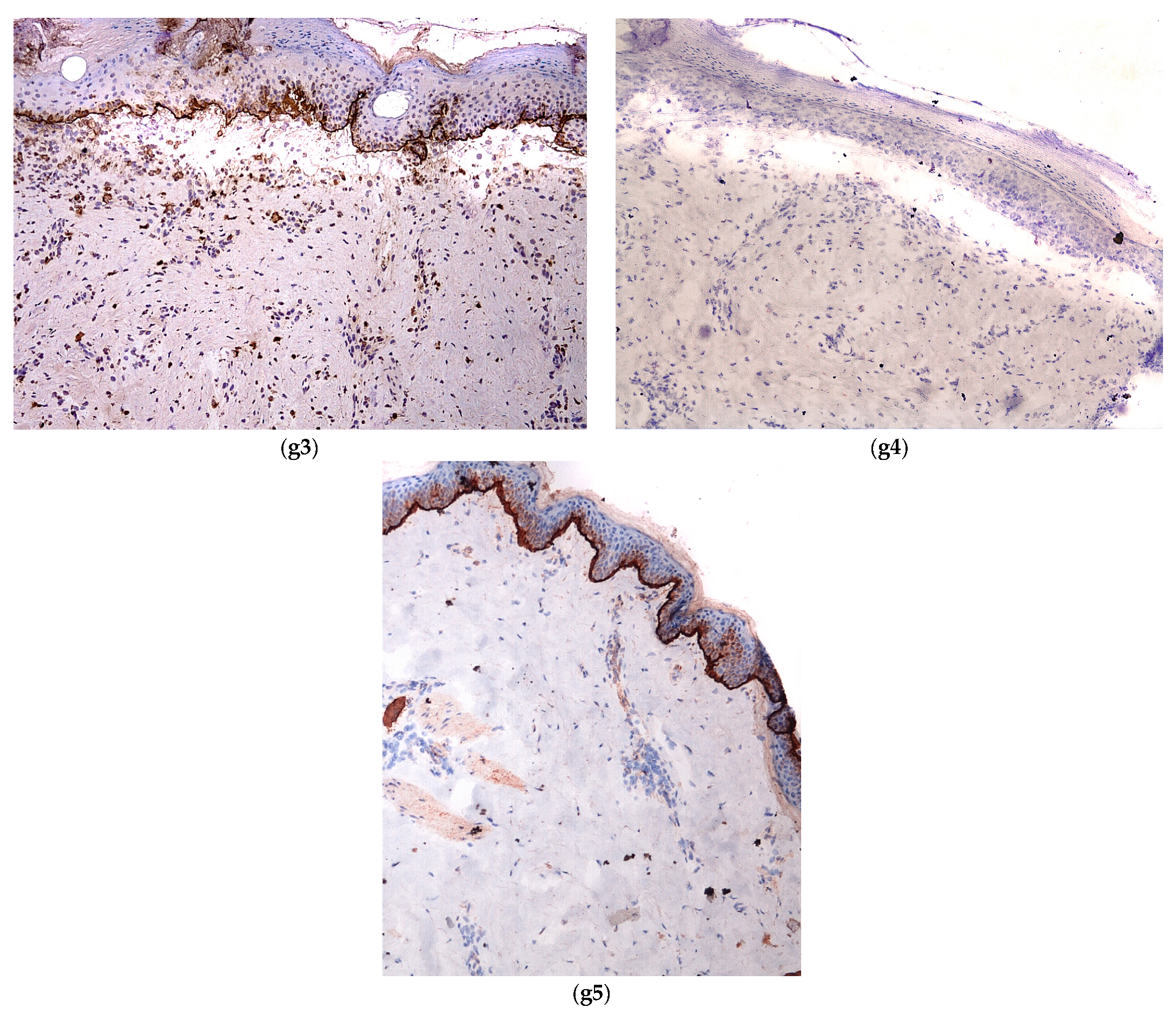
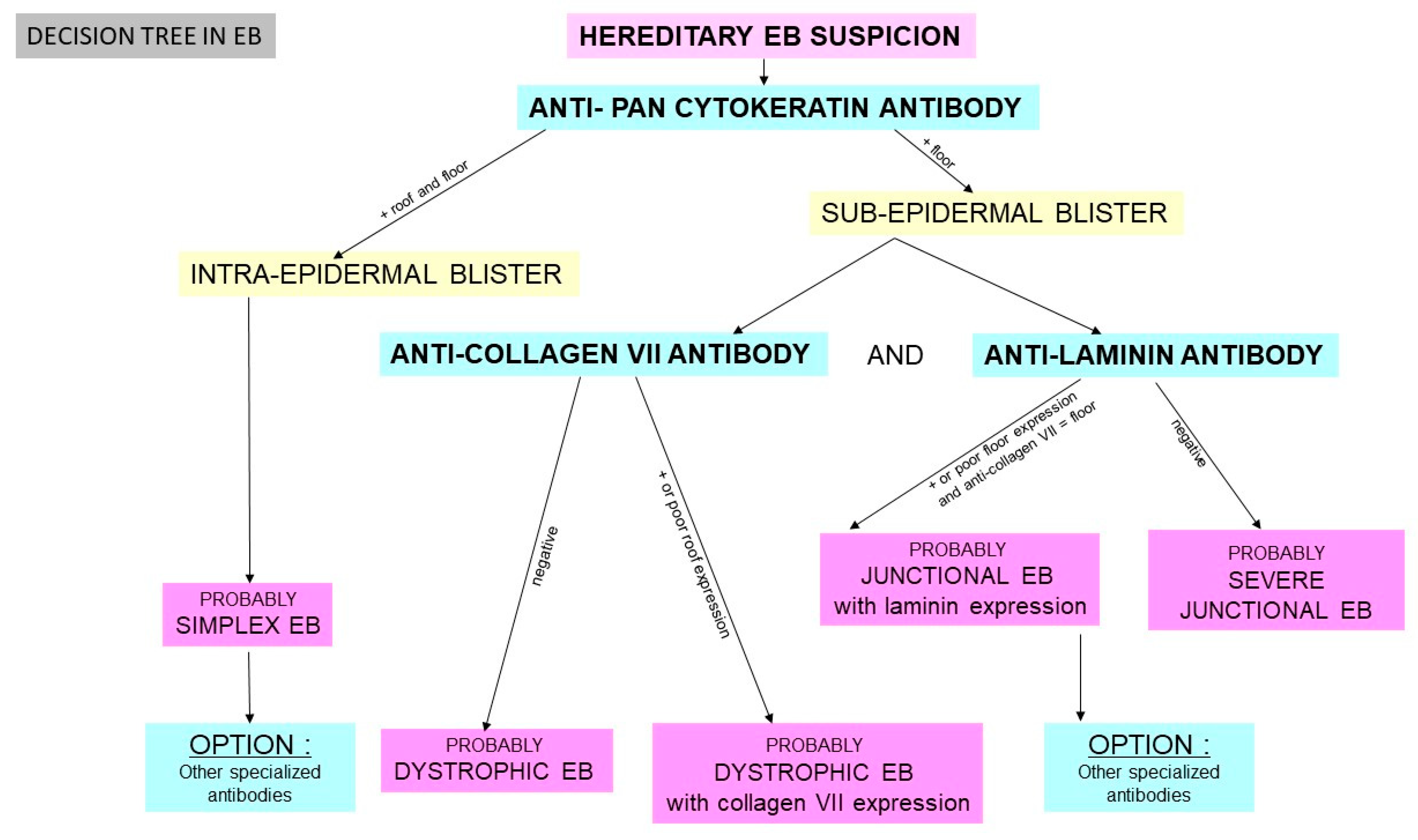
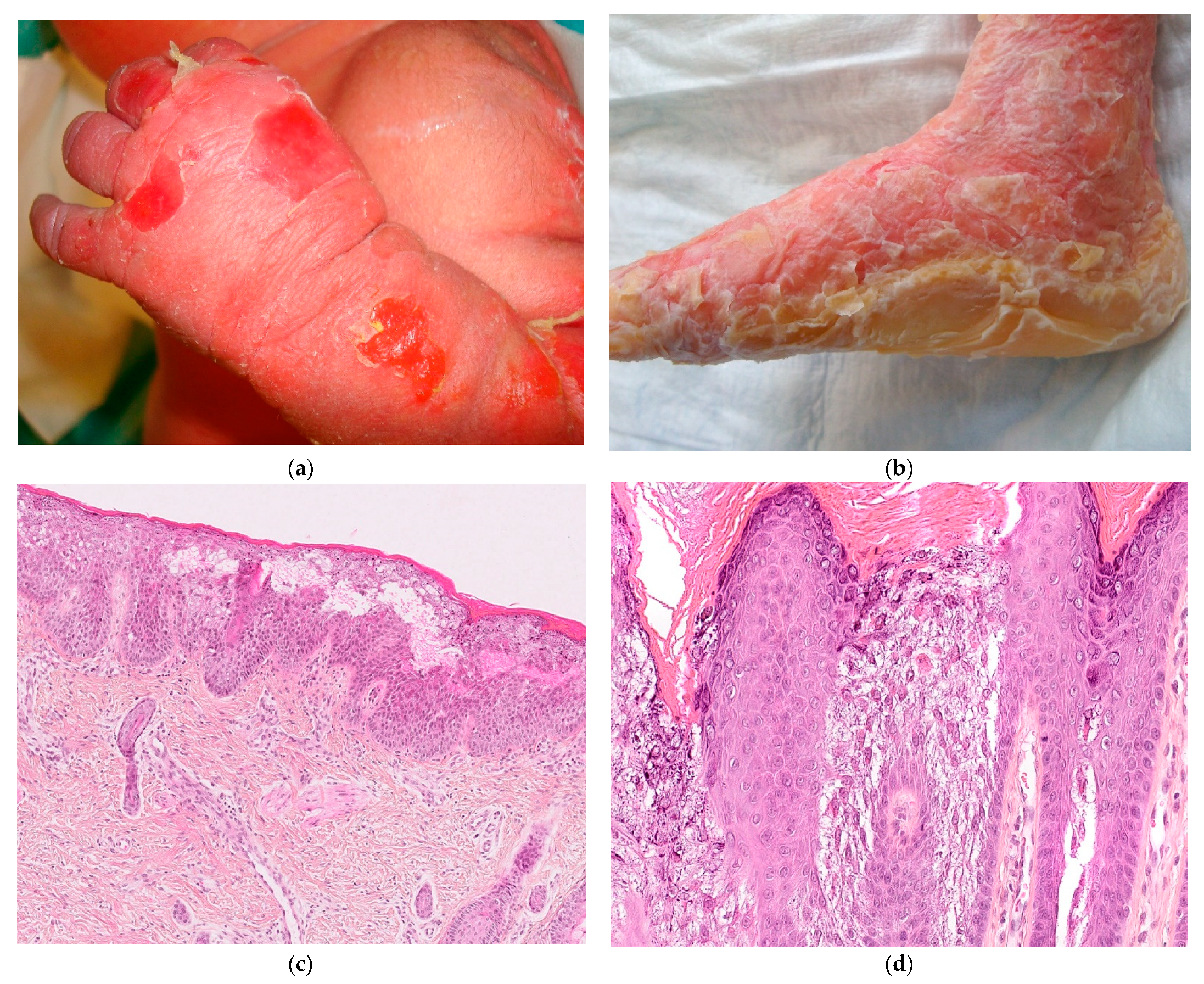
2.2.1. Hereditary Epidermolysis Bullosa
2.2.2. Histology
- -
- anti-pan cytokeratin AE1/AE3
- -
- anti-laminin antibody
- -
- anti-collagen VII antibody
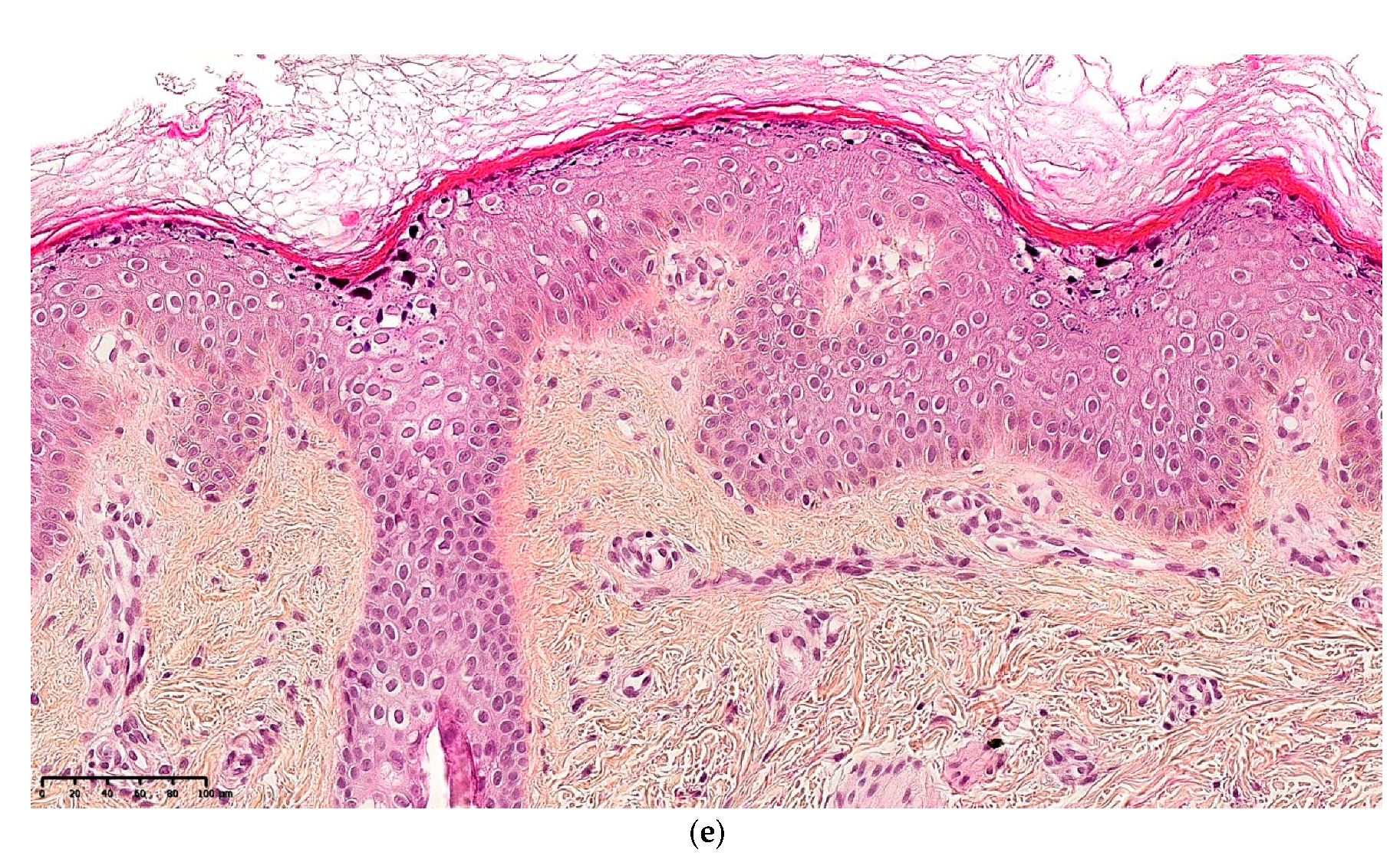
2.2.3. Keratinopathic Ichthyosis (KI)
2.2.4. Histology
2.2.5. Incontinentia Pigmenti (IP)
2.2.6. Histology
2.3. Cell Proliferation
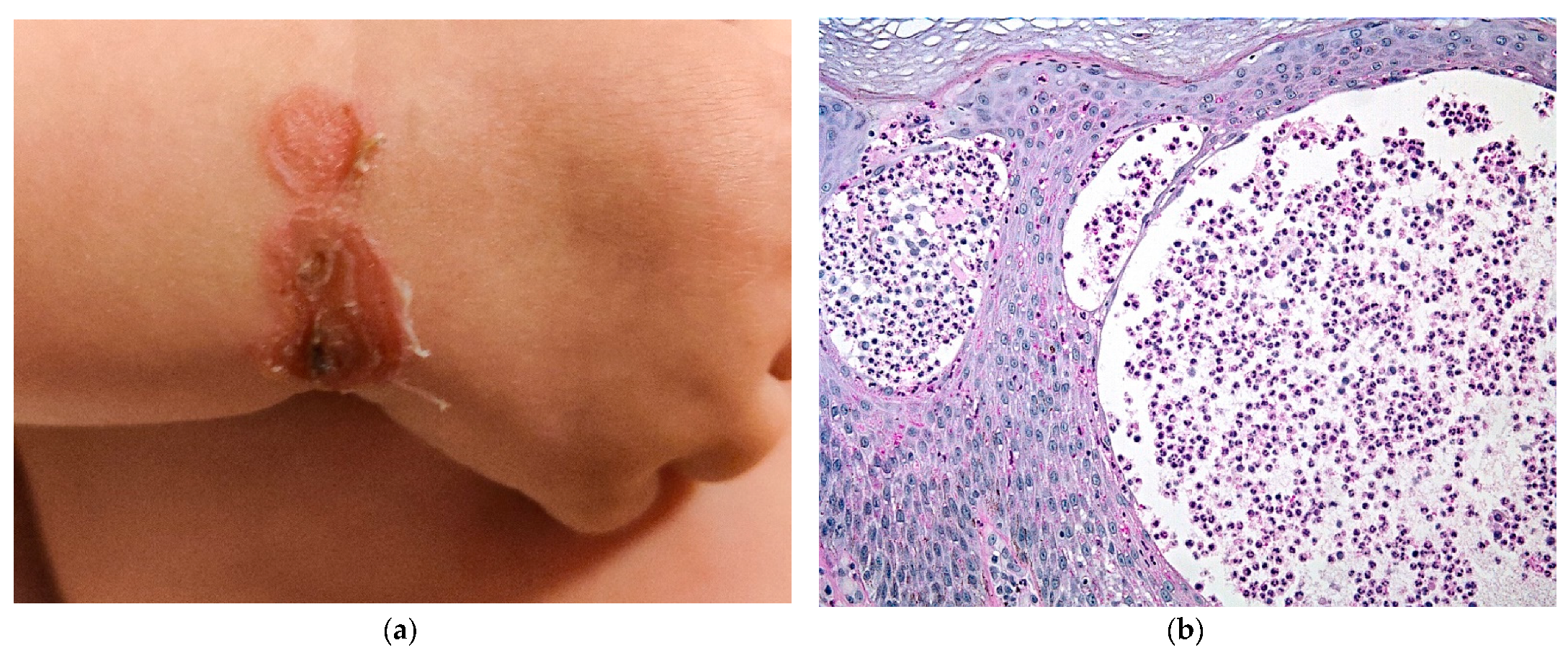
2.3.1. Mastocytosis
2.3.2. Histology
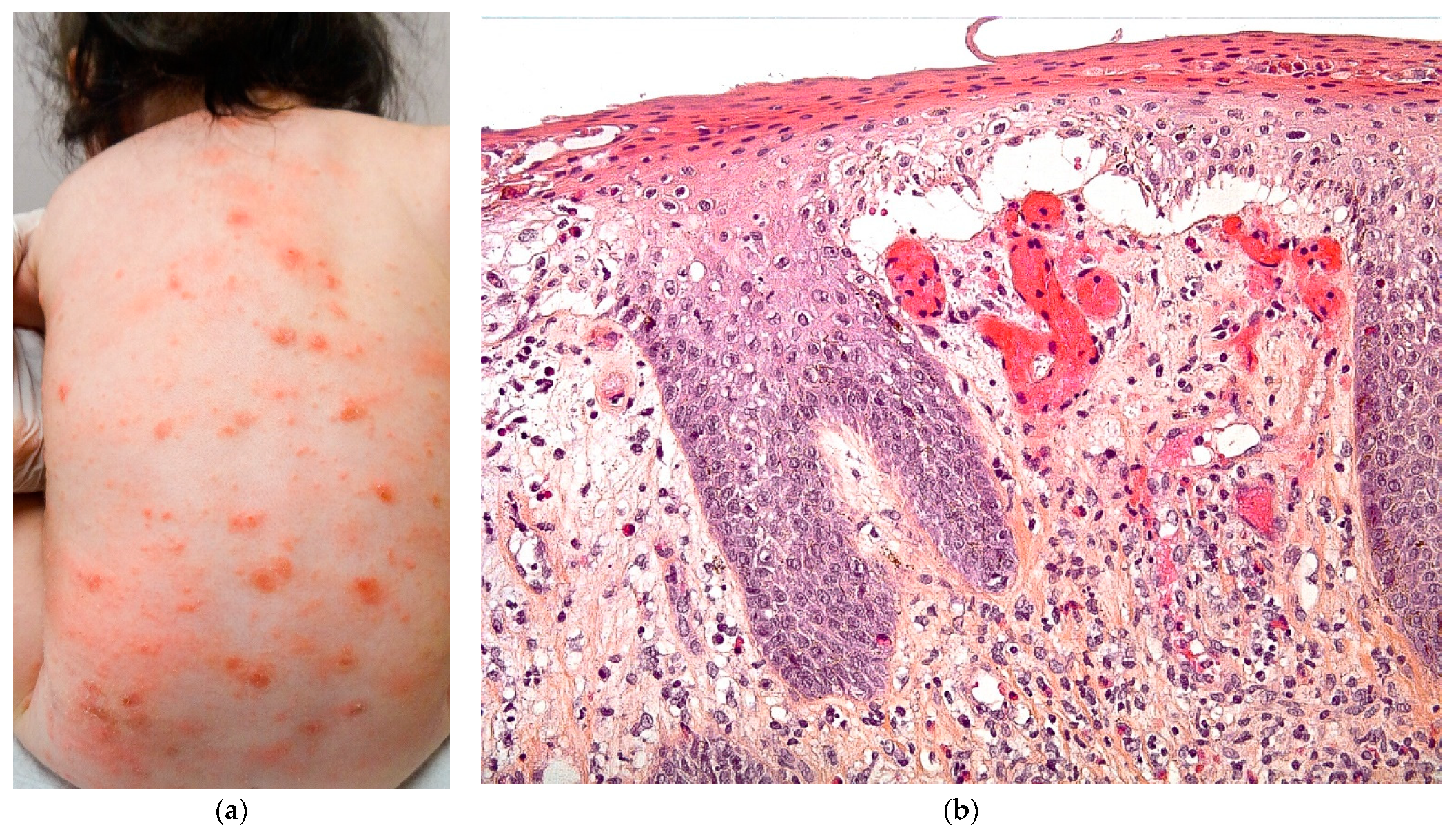
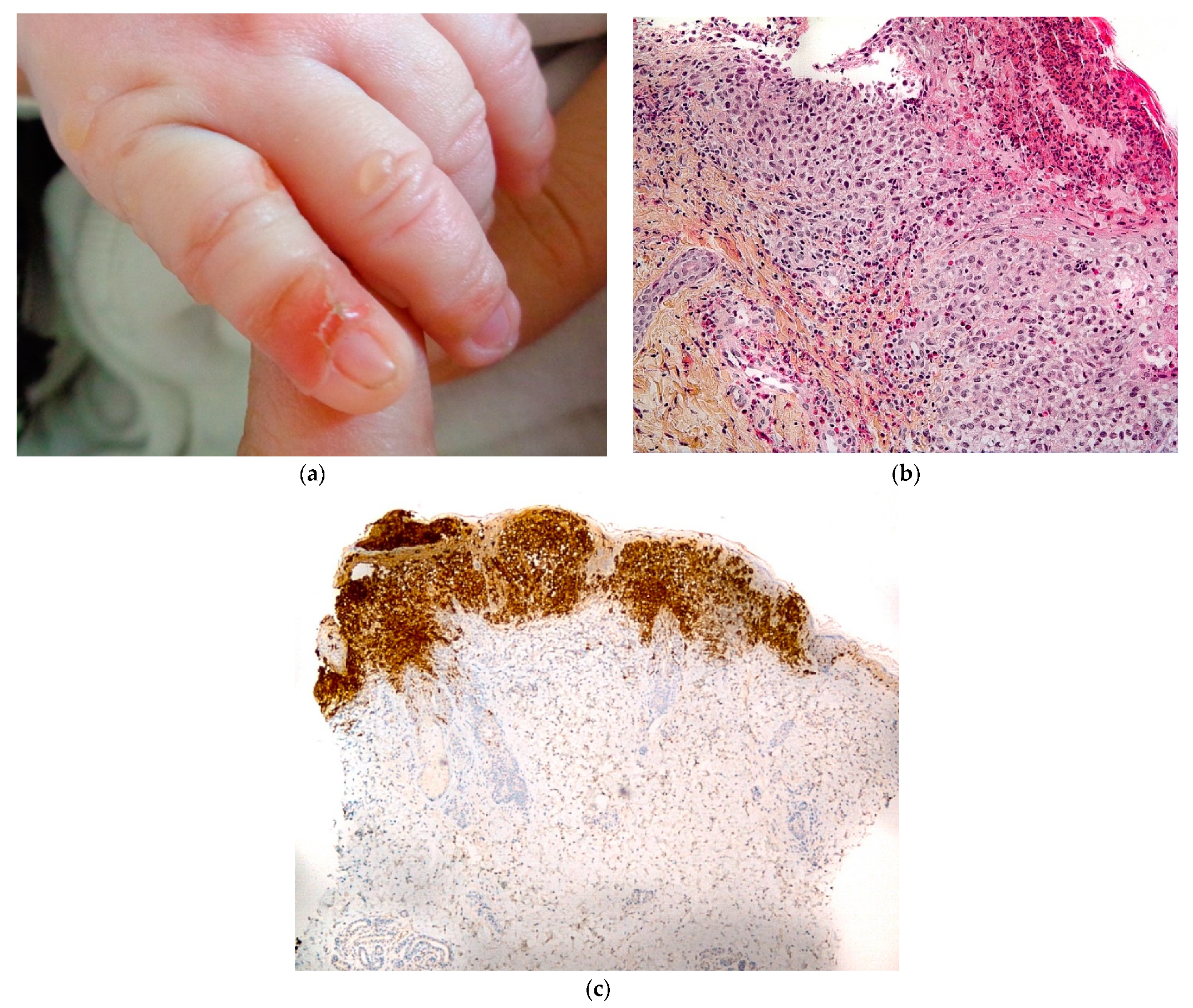
2.3.3. Langerhans Cell Histiocytosis (LCH)
2.3.4. Histology
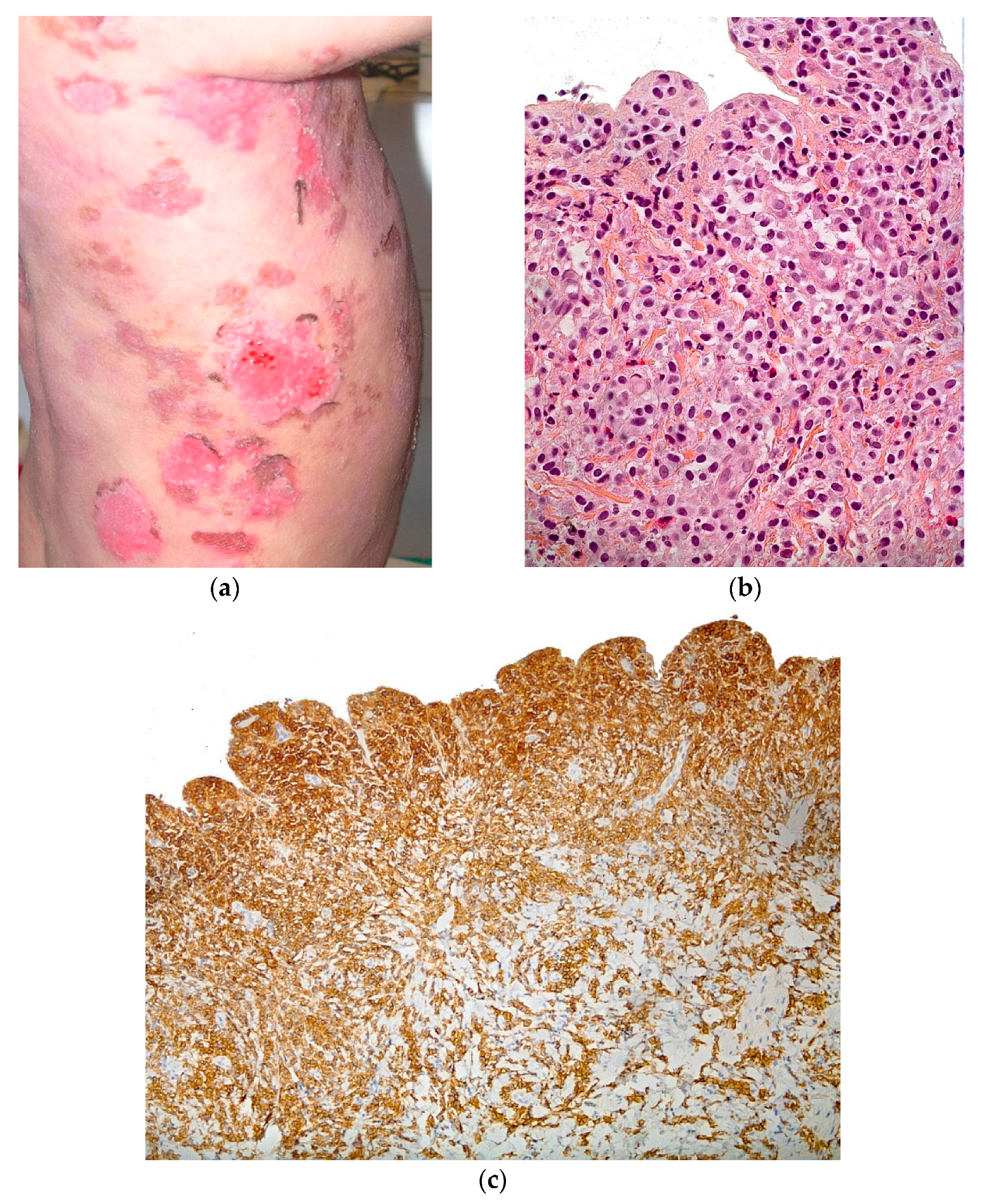
2.3.5. Auto-Immune Blistering Diseases
Bullous Pemphigoid (BP)
2.3.6. Histology
2.3.7. Other AIBDs
2.3.8. Drug Reactions
3. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Cazes, A.; Prost-Squarcioni, C.; Bodemer, C.; Heller, M.; Brousse, N.; Fraitag, S. Histologic cutaneous modifications after the use of EMLA cream, a diagnostic pitfall: Review of 13 cases. Arch. Dermatol. 2007, 143, 1073–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liy-Wong, C.; Pope, E.; Weinstein, M.; Lara-Corrales, I. Staphylococcal scalded skin syndrome: An epidemiological and clinical review of 84 cases. Pediatr. Dermatol. 2021, 38, 149–153. [Google Scholar] [CrossRef]
- Amon, R.B.; Dimond, R.L. Toxic epidermal necrolysis. Rapid differentiation between staphylococcal and drug induced disease. Arch. Dermatol. 1975, 111, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Mahon, C.; Martinez, A.E. Vesiculopustular, bullous and erosive diseases of the neonate. In Harper’s Textbook of Pediatric Dermatology; Hoeger, P., Kinsler, V., Yan, A., Harper, J., Oranje, A., Bodemer, C., Larralde, M., Luk, D., Mendiratta, V., Purvis, D., Eds.; Wiley Global Research: Hoboken, NJ, USA, 2019; Volume 1, pp. 134–153. [Google Scholar]
- Grayson, W.; Calonje, E. Infectious diseases of the skin. In McKee’s Pathology of the Skin, 5th ed.; Calonje, J.E., Brenn, T., Lazar, A., Billings, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 1, pp. 858–862. [Google Scholar]
- Has, C.; Bauer, J.; Bodemer, C.; Bolling, M.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.-D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.; et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 2020, 183, 614–627. [Google Scholar] [CrossRef] [Green Version]
- Fine, J.-D.; Bruckner-Tuderman, L.; Eady, R.A.; Bauer, E.A.; Bauer, J.W.; Has, C.; Heagerty, A.; Hintner, H.; Hovnanian, A.; Jonkman, M.F.; et al. Inherited epidermolysis bullosa: Updated recommendations on diagnosis and classification. J. Am. Acad. Dermatol. 2014, 70, 1103–1126. [Google Scholar] [CrossRef] [PubMed]
- Has, C.; Liu, L.; Bolling, M.; Charlesworth, A.; El Hachem, M.; Escámez, M.J.; Fuentes, I.; Büchel, S.; Hiremagalore, R.; Pohla-Gubo, G.; et al. Clinical practice guidelines for laboratory diagnosis of epidermolysis bullosa. Br. J. Dermatol. 2019, 182, 574–592. [Google Scholar] [CrossRef]
- Oji, V.; Tadini, G.; Akiyama, M.; Bardon, C.B.; Bodemer, C.; Bourrat, E.; Coudiere, P.; DiGiovanna, J.J.; Elias, P.; Fischer, J.; et al. Revised nomenclature and classification of inherited ichthyoses: Results of the First Ichthyosis Consensus Conference in Sorèze 2009. J. Am. Acad. Dermatol. 2010, 63, 607–641. [Google Scholar] [CrossRef] [Green Version]
- Ross, R.; DiGiovanna, J.J.; Capaldi, L.; Argenyi, Z.; Fleckman, P.; Robinson-Bostom, L. Histopathologic characterization of epidermolytic hyperkeratosis: A systematic review of histology from the National Registry for Ichthyosis and Related Skin Disorders. J. Am. Acad. Dermatol. 2008, 59, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Metze, D.; Oji, V. Disorders of keratinization. In McKee’s Pathology of the Skin, 5th ed.; Calonje, J.E., Brenn, T., Lazar, A., Billings, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 1, pp. 63–66. [Google Scholar]
- Bergman, R.; Khamaysi, Z.; Sprecher, E. A unique pattern of dyskeratosis characterizes epidermolytic hyperkeratosis and epidermolytic palmoplantar keratoderma. Am. J. Dermatopathol. 2008, 30, 101–105. [Google Scholar] [CrossRef]
- Galler, B.; Bowen, C.; Arnold, J.; Kobayashi, T.; Dalton, S.R. Use of the frozen section ‘jelly-roll’ technique to aid in the diagnosis of bullous congenital ichthyosiform erythroderma (epidermolytic hyperkeratosis). J. Cutan. Pathol. 2016, 43, 434–437. [Google Scholar] [CrossRef]
- McLean, W.H.I.; Morley, S.M.; Lane, E.B.; Eady, R.A.J.; Griffiths, W.A.D.; Paige, D.G.; Harper, J.I.; Higgins, C.; Leigh, I.M. Ichthyosis bullosa of Siemens–A disease involving keratin 2e. J. Investig. Dermatol. 1994, 103, 277–281. [Google Scholar] [CrossRef] [Green Version]
- Hadj-Rabia, S.; Froidevaux, D.; Bodak, N.; Hamel-Teillac, D.; Smahi, A.; Touil, Y.; Fraitag, S.; De Prost, Y.; Bodemer, C. Clinical study of 40 cases of incontinentia pigmenti. Arch. Dermatol. 2003, 139, 1163–1170. [Google Scholar] [CrossRef] [Green Version]
- Bodemer, C.; Diociaiuti, A.; Hadj-Rabia, S.; Robert, M.P.; Desguerre, I.; Manière, M.C.; de la Dure-Molla, M.; De Liso, P.; Federici, A.; Galeotti, A.; et al. Multidisciplinary consensus recommendations from a European network for the diagnosis and practical management of patients with incontinentia pigmenti. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Fraitag, S.; Rimella, A.; de Prost, Y.; Brousse, N.; Hadj-Rabia, S.; Bodemer, C. Skin biopsy is helpful for the diagnosis of incontinentia pigmenti at late stage (IV): A series of 26 cutaneous biopsies. J. Cutan. Pathol. 2009, 36, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Polivka, L.; Bodemer, C. Paediatric mastocytosis. In Harper’s Textbook of Pediatric Dermatology; Hoeger, P., Kinsler, V., Yan, A., Harper, J., Oranje, A., Bodemer, C., Larralde, M., Luk, D., Mendiratta, V., Purvis, D., Eds.; Wiley Global Research: Hoboken, NJ, USA, 2019; Volume 1, pp. 1097–1108. [Google Scholar]
- Goodlad, J.; Calonje, E. Cutaneous lymphoproliferative diseases and related disorders (mastocytosis). In McKee’s Pathology of the Skin, 5th ed.; Calonje, J.E., Brenn, T., Lazar, A., Billings, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 2, pp. 1515–1519. [Google Scholar]
- Kamat, D.; Chatterjee, D.; Vinay, K. Recurrent blistering in an infant. JAMA Dermatol. 2020, 156, 212. [Google Scholar] [CrossRef] [PubMed]
- Morren, M.A.; Broecke, K.V.; Vangeebergen, L.; Sillevis-Smitt, J.H.; Van Den Berghe, P.; Hauben, E.; Jacobs, S.; Van Gool, S.W. Diverse cutaneous presentations of langerhans cell histiocytosis in children: A retrospective cohort study. Pediatr. Blood Cancer 2016, 63, 486–492. [Google Scholar] [CrossRef]
- Chan, M.M.H.; Tan, D.J.A.; Koh, M.J.-A.; Tan, L.S. Blistering Langerhans cell histiocytosis. Lancet Oncol. 2018, 19, e500. [Google Scholar] [CrossRef]
- Emile, J.-F.; Abla, O.; Fraitag, S.; Horne, A.; Haroche, J.; Donadieu, J.; Requena-Caballero, L.; Jordan, M.B.; Abdel-Wahab, O.; Allen, C.E.; et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 2016, 127, 2672–2681. [Google Scholar] [CrossRef] [Green Version]
- Fraitag, S.; Donadieu, J. Langerhans Cell Histiocytosis. In Harper’s Textbook of Pediatric Dermatology; Hoeger, P., Kinsler, V., Yan, A., Harper, J., Oranje, A., Bodemer, C., Larralde, M., Luk, D., Mendiratta, V., Purvis, D., Eds.; Wiley Global Research: Hoboken, NJ, USA, 2019; Volume 1, pp. 1071–1077. [Google Scholar]
- Hill, S.F.; Murrell, D.F. Differential diagnosis of vesiculobullous lesions. In Harper’s Textbook of Pediatric Dermatology; Hoeger, P., Kinsler, V., Yan, A., Harper, J., Oranje, A., Bodemer, C., Larralde, M., Luk, D., Mendiratta, V., Purvis, D., Eds.; Wiley Global Research: Hoboken, NJ, USA, 2019; Volume 1, pp. 868–897. [Google Scholar]
- Welfringer-Morin, A.; Bekel, L.; Bellon, N.; Gantzer, A.; Boccara, O.; Hadj-Rabia, S.; Leclerc-Mercier, S.; Frassati-Biaggi, A.; Fraitag, S.; Bodemer, C. Long-term evolving profile of childhood autoimmune blistering diseases: Retrospective study on 38 children. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 1158–1163. [Google Scholar] [CrossRef]
- Marathe, K.; Lu, J.; Morel, K.D. Bullous diseases: Kids are not just little people. Clin. Dermatol. 2015, 33, 644–656. [Google Scholar] [CrossRef]
- Zhao, C.Y.; Chiang, Y.Z.; Murrell, D.F. Neonatal autoimmune blistering disease: A systematic review. Pediatr. Dermatol. 2016, 33, 367–374. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Infections | Staphylococcal scalded skin syndrome |
| Impetigo | |
| Scabies | |
| Genodermatosis | Hereditary epidermolysis bullosa |
| Keratinopathic ichthyosis | |
| Incontinentia pigmenti | |
| Cell proliferation | Mastocytosis |
| Langerhans cell histiocytosis | |
| Autoimmune blistering diseases | Bullous pemphigoid and others |
| Drug reactions | SJS, TEN, etc. |
| Level of Skin Cleavage | EB Type | Inheritance | Mutated Gene (s) | Targeted Protein (s) |
|---|---|---|---|---|
| Intraepidermal | EB simplex | Autosomal dominant | KRT5, KRT14 | Keratin 5, keratin 14 |
| PLEC | Plectin | |||
| KLHL24 | Kelch-like member 24 | |||
| Autosomal recessive | KRT5, KRT14 | Keratin 5, keratin 14 | ||
| DST | Bullous pemphigoid antigen 230 (BP230) (syn. BPAG1e, dystonin) | |||
| EXPH5 (syn. SLAC2B) | Exophilin-5 (syn. synaptotagmin-like protein homolog lacking C2 domains b, Slac2-b) | |||
| PLEC | Plectin | |||
| CD151(syn. TSPAN24) | CD151 antigen (syn. tetraspanin 24) | |||
| Junctional | Junctional EB | Autosomal recessive | LAMA3, LAMB3, LAMC2 | Laminin 332 |
| COL17A1 | Type XVII collagen | |||
| ITGA6, ITGB4 | Integrin a6b4 | |||
| ITGA3 | Integrin a3 subunit | |||
| Dermal | Dystrophic EB | Autosomal dominant | COL7A1 | Type VII collagen |
| Autosomal recessive | COL7A1 | Type VII collagen | ||
| Mixed | Kindler EB | Autosomal recessive | FERMT1 (syn. KIND1) | Fermitin family homolog 1 (syn. kindlin-1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leclerc-Mercier, S. How to Deal with Skin Biopsy in an Infant with Blisters? Dermatopathology 2021, 8, 159-175. https://doi.org/10.3390/dermatopathology8020022
Leclerc-Mercier S. How to Deal with Skin Biopsy in an Infant with Blisters? Dermatopathology. 2021; 8(2):159-175. https://doi.org/10.3390/dermatopathology8020022
Chicago/Turabian StyleLeclerc-Mercier, Stéphanie. 2021. "How to Deal with Skin Biopsy in an Infant with Blisters?" Dermatopathology 8, no. 2: 159-175. https://doi.org/10.3390/dermatopathology8020022
APA StyleLeclerc-Mercier, S. (2021). How to Deal with Skin Biopsy in an Infant with Blisters? Dermatopathology, 8(2), 159-175. https://doi.org/10.3390/dermatopathology8020022