
What to Look Out for in a Newborn with Multiple Papulonodular Skin Lesions at Birth


Abstract
:1. Introduction
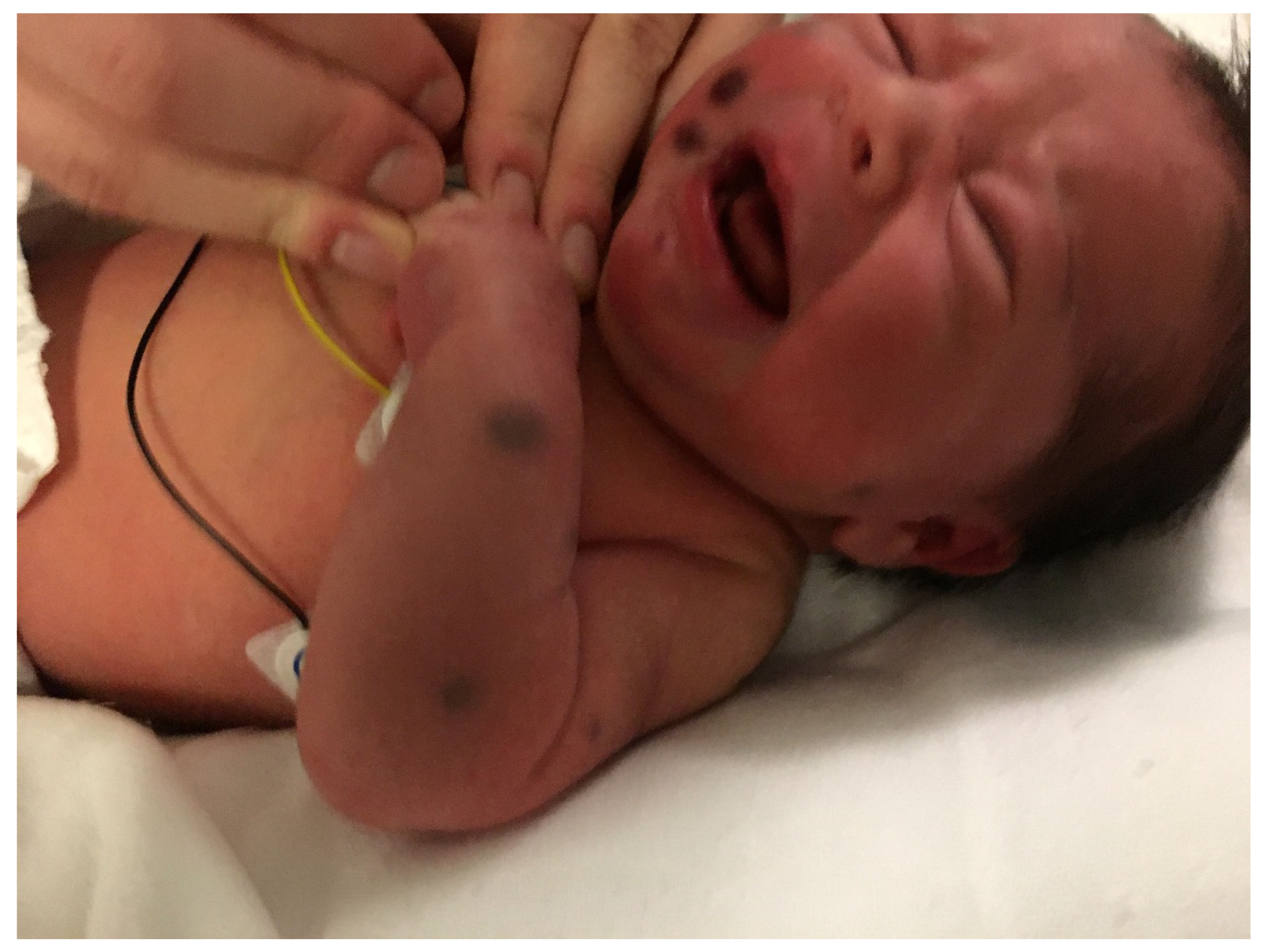
2. Blueberry Muffin Rash

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
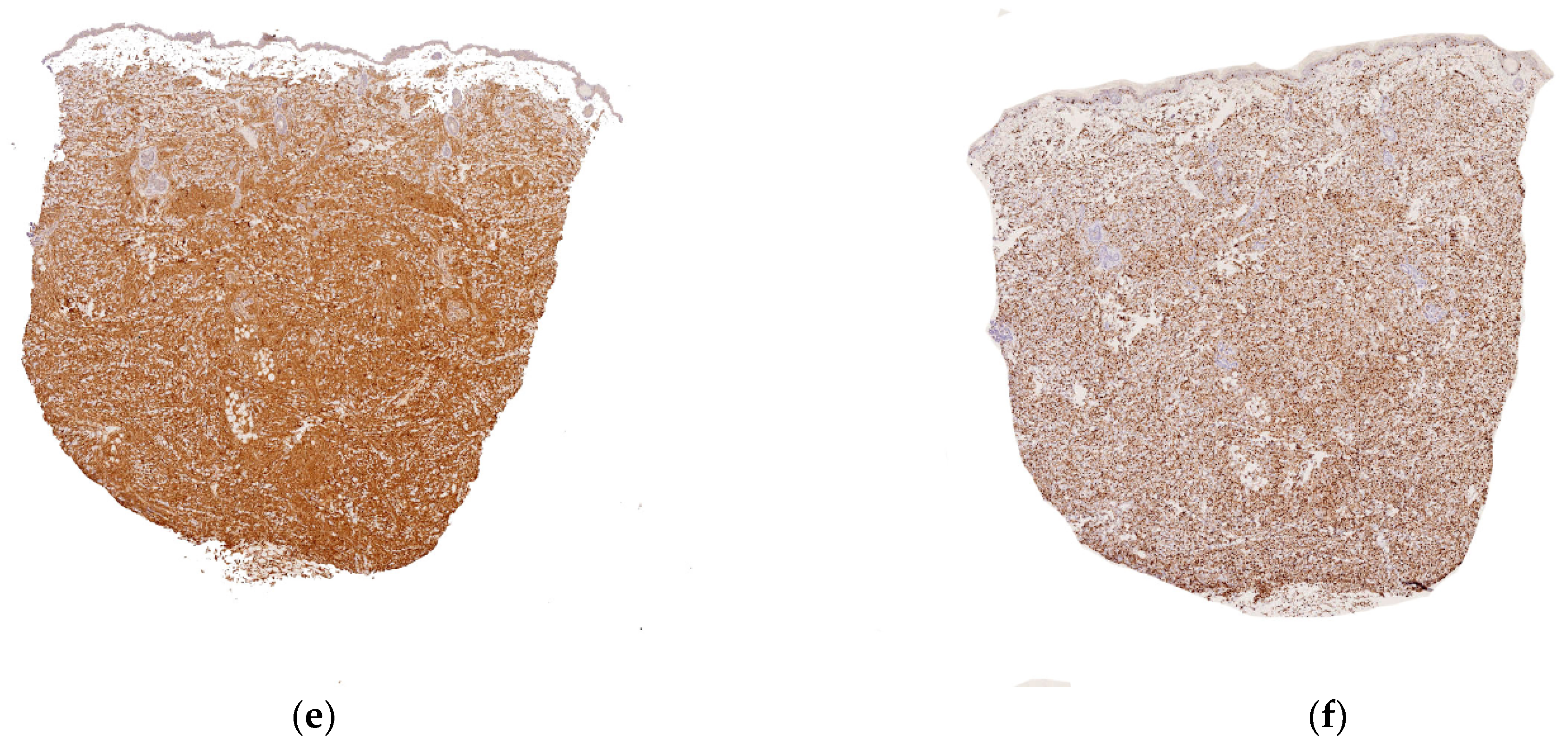{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dermal Erythropoiesis |
| • Congenital infections |
| Toxoplasmosis |
| Rubella |
| Cytomegalovirus |
| Herpes simplex virus |
| Coxsackie B2 virus |
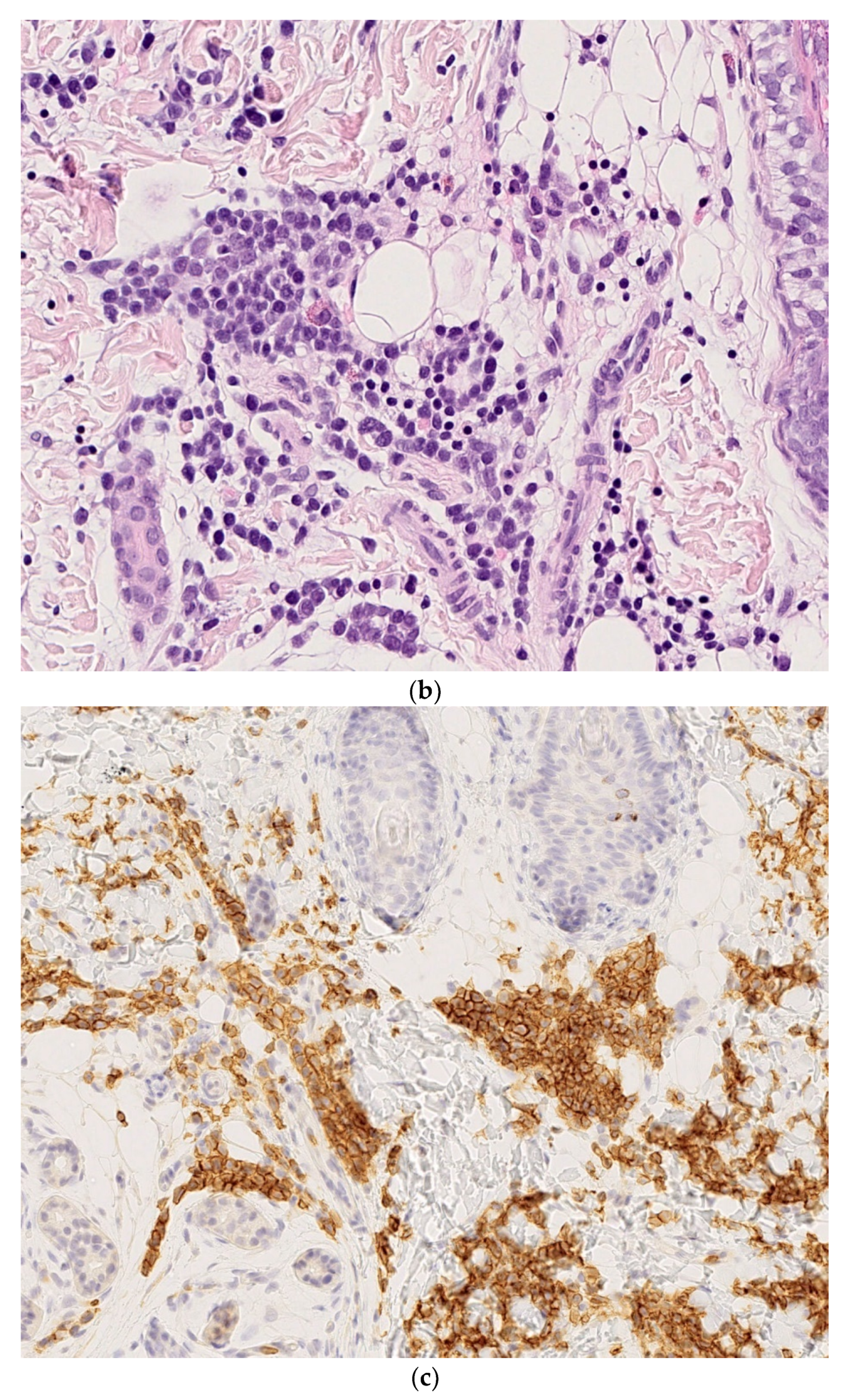
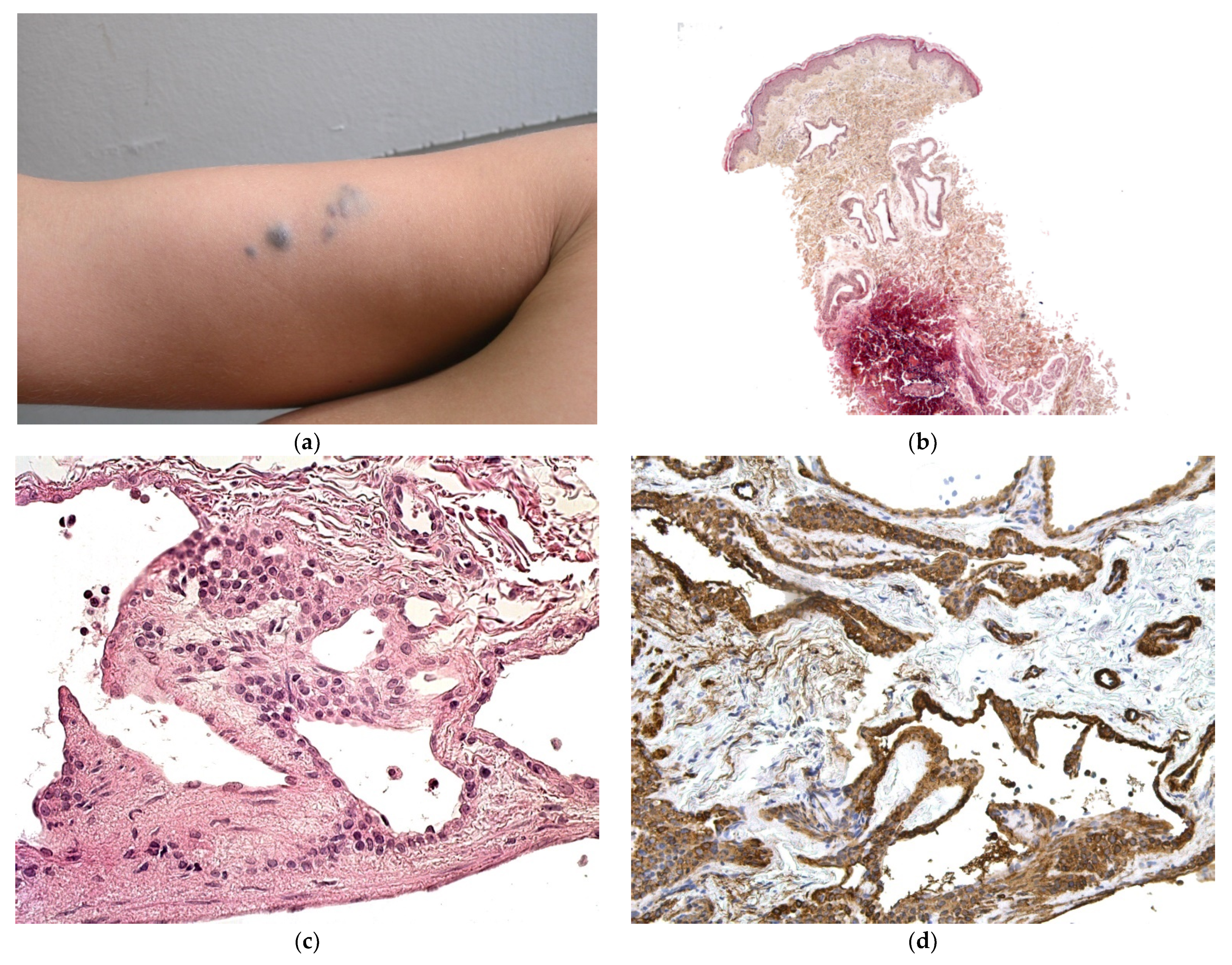
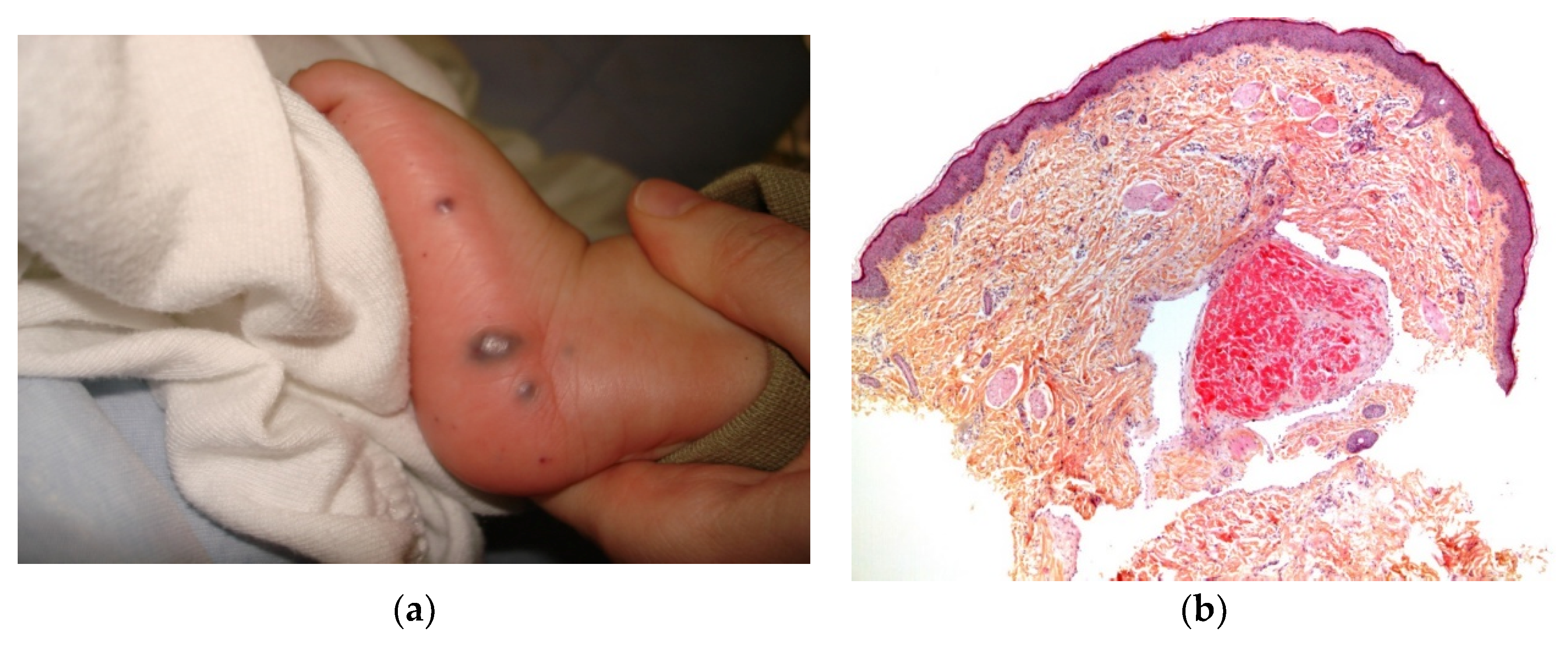
| Syphilis (Figure 2a–d) |
| • Hemolytic disease of the newborn |
| Rhesus and ABO incompatibility |
| Hereditary spherocytosis |
| Twin-twin transfusion syndrome |
| Neoplastic Diseases |
| • Congenital leukemia |
| • Metastatic neuroblastoma |
| • Transitory myeloproliferative disease |
| • Langerhans cell histiocytosis |
| • Congenital metastatic rhabdomyosarcoma |
| • Metastatic rhabdoid tumor |
| • Disseminated juvenile xanthogranuloma |
| • Multicentric infantile myofibromatosis |
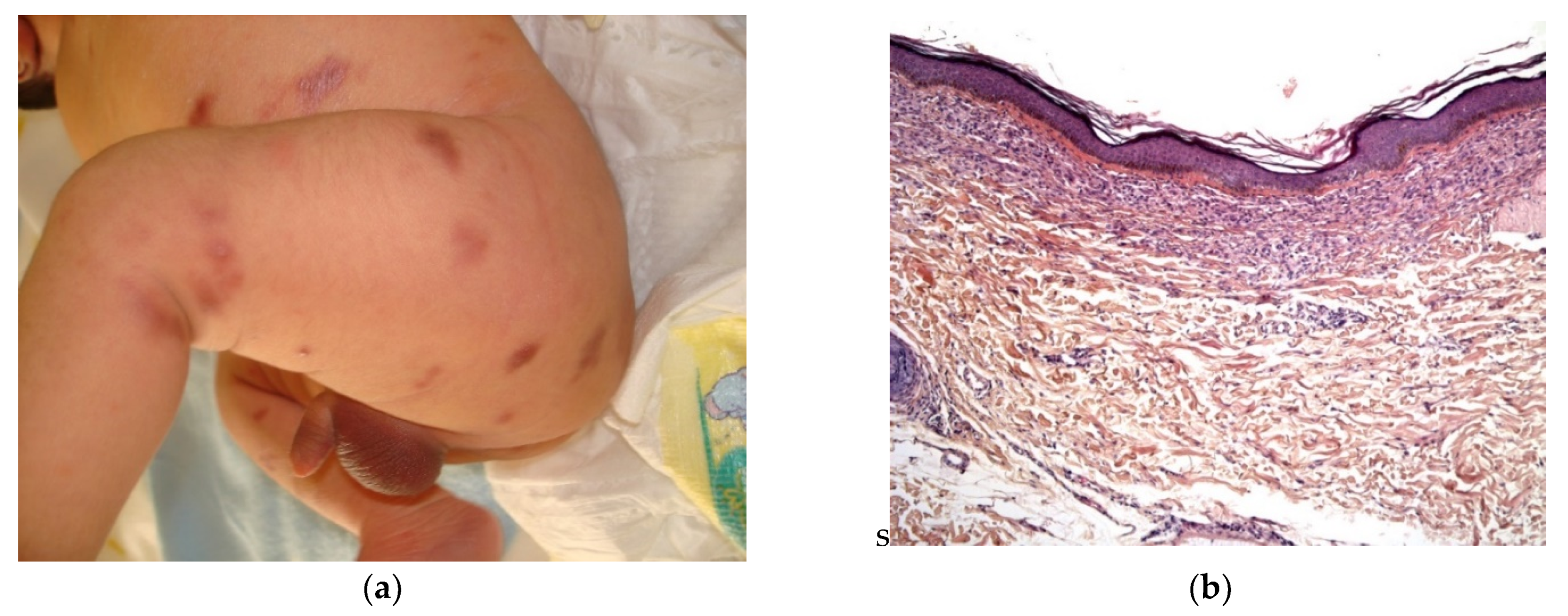
2.1. Dermal Erythropoiesis
2.2. Neoplastic Diseases
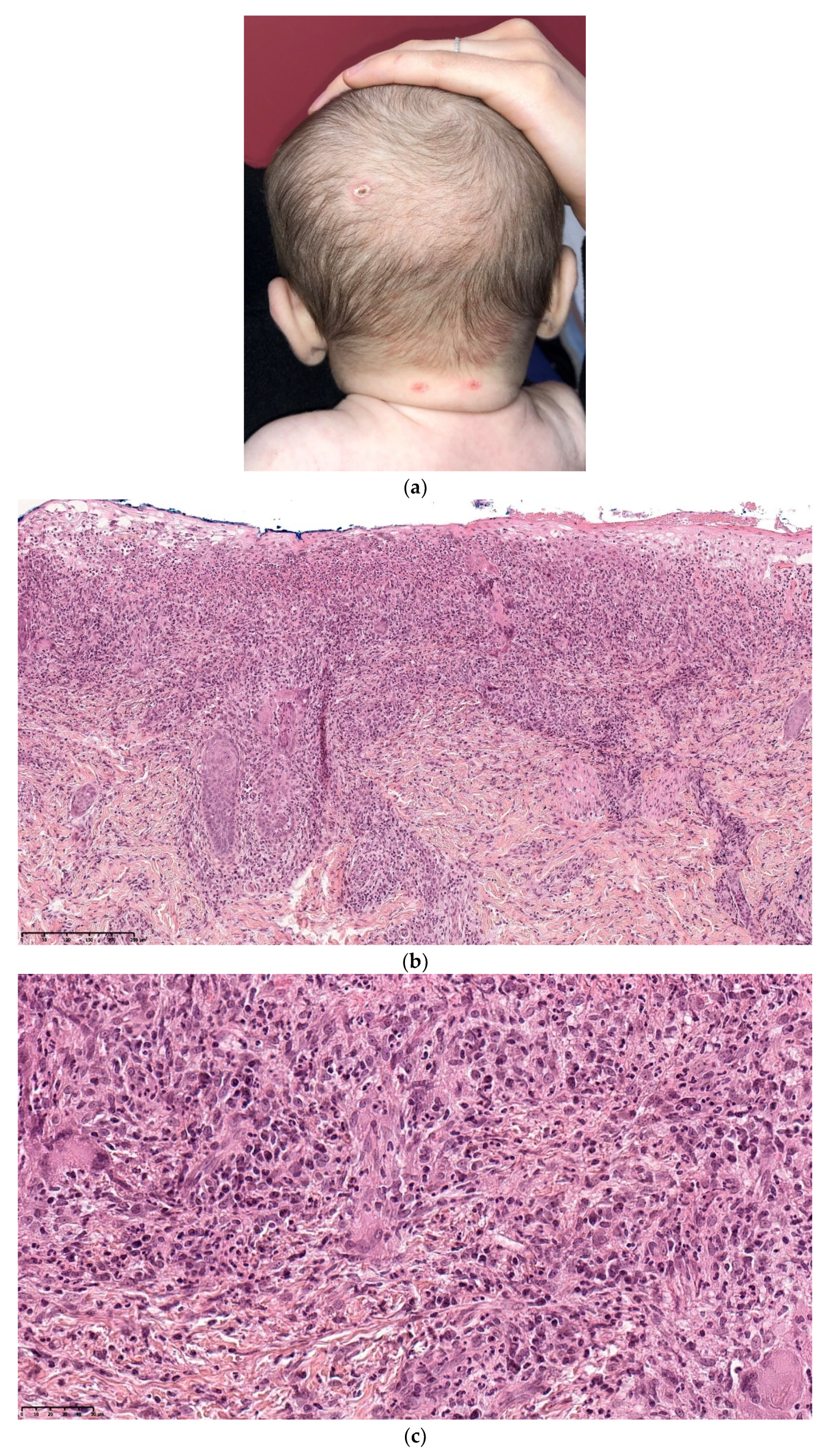
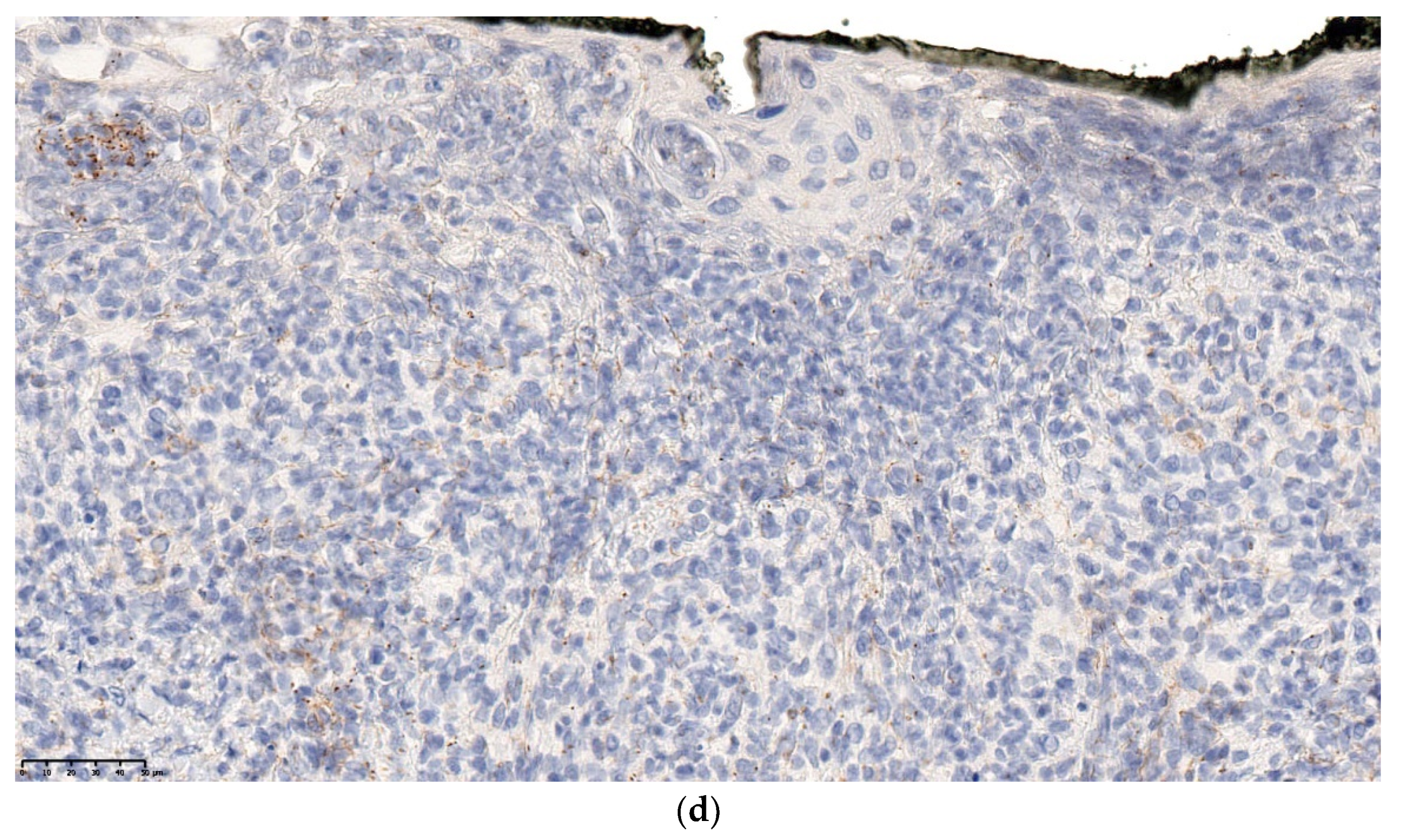
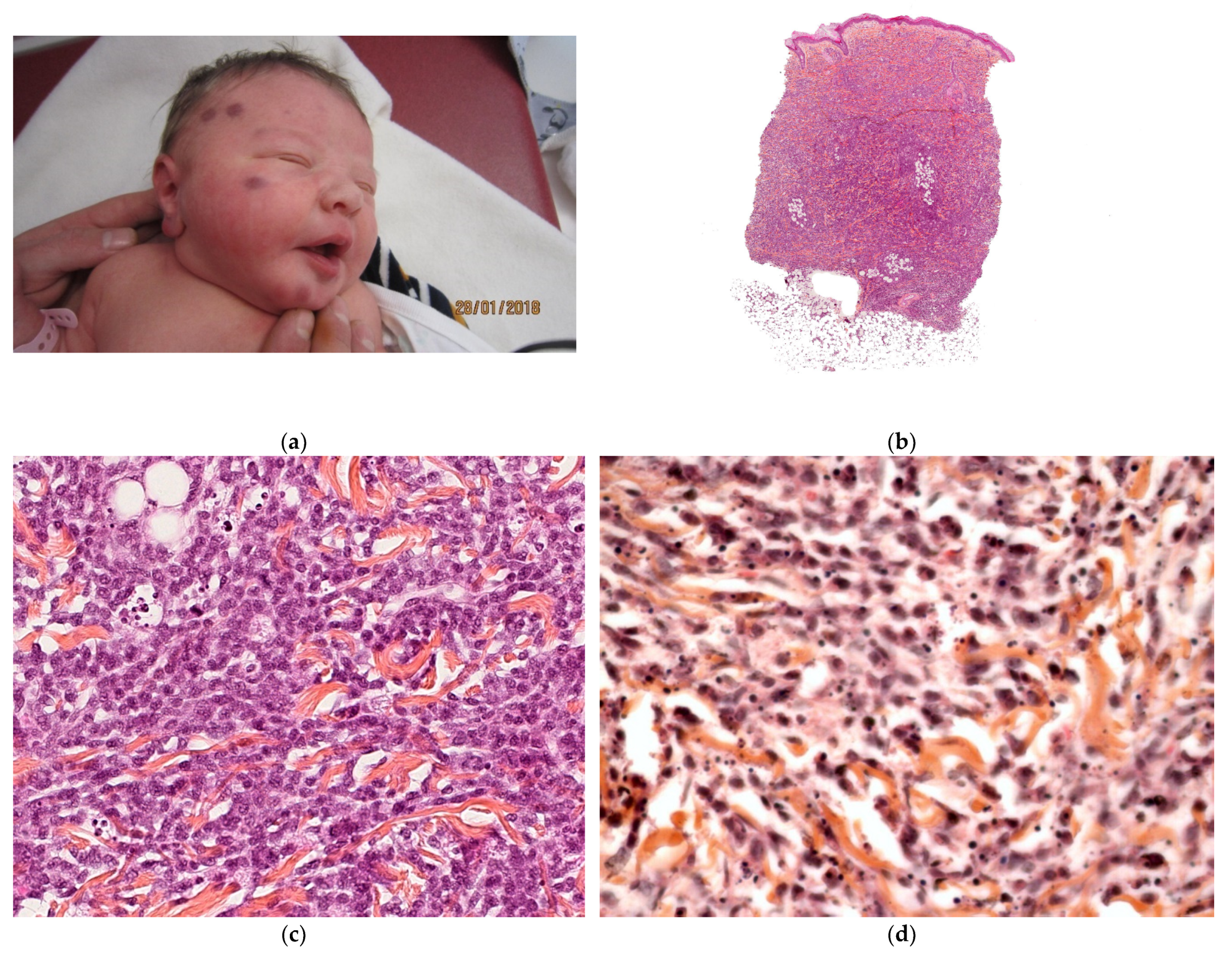
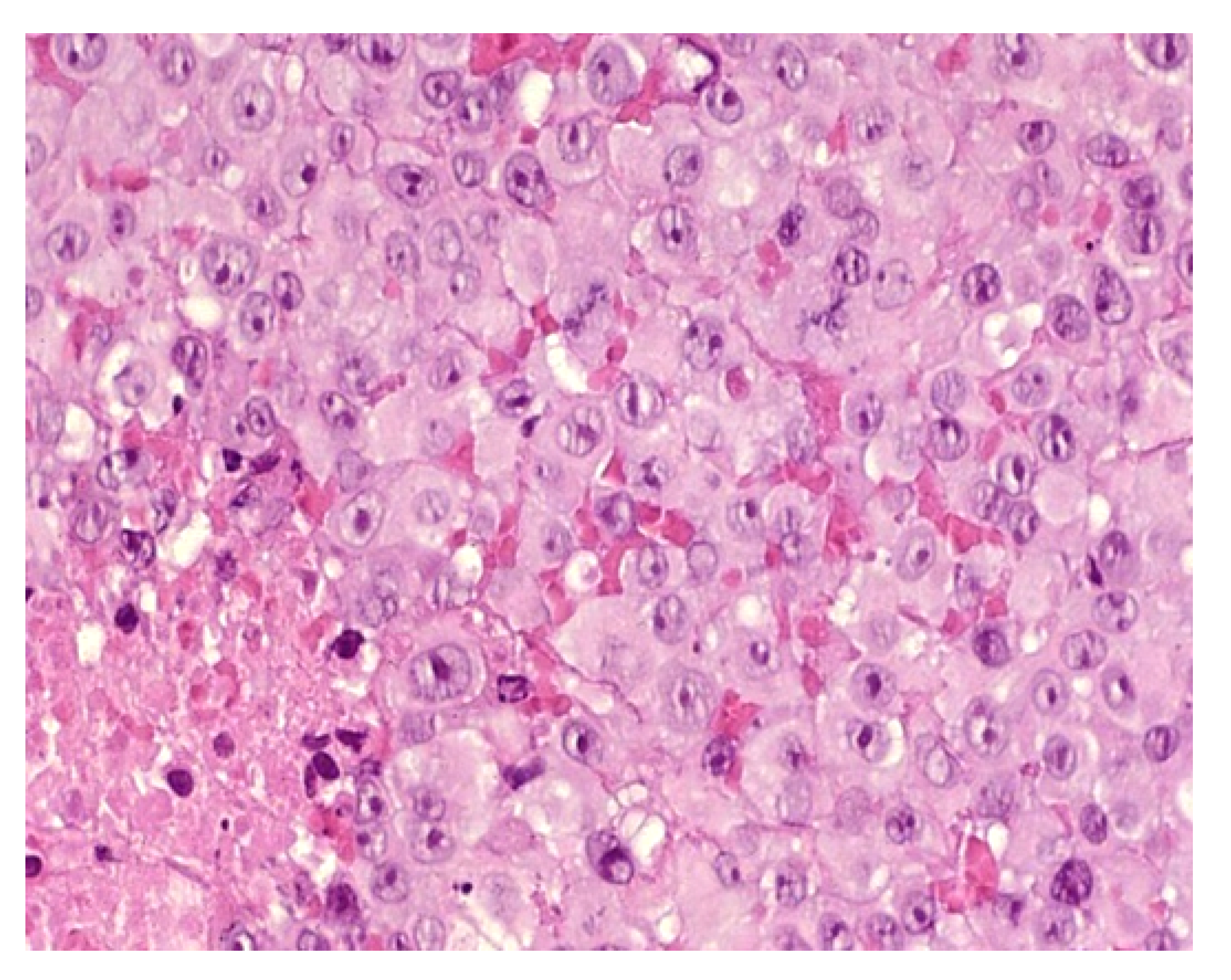
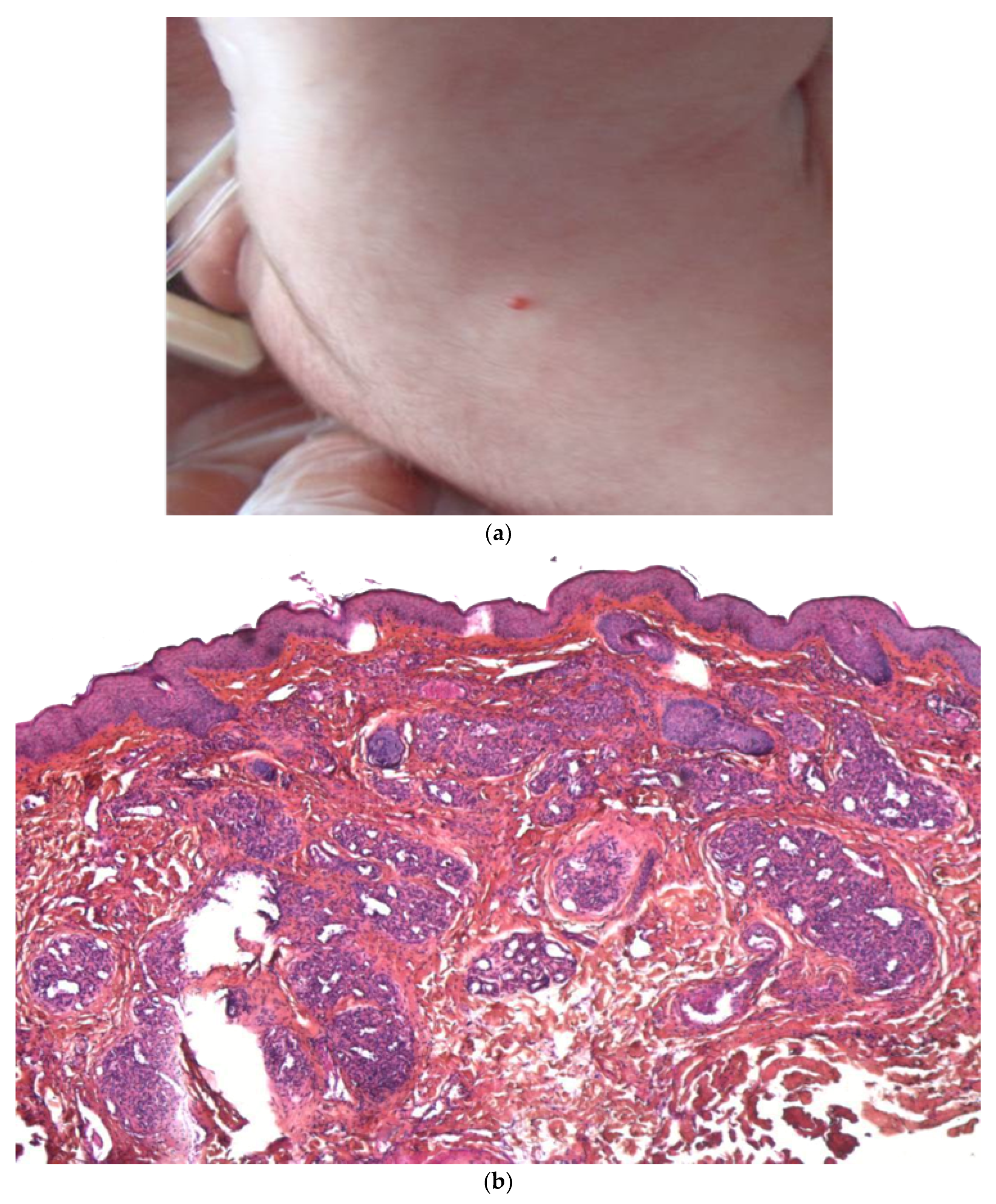
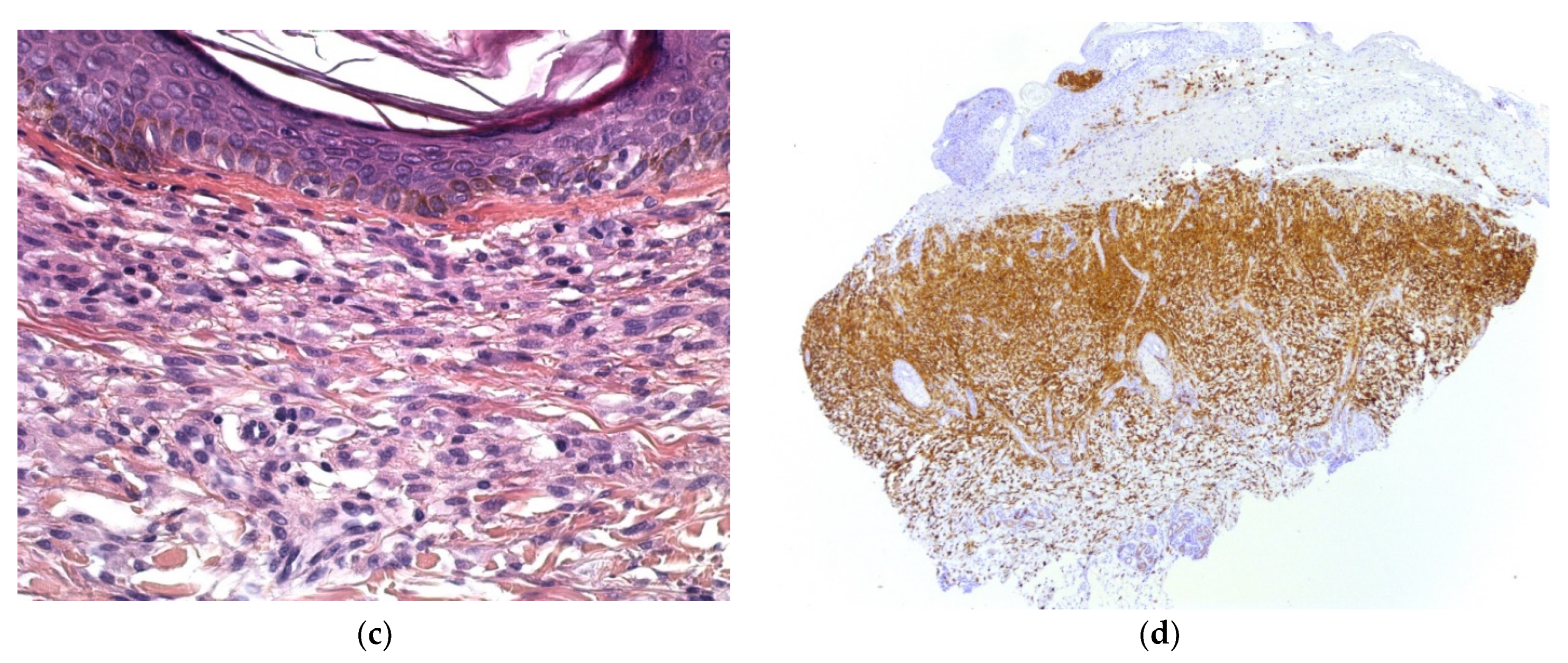
2.2.1. Leukemia Cutis
Histology
Outcome
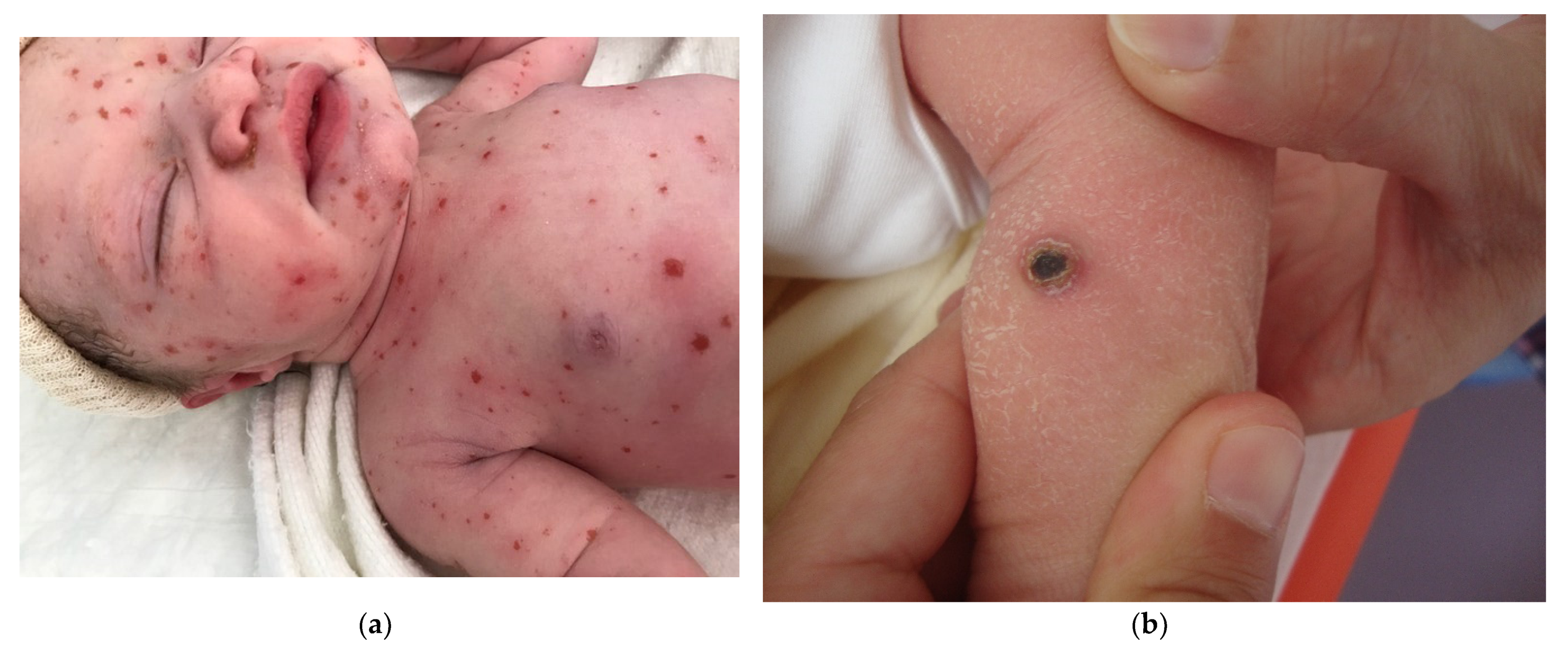
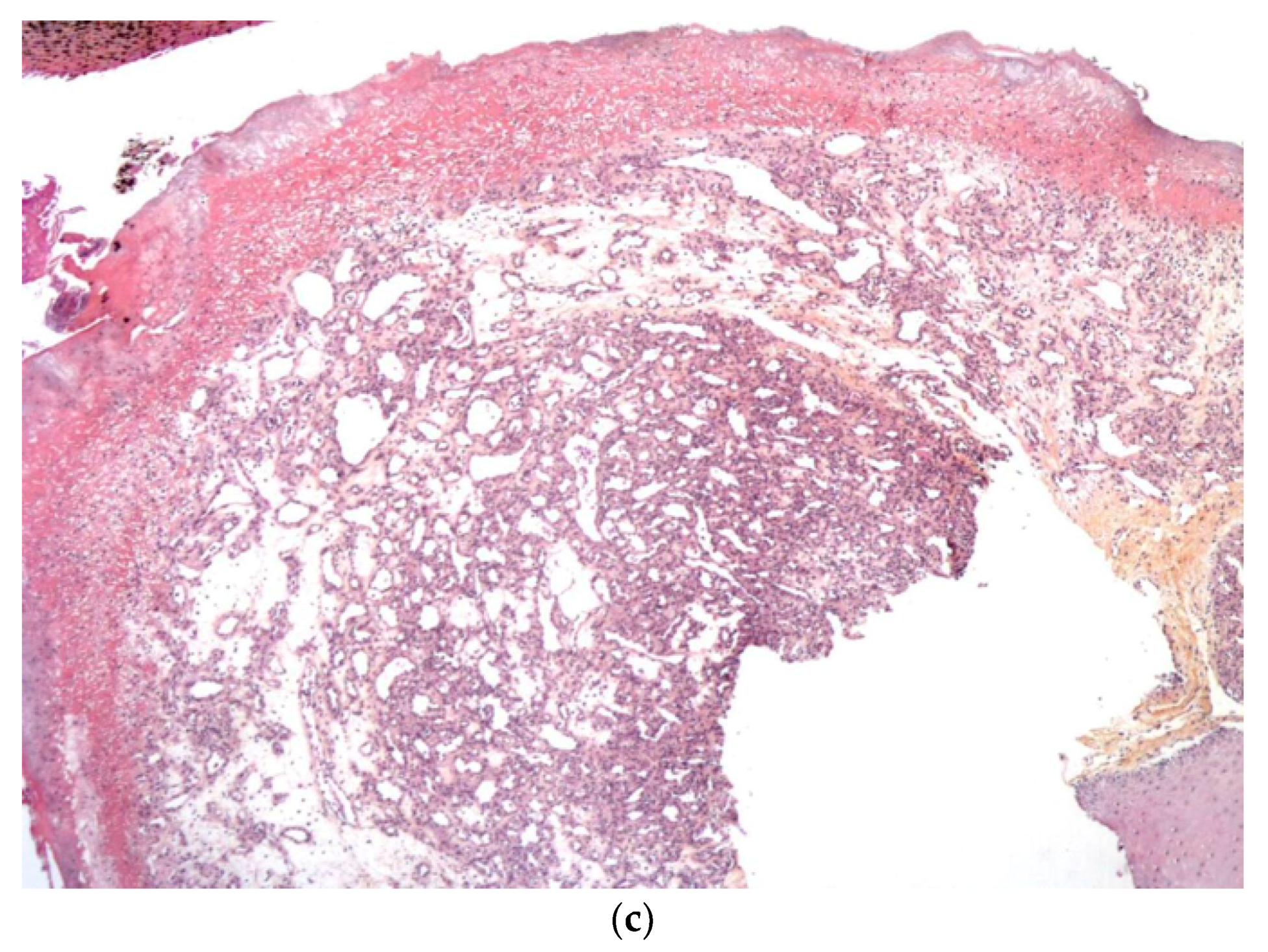
2.2.2. Metastatic Neuroblastoma
Histology
Outcome
2.2.3. Transitory Myeloproliferative Disease
Metastatic Rhabdomyosarcoma
Histology
Outcome
Metastatic Rhabdoid Tumor
Histology
Outcome
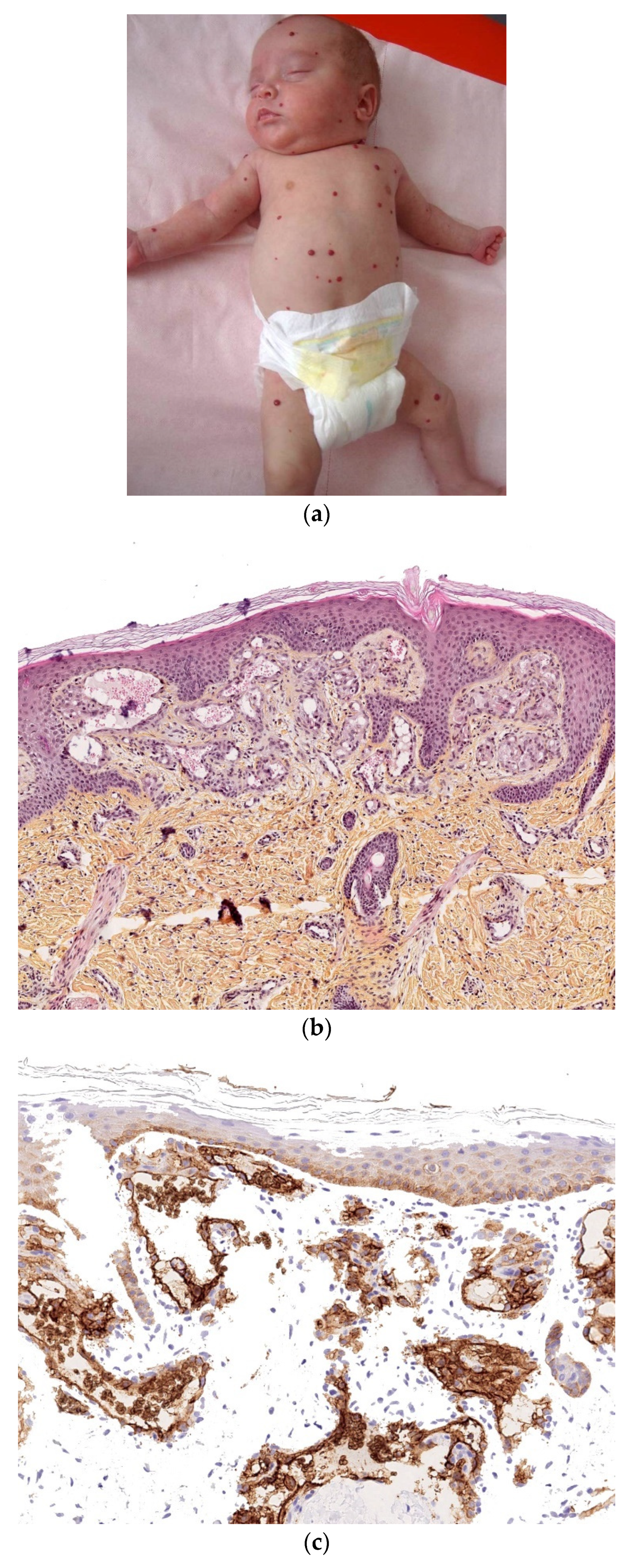
2.2.4. Congenital Histiocytosis
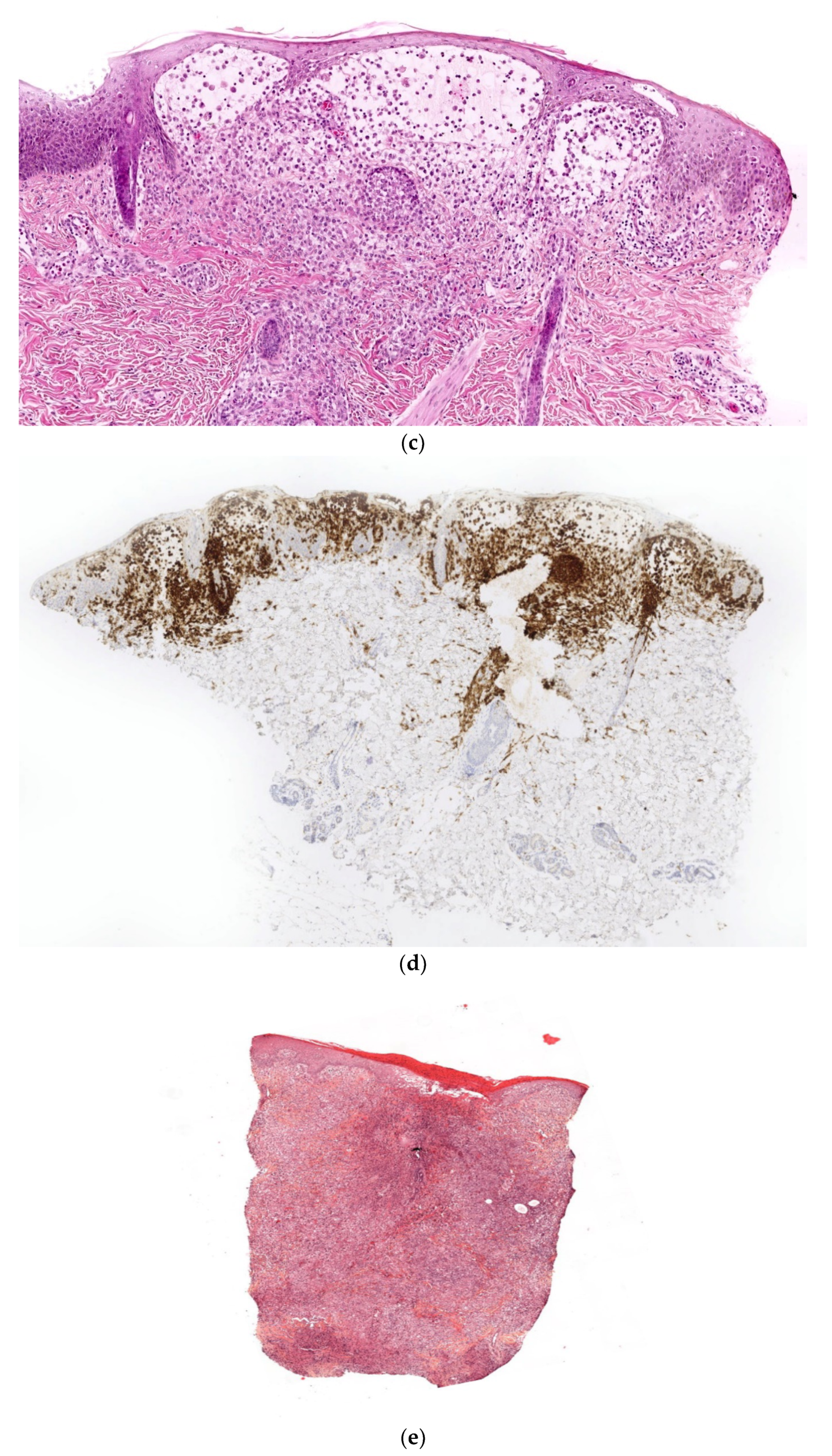
Congenital Langerhans Cell Histiocytosis (LCH)
Histology
Outcome
Multiple Xanthogranuloma
Histology
Outcome
2.2.5. Transplacentally Acquired Tumors
Transplacental Melanoma
Histology
Outcome
Transplacental Choriocarcinoma
Histology
Outcome
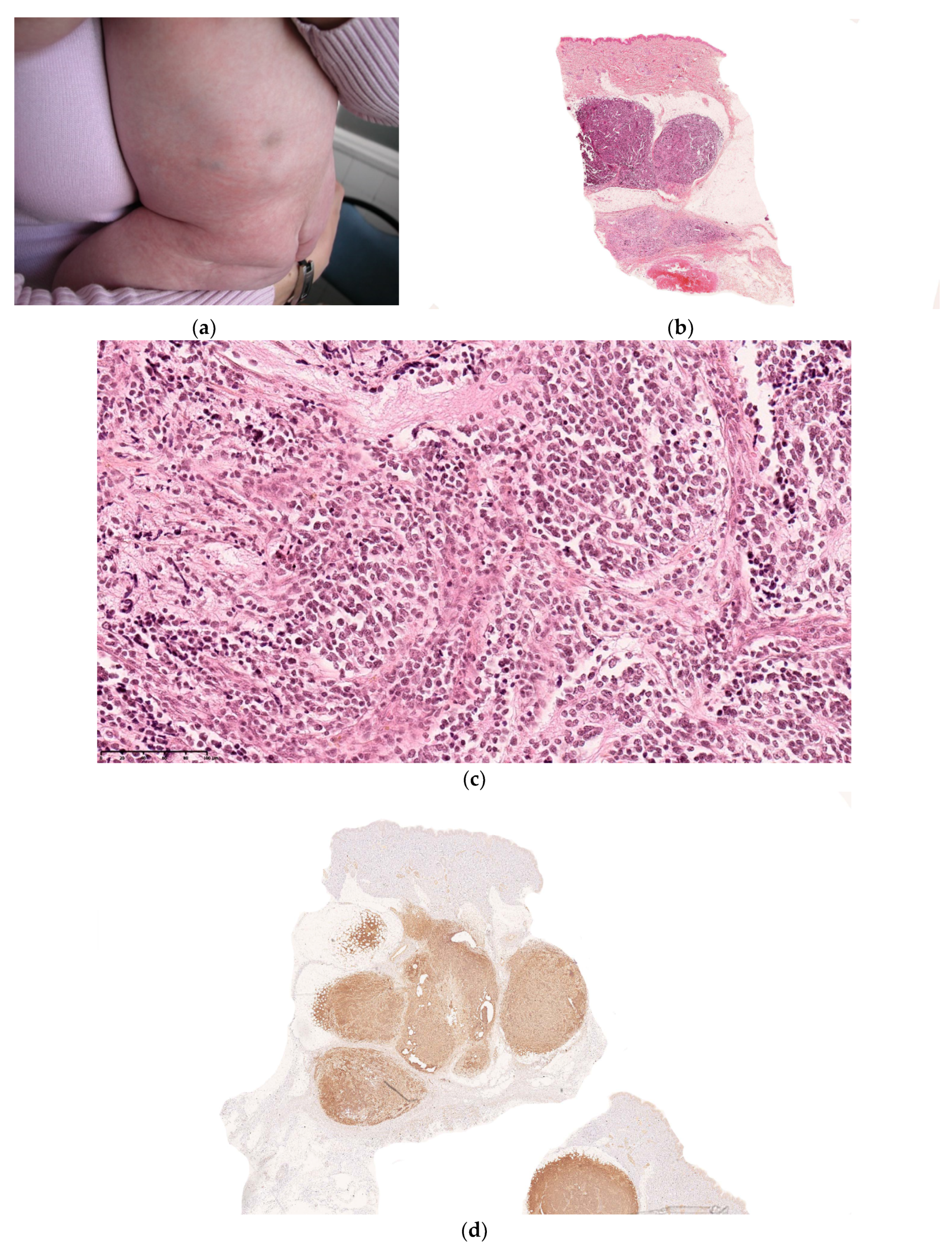
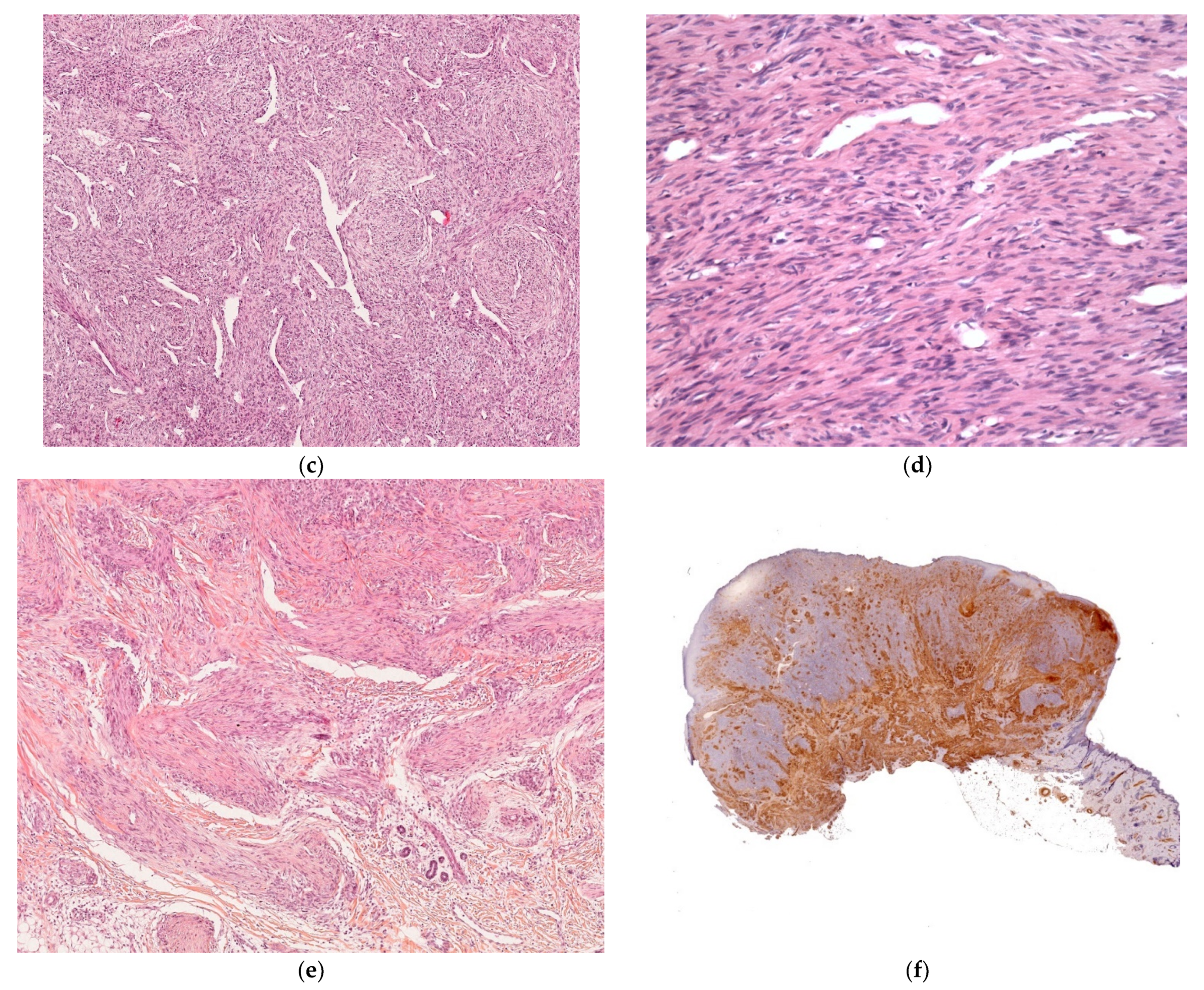
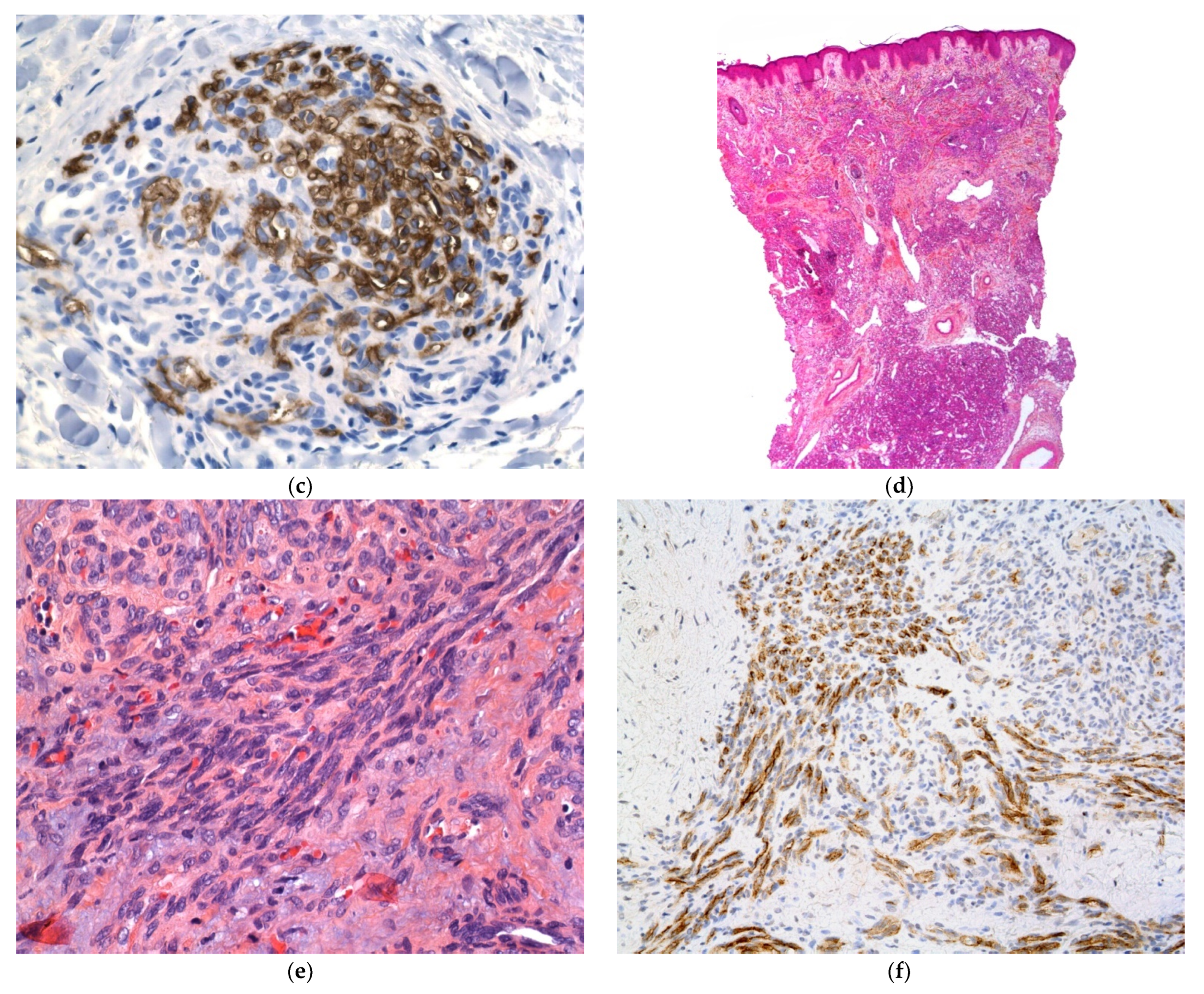
2.2.6. Infantile Myofibromatosis
Histology
Outcome
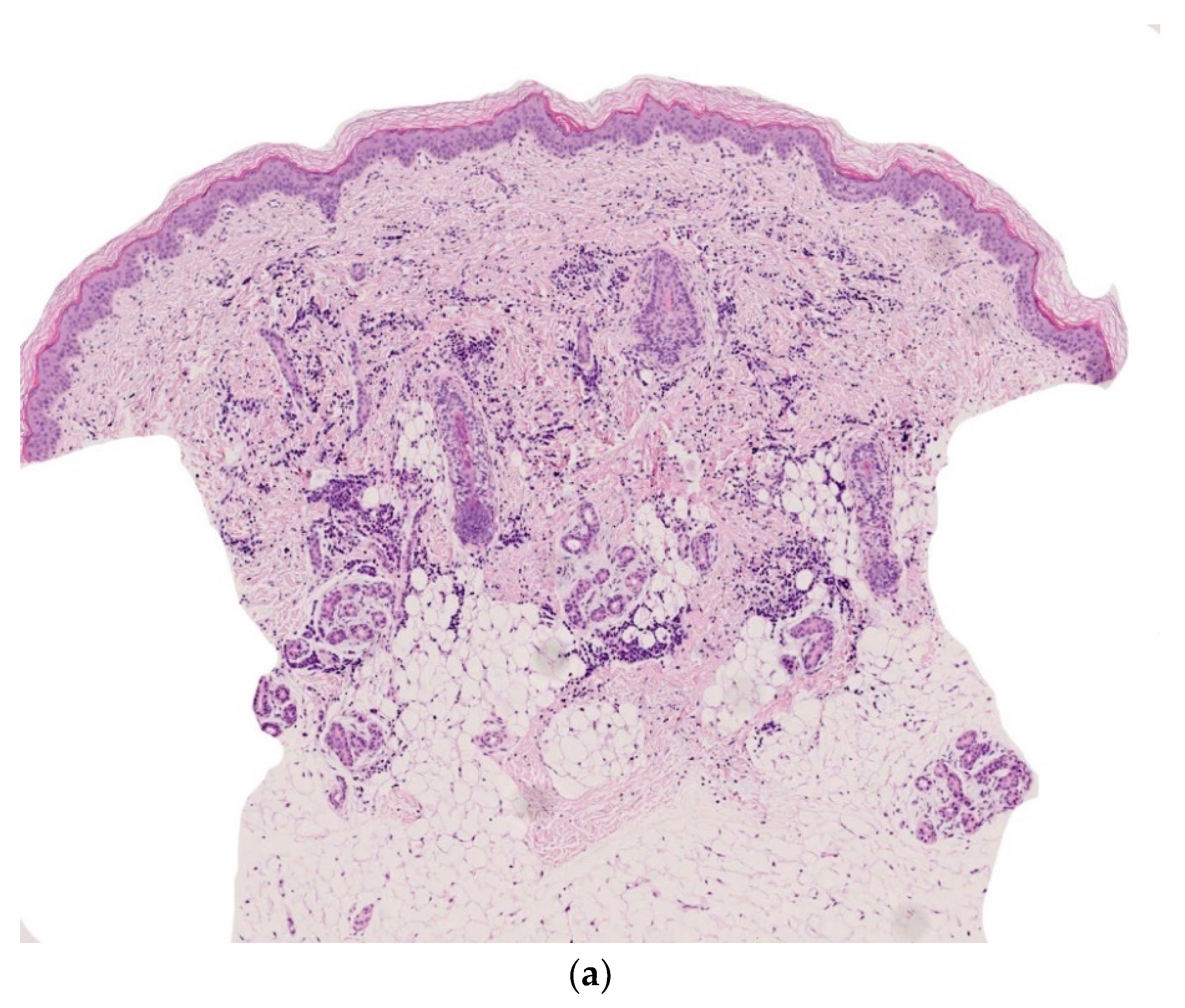
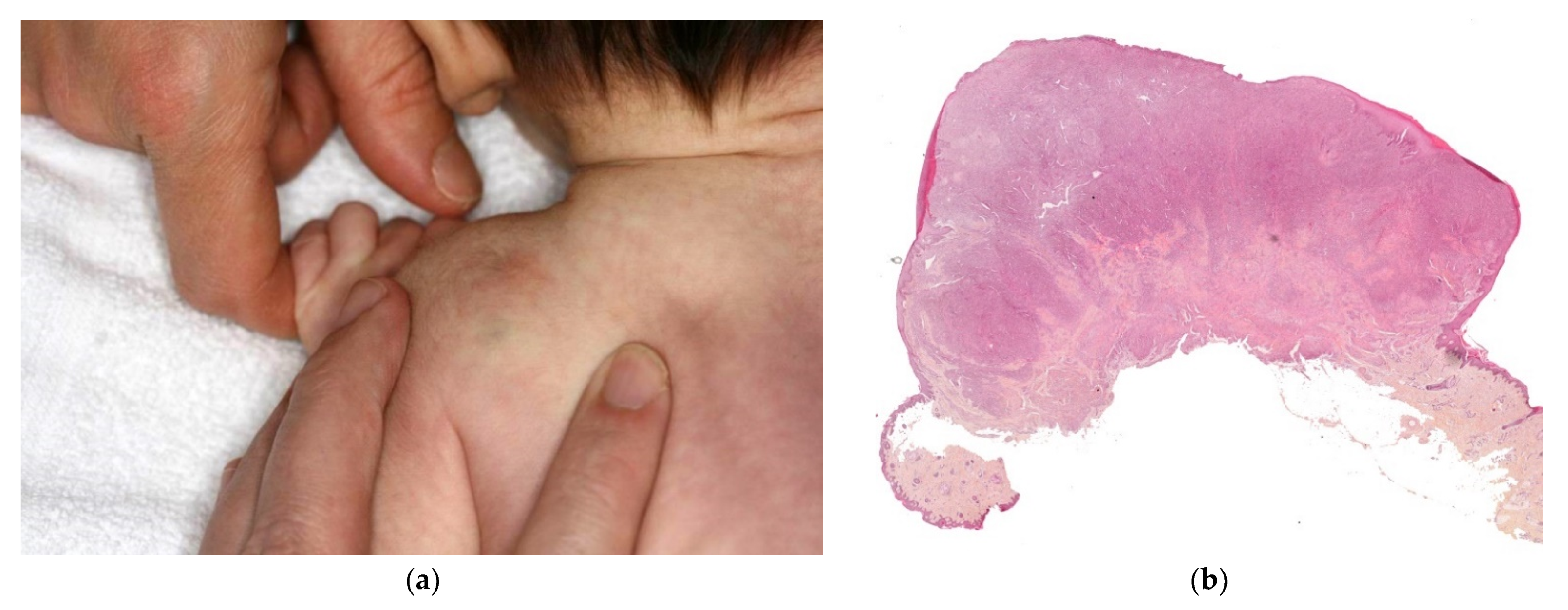
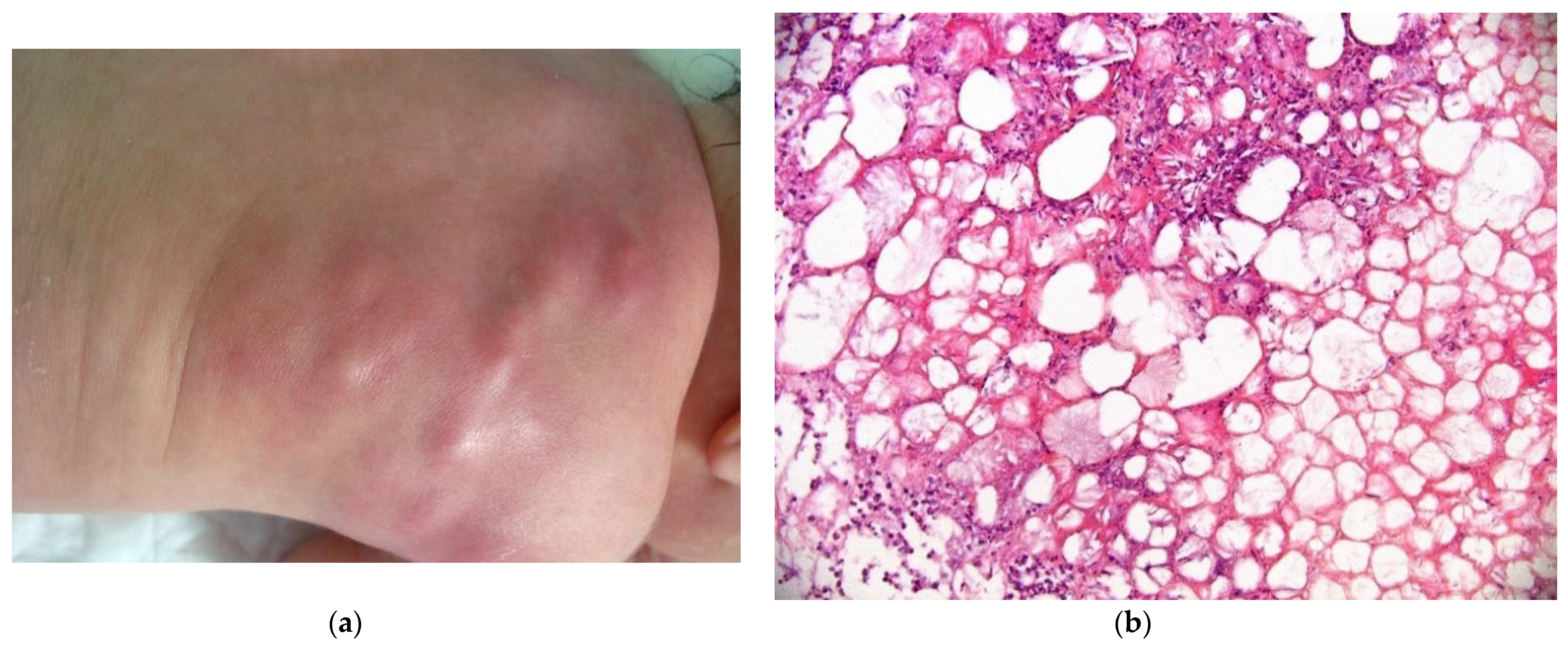
2.3. Non-Neoplastic Disorder: Subcutaneous Fat Necrosis of the Newborn
2.3.1. Histology
2.3.2. Outcome
3. Multifocal Vascular Skin Lesions
3.1. Multifocal Infantile Hemangioma
3.1.1. Histology
3.1.2. Outcome
3.2. Multifocal Lymphangiomatosis with Thrombocytopenia
3.2.1. Histology
3.2.2. Outcome
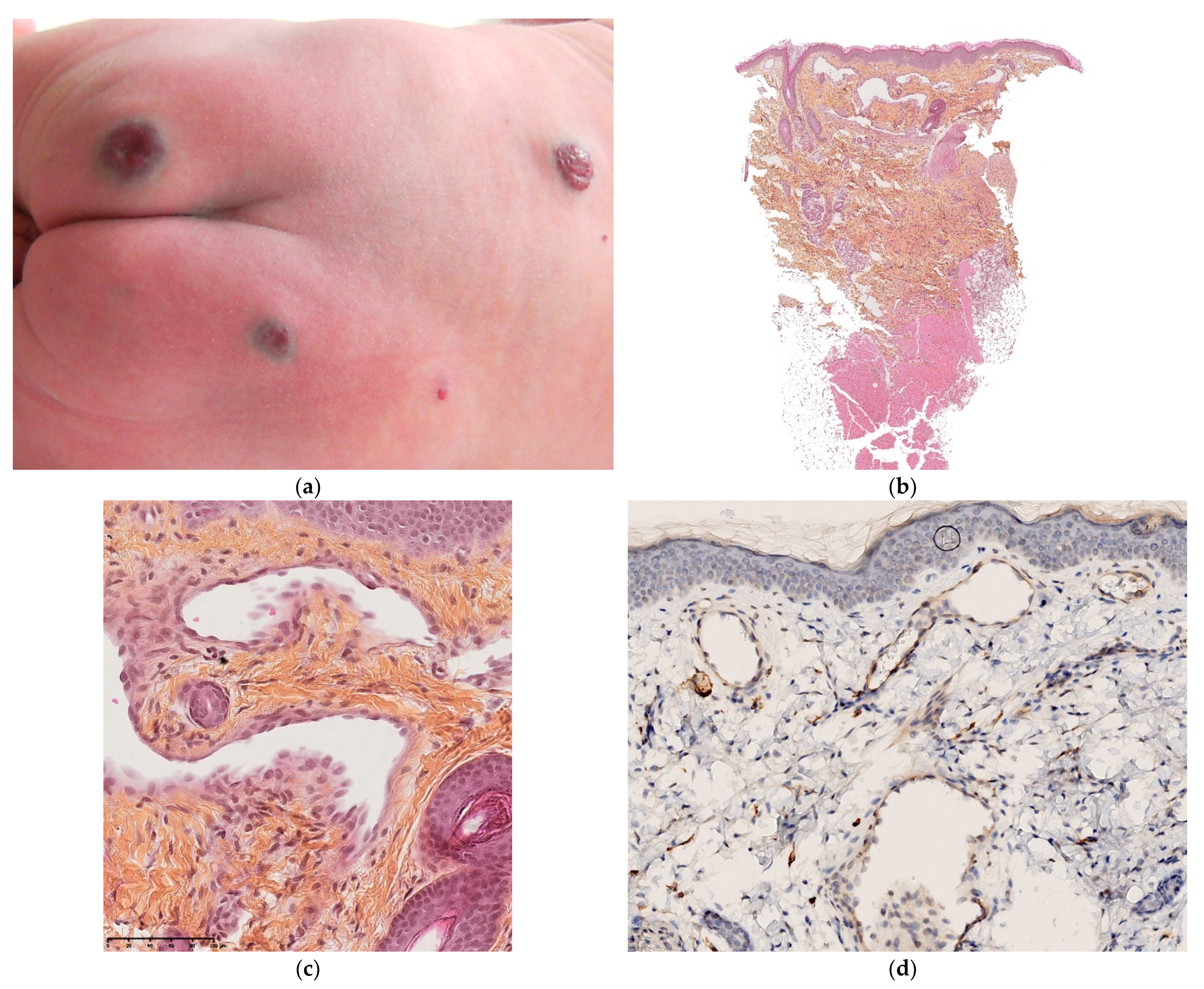
3.3. Multiple Neonatal Pyogenic Granulomas
3.3.1. Histology
3.3.2. Outcome
3.4. Multiple Tufted Angiomas/Kaposiform Hemangioendotheliomatosis
Histology
3.5. Venous Malformations
3.5.1. Glomuvenous Malformation
Histology
3.5.2. Blue Rubber Bleb Naevus Syndrome (Bean’s Syndrome)
Histology
Outcome
4. Cutaneous Mastocytosis
4.1. Histology
4.2. Outcome
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Isaacs, H., Jr. Cutaneous metastases in neonates: A review. Pediatr. Dermatol. 2011, 28, 85–93. [Google Scholar] [CrossRef]
- Brough, A.J.; Jones, D.; Page, R.H.; Mizukami, I. Dermal erythropoïesis in neonatal infants: A manifestation of intrauterine viral disease. Pediatrics 1967, 40, 627–635. [Google Scholar]
- Bowden, J.B.; Hebert, A.A.; Rapini, R.P. Dermal hematopoiesis in neonates: Report of 5 cases. J. Am. Acad. Dermatol. 1989, 20, 1104–1110. [Google Scholar] [CrossRef]
- Gottesfeld, E.; Silverman, R.A.; Coccia, P.F.; Jacobs, G.; Zaim, M.T. Transient blueberry muffin appearance of a newborn with congenital monoblastic leukemia. J. Am. Acad. Dermatol. 1989, 21, 347–351. [Google Scholar] [CrossRef]
- Eberst, E.; Michel, B.; Stoebner, P.; Dandurand, M.; Meunier, L. Spontaneous remission of congenital leukemia cutis. Ann. Dermatol. Venereol. 2011, 138, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Groet, J.; McElwaine, S.; Spinelli, M.; Rinaldi, A.; Burtscher, I.; Mulligan, C.; Mensah, A.; Cavani, S.; Dagna-Bricarelli, F.; Basso, G.; et al. Acquired mutations in GATA1 in neonates with Down’s syndrome with transient myeloid disorder. Lancet 2003, 361, 1617–1620. [Google Scholar] [CrossRef]
- Isaacs, H., Jr. Fetal and neonatal rhabdoid tumor. J. Pediatr. Surg. 2010, 45, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, H., Jr. Pathology of skin diseases. In Potter’s Pathology of the Fetus Infant and Child, 2nd ed.; Gilbert-Barness, E., Ed.; Mosby Elsevier: Philadelphia, PA, USA, 2007; Volume 2, pp. 1743–1795. [Google Scholar]
- Grundy, R.; Anderson, J.; Gaze, M.; Gerrard, M.; Glaser, A.; Gordon, A.; Malone, M.; Pritchard-Jones, K.; Michalski, A. Congenital alveolar rhabdomyosarcoma: Clinical and molecular distinction from alveolar rhabdomyosarcoma in older children. Cancer 2001, 91, 606–612. [Google Scholar] [CrossRef]
- Hayes-Jordan, A.; Andrassy, R. Rhabdomyosarcoma in children. Curr. Opin. Pediatr. 2009, 21, 373–378. [Google Scholar] [CrossRef]
- Morren, M.A.; Broecke, K.V.; Vangeebergen, L.; Sillevis-Smitt, J.H.; Van Den Berghe, P.; Hauben, E.; Jacobs, S.; Van Gool, S.W. Diverse Cutaneous Presentations of Langerhans Cell Histiocytosis in Children: A Retrospective Cohort Study. Pediatr. Blood Cancer 2016, 63, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Larralde, M.; Rositto, A.; Giardelli, M.; Gatti, C.F.; Muñoz, A.S. Congenital self-healing histiocytosis (Hashimoto–Pritzker). Int. J. Dermatol. 1999, 38, 693–696. [Google Scholar] [CrossRef]
- Kapur, P.; Erickson, C.; Rakheja, D.; Carder, K.R.; Hoang, M.P. Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease): Ten-year experience at Dallas Children’s Medical Center. J. Am. Acad. Dermatol. 2007, 56, 290–294. [Google Scholar] [CrossRef]
- Dupeux, M.; Boccara, O.; Frassati-Biaggi, A.; Hélias-Rodzewicz, Z.; Leclerc-Mercier, S.; Bodemer, C.; Molina, T.J.; Emile, J.F.; Fraitag, S. Langerhans Cell Histiocytoma: A Benign Histiocytic Neoplasm of Diverse Lines of Terminal Differentiation. Am. J. Dermatopathol. 2019, 41, 29–36. [Google Scholar] [CrossRef]
- Battistella, M.; Fraitag, S.; Teillac, D.H.; Brousse, N.; de Prost, Y.; Bodemer, C. Neonatal and early infantile cutaneous Langerhans cell histiocytosis: Comparison of self-regressive and non-self-regressive forms. Arch. Dermatol. 2010, 146, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Haughton, A.M.; Horii, K.A.; Shao, L.; Daniel, J.; Nopper, A.J. Disseminated juvenile xanthogranulomatosis in a newborn resulting in liver transplantation. J. Am. Acad. Dermatol. 2008, 58, S12–S15. [Google Scholar] [CrossRef]
- Paxton, C.N.; O’Malley, D.P.; Bellizzi, A.M.; Alkapalan, D.; Fedoriw, Y.; Hornick, J.L.; Perkins, S.L.; South, S.T.; Andersen, E.F. Genetic evaluation of juvenile xanthogranuloma: Genomic abnormalities are uncommon in solitary lesions, advanced cases may show more complexity. Mod. Pathol. 2017, 30, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Dehner, L.P. Juvenile xanthogranulomas in the first two decades of life: A clinicopathologic study of 174 cases with cutaneous and extracutaneous manifestations. Am. J. Surg. Pathol. 2003, 27, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.M.; Maceira, J.M.; Coelho, J.M. Melanoma and pregnancy with placental metastases. Report of a case. Am. J. Dermatopathol. 1998, 20, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Menada, M.V.; Moioli, M.; Garaventa, A.; Nozza, P.; Foppiano, M.; Trimarchi, N.; Fulcheri, E. Spontaneous regression of transplacental metastases from maternal melanoma in a newborn: Case report and review of the literature. Melanoma Res. 2010, 20, 443–449. [Google Scholar] [CrossRef]
- Alomari, A.K.; Glusac, E.J.; Choi, J.; Hui, P.; Seeley, E.H.; Caprioli, R.M.; Watsky, K.L.; Urban, J.; Lazova, R. Congenital nevi versus metastatic melanoma in a newborn to a mother with malignant melanoma—Diagnosis supported by sex chromosome analysis and Imaging Mass Spectrometry. J. Cutan. Pathol. 2015, 42, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Brooks, T.; Nolting, L. Cutaneous manifestation of metastatic infantile choriocarcinoma.Case reports in pediatrics. Case Rep. Pediatr. 2014, 2014, 104652. [Google Scholar]
- Blohm, M.E.; Göbel, U. Unexplained anaemia and failure to thrive as initial symptoms of infantile choriocarcinoma: A review Unexplained anaemia and failure to thrive as initial symptoms of infantile choriocarcinoma: A review. Eur. J. Pediatr. 2004, 163, 1–6. [Google Scholar] [CrossRef]
- Getrajdman, J.; Kolev, V.; Brody, E.; Chuang, L. Case of maternal and infantile choriocarcinoma following normal pregnancy. Gynecol. Oncol. Case Rep. 2012, 2, 102–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashiah, J.; Hadj-Rabia, S.; Dompmartin, A.; Harroche, A.; Laloum-Grynberg, E.; Wolter, M.; Amoric, J.C.; Hamel-Teillac, D.; Guero, S.; Fraitag, S.; et al. Infantile myofibromatosis: A series of 28 cases. J. Am. Acad. Dermatol. 2014, 71, 264–270. [Google Scholar] [CrossRef]
- Wiswell, T.E.; Davis, J.; Cunningham, B.E.; Solenberger, R.; Thomas, P.J. Infantile myofibromatosis: The most common fibrous tumor of infancy. J. Pediatr. Surg. 1988, 23, 315–318. [Google Scholar] [CrossRef]
- Dachy, G.; de Krijger, R.R.; Fraitag, S.; Théate, I.; Brichard, B.; Hoffman, S.B.; Libbrecht, L.; Arts, F.A.; Brouillard, P.; Vikkula, M.; et al. Association of PDGFRB Mutations With Pediatric Myofibroma and Myofibromatosis. JAMA Dermatol. 2019, 155, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Proust, S.; Benchimol, G.; Fraitag, S.; Starck, J.; Giacobbi, V.; Pierron, G.; Bodemer, C.; Orbach, D. Major response to imatinib and chemotherapy in a newborn patient prenatally diagnosed with generalized infantile. Pediatr. Blood Cancer 2021, 68, e28576. [Google Scholar] [CrossRef]
- Mahé, E.; Girszyn, N.; Hadj-Rabia, S.; Bodemer, C.; Hamel-Teillac, D.; De Prost, Y. Subcutaneous fat necrosis of the newborn: A systematic evaluation of risk factors, clinical manifestations, complications and outcome of 16 children. Br. J. Dermatol. 2007, 156, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Wessman, L.L.; Mori, W.S.; Totoraitis, K.; Murati, M.; Maguiness, S. Multifocal infantile hemangiomas associated with concomitant giant hepatic rapidly involuting congenital hemangioma. Pediatr. Dermatol. 2020, 37, 972–973. [Google Scholar] [CrossRef]
- Ferrandiz, L.; Toledo-Pastrana, T.; Moreno-Ramirez, D.; Bardallo-Cruzado, L.; Perez-Bertolez, S.; Luna-Lagares, S.; Rios-Martin, J.J. Diffuse neonatal hemangiomatosis with partial response to propranolol. Int. J. Dermatol. 2014, 53, e247–e250. [Google Scholar] [CrossRef]
- North, P.E.; Kahn, T.; Cordisco, M.R.; Dadras, S.S.; Detmar, M.; Frieden, I.J. Multifocal lymphangioendotheliomatosis with thrombocytopenia: A newly recognized clinicopathological entity. Arch. Dermatol. 2004, 140, 599–606. [Google Scholar] [CrossRef] [Green Version]
- Prasad, V.; Fishman, S.J.; Mulliken, J.B.; Fox, V.L.; Liang, M.G.; Klement, G.; Kieran, M.W.; Burrows, P.E.; Waltz, D.A.; Powell, J.; et al. Cutaneovisceral angiomatosis with thrombocytopenia. Pediatr. Dev. Pathol. 2005, 8, 407–419. [Google Scholar] [CrossRef]
- Droitcourt, C.; Boccara, O.; Fraitag, S.; Favrais, G.; Dupuy, A.; Maruani, A. Multifocal lymphangioendotheliomatosis with thrombocytopenia: Clinical features and response to sirolimus. Pediatrics 2015, 136, e517–e522. [Google Scholar] [CrossRef] [Green Version]
- Alomari, M.H.; Kozakewich, H.P.W.; Kerr, C.L.; Uller, W.; Davis, S.L.; Chaudry, G.; Liang, M.G.; Orbach, D.B.; Mulliken, J.B.; Greene, A.K.; et al. Congenital Disseminated Pyogenic Granuloma: Characterization of an Aggressive Multisystemic Disorder. J. Pediatr. 2020, 2, 157–166. [Google Scholar] [CrossRef]
- Mallet, S.; Rebelle, C.; Ligi, I.; Scavarda, D.; Bouvier, C.; Petit, P.; Fraitag, S.; Wassef, M.; Gaudy-Marqueste, C.; Hesse, S.; et al. Congenital and disseminated pyogenic granuloma-like vascular lesions. Acta Derm. Venereol. 2015, 95, 860–861. [Google Scholar] [CrossRef] [Green Version]
- Maronn, M.; Chamlin, S.; Metry, D. Multifocal tufted angiomas in 2 infants. Arch. Dermatol. 2009, 145, 847–848. [Google Scholar] [CrossRef] [PubMed]
- Gianotti, R.; Gelmetti, C.; Alessi, E. Congenital cutaneous multifocal kaposiform hemangioendothelioma. Am. J. Dermatopathol. 1999, 21, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Mallory, S.B.; Enjolras, O.; Boon, L.M.; Rogers, E.; Berk, D.R.; Blei, F.; Baselga, E.; Ros, A.M.; Vikkula, M. Congenital plaque-type glomuvenous malformations presenting in childhood. Arch. Dermatol. 2006, 142, 892–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, D.; Teichler, A.; Hoeger, P.H. Long-term sirolimus treatment in blue rubber bleb nevus syndrome: Case report and review of the literature. Pediatr. Dermatol. 2021, 38, 464–468. [Google Scholar] [CrossRef]
- Meni, C.; Georgin-Lavialle, S.; Le Saché de Peufeilhoux, L.; Jais, J.P.; Hadj-Rabia, S.; Bruneau, J.; Fraitag, S.; Hanssens, K.; Dubreuil, P.; Hermine, O.; et al. Paediatric mastocytosis: Long-term follow-up of 53 patients with whole sequencing of KIT. A prospective study. Br. J. Dermatol. 2018, 179, 925–932. [Google Scholar] [CrossRef]



























| Multifocal infantile hemangioma |
|---|
| Multifocal lymphangiomatosis with thrombocytopenia |
| Multiple neonatal pyogenic granuloma |
| Multiple tufted angioma/kaposiform hemangioendothelioma |
| Venous malformations: |
| glomuvenous malformation |
| blue rubber bleb naevus syndrome (Bean’s syndrome) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fraitag, S.; Boccara, O. What to Look Out for in a Newborn with Multiple Papulonodular Skin Lesions at Birth. Dermatopathology 2021, 8, 390-417. https://doi.org/10.3390/dermatopathology8030043
Fraitag S, Boccara O. What to Look Out for in a Newborn with Multiple Papulonodular Skin Lesions at Birth. Dermatopathology. 2021; 8(3):390-417. https://doi.org/10.3390/dermatopathology8030043
Chicago/Turabian StyleFraitag, Sylvie, and Olivia Boccara. 2021. "What to Look Out for in a Newborn with Multiple Papulonodular Skin Lesions at Birth" Dermatopathology 8, no. 3: 390-417. https://doi.org/10.3390/dermatopathology8030043
APA StyleFraitag, S., & Boccara, O. (2021). What to Look Out for in a Newborn with Multiple Papulonodular Skin Lesions at Birth. Dermatopathology, 8(3), 390-417. https://doi.org/10.3390/dermatopathology8030043