
Investigation of Imidazolium-Based Ionic Liquids as Additives for the Separation of Urinary Biogenic Amines via Capillary Electrophoresis

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemical Reagents
2.2. Analytical Equipment
2.3. Preparation of Standard and Working Solutions of BAs
2.4. Instrumentation and Separation Conditions
2.5. Biological Sample Collection and Pretreatment
2.5.1. Sample Storage and Source
2.5.2. The Preparation of Human Urine Samples
2.5.3. Sample Pretreatment Procedure
2.6. Validation Procedure
3. Results
3.1. The Separation Buffer Composition
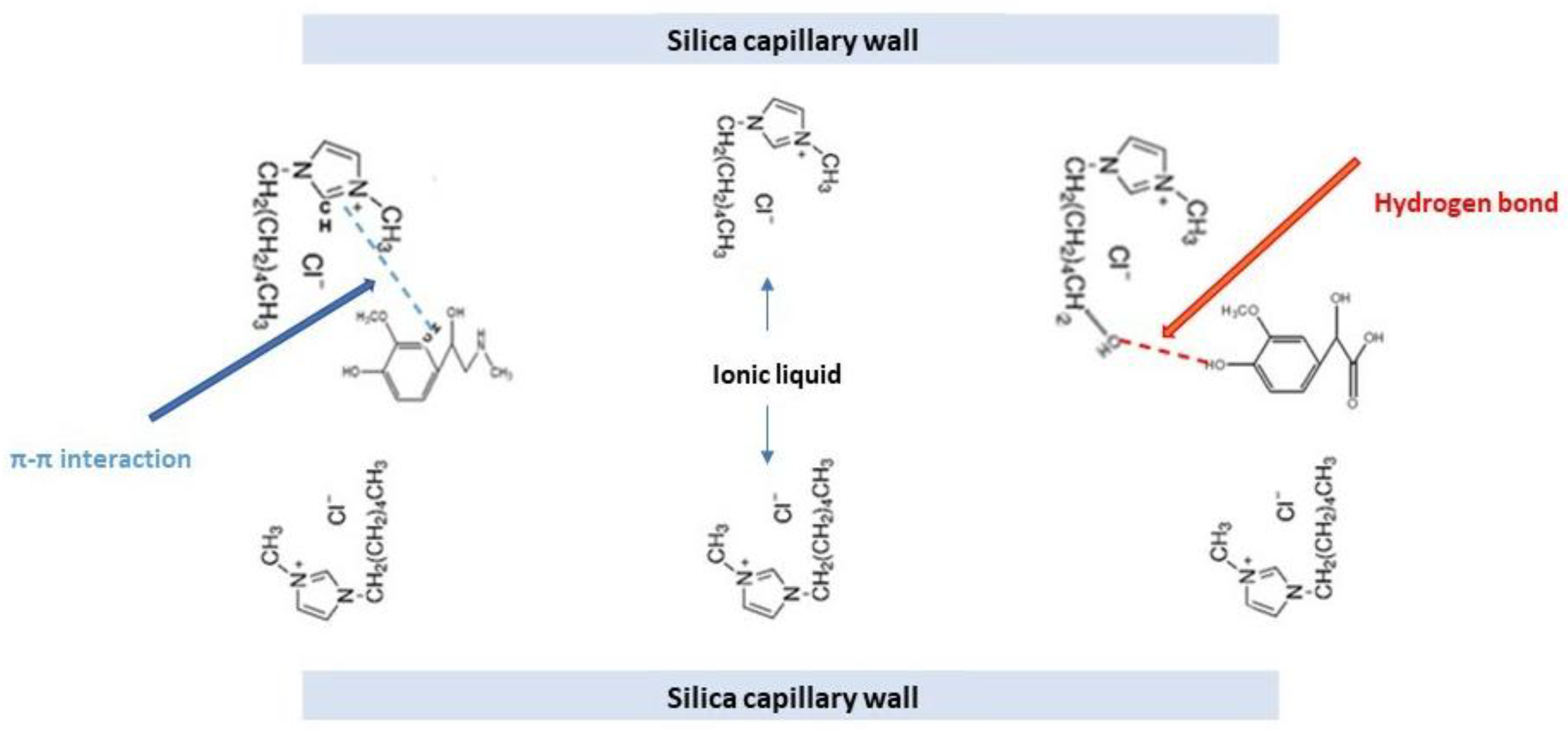
3.2. Dynamic Double Coating Effect of Examined ILs as Additives to Separation Buffer
3.3. Optimization of Sample Preparation Procedure
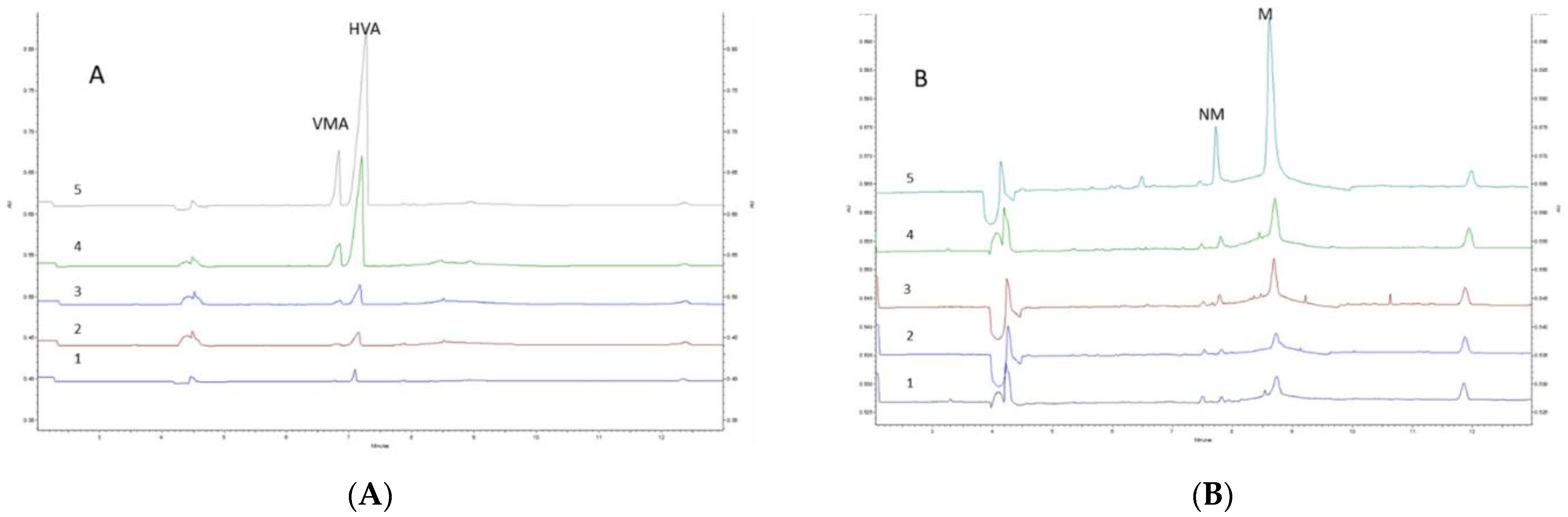
3.4. Validation of the Developed Method
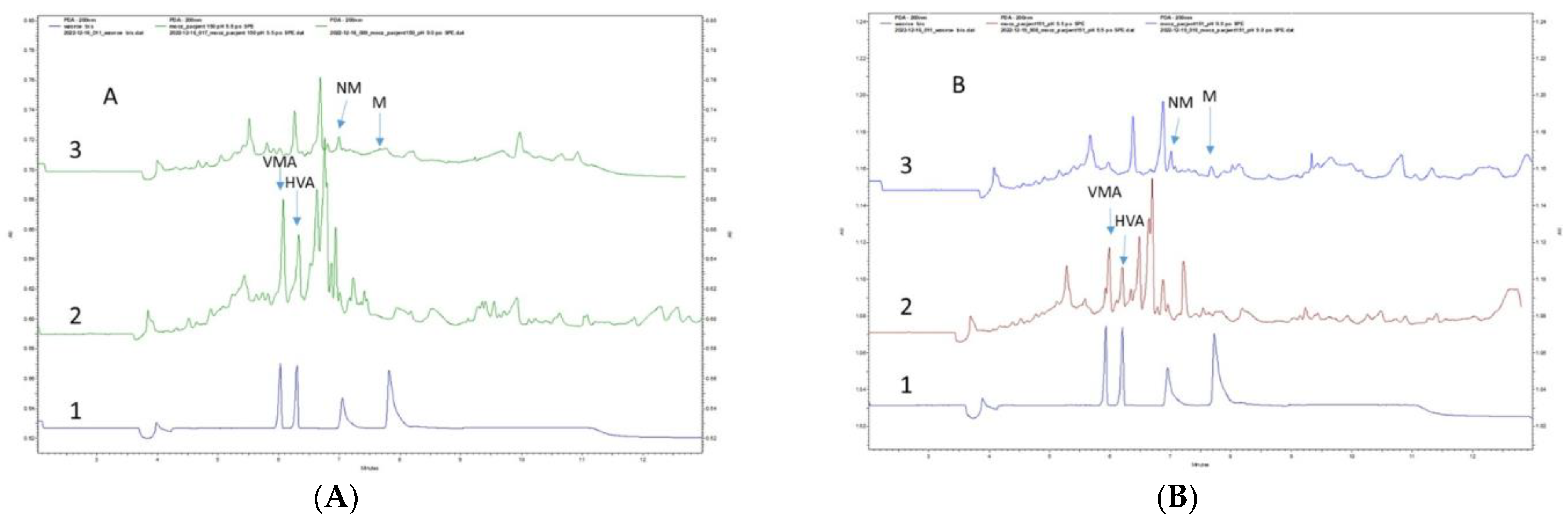
3.5. Application of the Developed Method in Real Urine Samples
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Y.; Du, Y.; Yu, T.; Feng, Z.; Chen, J. Investigation of dextrin-based synergistic system with chiral ionic liquids as additives for enantiomeric separation in capillary electrophoresis. J. Pharm. Biomed. Anal. 2019, 164, 413–420. [Google Scholar] [CrossRef]
- Wang, X.; Li, K.; Yao, L.; Wang, C.; Schepdael, A. Recent advances in vitamins analysis by capillary electrophoresis. J. Pharm. Biomed. Anal. 2018, 147, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Chen, Z. Recent advances in screening of enzymes inhibitors based on capillary electrophoresis. J. Pharm. Anal. 2018, 8, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarczyk, N.; Ciżewska, J.; Treder, N.; Miękus, N.; Plenis, A.; Kowalski, P.; Roszkowska, A.; Bączek, T.; Olędzka, I. The critical evaluation of the effects of imidazolium-based ionic liquids on the separation efficiency of selected biogenic amines and their metabolites during MEKC analysis. Talanta 2022, 238, 122997. [Google Scholar] [CrossRef]
- Bessonova, E.A.; Kartsova, L.A.; Moskvichev, D.O. Ionic Liquids in Electrophoretic Separation and Preconcentration Processes. J. Anal. Chem. 2021, 76, 1111–1118. [Google Scholar] [CrossRef]
- Mohamed, A.H.; Noorhisham, N.A.; Bakar, K.; Yahaya, N.; Mohamad, S.; Kamaruzaman, S.; Osman, H. Synthesis of imidazolium-based poly(ionic liquids) with diverse substituents and their applications in dispersive solid-phase extraction. Microchem. J. 2022, 178, 107363. [Google Scholar] [CrossRef]
- Ma, X.; Li, J.; Li, X.; Feng, Z.; Yang, X.; Liu, J.; Du, Y. L-Histidinium Chiral Ionic Liquid Functionalized β-Cyclodextrin as Chiral Selector in Capillary Electrophoresis. J. Chromatogr. Sci. 2021, 59, 388–395. [Google Scholar] [CrossRef]
- Guo, Y.; Yin, Z.; Sun, Y.; Yu, H. Separation and indirect ultraviolet detection of common fluorine-containing anions by ionic liquids in reversed-phase chromatography. J. Liq. Chromatogr. Relat. Technol. 2020, 43, 597–603. [Google Scholar] [CrossRef]
- Treder, N.; Bączek, T.; Wychodnik, K.; Rogowska, J.; Wolska, L.; Plenis, A. The influence of ionic liquids on the effectiveness of analytical methods used in the monitoring of human and veterinary pharmaceuticals in biological and environmental samples—Trends and perspectives. Molecules 2020, 25, 286. [Google Scholar] [CrossRef]
- Dharaskar, S.A.; Varma, M.N.; Shende, D.Z.; Yoo, C.K.; Wasewar, K.L. Synthesis, characterization and application of 1-butyl-3 methylimidazolium chloride as green material for extractive desulfurization of liquid fuel. Sci. World J. 2013, 2013, 395274. [Google Scholar] [CrossRef]
- Calabuig-Hernández, S.; Peris-García, E.; García-Alvarez-Coque, M.C.; Ruiz-Angel, M.J. Suitability of 1-hexyl-3-methylimidazolium ionic liquids for the analysis of pharmaceutical formulations containing tricyclic antidepressants. J. Chromatogr. A 2018, 1559, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Suhail, M.; Locatelli, M.; Ali, S.; Aboul-Enein, H.Y. Role of Ionic Liquids in Capillary Electrophoresis. Analytica 2022, 3, 236–250. [Google Scholar] [CrossRef]
- Flieger, J.; Flieger, M. Ionic liquids toxicity—Benefits and threats. Int. J. Mol. Sci. 2020, 21, 6267. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Li, S.F.Y. Electrophoresis of DNA in ionic liquid coated capillary. Analyst 2003, 128, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Bessonova, E.; Kartsova, L.; Gallyamova, V. Ionic liquids based on imidazole for online concentration of catecholamines in capillary electrophoresis. J. Sep. Sci. 2017, 40, 2304–2311. [Google Scholar] [CrossRef]
- Kolobova, E.; Kartsova, L.; Kravchenko, A.; Bessonova, E. Imidazolium ionic liquids as dynamic and covalent modifiers of electrophoretic systems for determination of catecholamines. Talanta 2018, 188, 183–191. [Google Scholar] [CrossRef]
- Hussain, A.; AlAjmi, M.F.; Hussain, I.; Ali, I. Future of Ionic Liquids for Chiral Separations in High-Performance Liquid Chromatography and Capillary Electrophoresis. Crit. Rev. Anal. Chem. 2019, 49, 289–305. [Google Scholar] [CrossRef]
- Jha, R.R.; Singh, C.; Pant, A.B.; Patel, D.K. Ionic liquid-based ultrasound assisted liquid-liquid microextraction for simultaneous determination of 15 neurotransmitters in rat brain, plasma and cell samples. Anal. Chim. Acta 2018, 1005, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Kossakowska, N.; Olędzka, I.; Kowalik, A.; Miękus, N.; Kowalski, P.; Plenis, A.; Bień, E.; Kaczorowska, A.; Krawczyk, M.A.; Adamkiewicz-Drożyńska, E.; et al. Application of SPME supported by ionic liquids for the determination of biogenic amines by MEKC in clinical practice. J. Pharm. Biomed. Anal. 2019, 173, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Lew, B.L.; Sim, W.Y.; Hong, J.; Chung, B.C. Serial hydrolysis for the simultaneous analysis of catecholamines and steroids in the urine of patients with alopecia areata. Molecules 2021, 26, 2734. [Google Scholar] [CrossRef] [PubMed]
- Grouzmann, E.; Centeno, C.; Eugster, P.J. Quantification of vanillylmandelic acid, homovanilic acid and 5-hydroxyindoleacetic acid in urine using a dilute-and-shoot and ultra-high pressure liquid chromatography tandem mass spectrometry method. Clin. Chem. Lab. Med. 2018, 56, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Zhang, Y.; Zhao, R. Quantitative Measurement of Plasma Free Metanephrines by a Simple and Cost-Effective Microextraction Packed Sorbent with Porous Graphitic Carbon and Liquid Chromatography-Tandem Mass Spectrometry. J. Anal. Methods Chem. 2021, 2021, 8821276. [Google Scholar] [CrossRef] [PubMed]
- Lefeuvre, S.; Bois-Maublanc, J.; Mongeois, E.; Policarpo, V.; Farmaux, L.; Francia, T.; Billaud, E.M.; Got, L. Quantitation using HRMS: A new tool for rapid, specific and sensitive determination of catecholamines and deconjugated methanephrines metanephrines in urine. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2021, 1166, 122391. [Google Scholar] [CrossRef] [PubMed]
- Hwang, N.; Chong, E.; Oh, H.; Cho, H.W.; Lee, J.W.; Sung, K.W.; Lee, S. Application of an lc–ms/ms method for the simultaneous quantification of homovanillic acid and vanillylmandelic acid for the diagnosis and follow-up of neuroblastoma in 357 patients. Molecules 2021, 26, 3470. [Google Scholar] [CrossRef]
- Zhou, G.S.; Yuan, Y.-C.; Yin, Y.; Tang, Y.-P.; Xu, R.-J.; Liu, Y.; Chen, P.-D.; Yin, L.; Duan, J.-A. Hydrophilic interaction chromatography combined with ultrasound-assisted ionic liquid dispersive liquid–liquid microextraction for determination of underivatized neurotransmitters in dementia patients’ urine samples. Anal. Chim. Acta 2020, 1107, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Jong, W.H.A.; Eisenhofer, G.; Post, W.J.; Muskiet, F.A.J.; Vries, E.G.E.; Kema, I.P. Dietary influences on plasma and urinary metanephrines: Implications for diagnosis of catecholamine-producing tumors. J. Clin. Endocrinol. Metab. 2009, 94, 2841–2849. [Google Scholar] [CrossRef]
- FDA and CDER. Bioanalytical Method Validation Guidance for Industry Biopharmaceutics Bioanalytical Method Validation Guidance for Industry Biopharmaceutics Contains Nonbinding Recommendations. 2018. Available online: http://www.fda.gov/AnimalVeterinary/GuidanceComplianceEnforcement/GuidanceforIndustry/default.htm (accessed on 22 May 2018).
- European Medicines Agency. 2** Committee for Medicinal Products for Human Use (CHMP) Guideline on Bioanalytical Method Validation. Available online: www.ema.europa.eu/contact (accessed on 21 July 2011).
- Miękus, N.; Plenis, A.; Rudnicka, M.; Kossakowska, N.; Olędzka, I.; Kowalski, P.; Bączek, T. Extraction and preconcentration of compounds from the L-tyrosine metabolic pathway prior to their micellar electrokinetic chromatography separation. J. Chromatogr. A 2020, 1620, 461032. [Google Scholar] [CrossRef]
- Yang, X.; Du, Y.; Feng, Z.; Liu, Z.; Li, J. Establishment and molecular modeling study of maltodextrin-based synergistic enantioseparation systems with two new hydroxy acid chiral ionic liquids as additives in capillary electrophoresis. J. Chromatogr. A 2018, 1559, 170–177. [Google Scholar] [CrossRef]
- Zhang, Y.; Du, S.; Feng, Z.; Du, Y.; Yan, Z. Evaluation of synergistic enantioseparation systems with chiral spirocyclic ionic liquids as additives by capillary electrophoresis. Anal. Bioanal. Cem. 2016, 408, 2543–2555. [Google Scholar] [CrossRef]
- Woo, H.I.; Yang, J.S.; Oh, H.J.; Cho, Y.Y.; Kim, J.H.; Park, H.-D.; Lee, S.-Y. A simple and rapid analytical method based on solid-phase extraction and liquid chromatography-tandem mass spectrometry for the simultaneous determination of free catecholamines and metanephrines in urine and its application to routine clinical analysis. Clin. Biochem. 2016, 49, 573–579. [Google Scholar] [CrossRef]
- Strenger, V.; Kerbl, R.; Dornbusch, H.J.; Ladenstein, R.; Ambros, P.F.; Ambros, I.M.; Urban, C. Diagnostic and prognostic impact of urinary catecholamines in neuroblastoma patients. Pediatr. Blood Cancer 2007, 48, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Miekus, N.; Kowalski, P.; Olędzka, I.; Plenis, A.; Bień, E.; Miękus, A.; Krawczyk, M.; Adamkiewicz-Drożyńska, E.; Bączek, T. Cyclodextrin-modified MEKC method for quantification of selected acidic metabolites of catecholamines in the presence of various biogenic amines. Application to diagnosis of neuroblastoma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 1003, 27–34. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| BGE with IL | Migration Time [Min] | |||
|---|---|---|---|---|
| VMA | HVA | NM | M | |
| [HMIM+ Cl−] | ||||
| AVG | 6.53 | 6.86 | 7.63 | 8.52 |
| SD | 0.014 | 0.023 | 0.012 | 0.010 |
| RSD | 0.209 | 0.336 | 0.156 | 0.121 |
| [HMIM+ BF4−] | ||||
| AVG | 6.61 | 6.94 | 7.09 | 7.83 |
| SD | 0.024 | 0.024 | 0.023 | 0.025 |
| RSD | 0.366 | 0.345 | 0.319 | 0.326 |
| BGE without IL | BGE with [HMIM+CL−] | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Migration Time [Min] | Height | Migration Time [Min] | Height | ||||||||||
| VMA | AVG | SD | RSD | AVG | SD | RSD | VMA | AVG | SD | RSD | AVG | SD | RSD |
| 48,917.00 | 702.94 | 1.44 | 50,807 | 411.15 | 0.81 | ||||||||
| 28,806.00 | 806.72 | 2.80 | 29,006.33 | 540.73 | 1.86 | ||||||||
| 5627.00 | 271.76 | 4.83 | 6221.00 | 204.54 | 3.29 | ||||||||
| 6.66 | 0.11 | 1.71 | 4157.67 | 375.97 | 9.04 | 6.75 | 0.09 | 1.38 | 4956.67 | 283.15 | 5.71 | ||
| 2940.00 | 342.35 | 11.64 | 3317.67 | 257.28 | 7.75 | ||||||||
| 1371.33 | 108.57 | 7.92 | 1516.00 | 75.67 | 4.99 | ||||||||
| HVA | HVA | ||||||||||||
| 44,152.00 | 3073.96 | 6.96 | 46,098.67 | 2797.48 | 6.07 | ||||||||
| 23,941.33 | 2134.91 | 8.92 | 23,340.33 | 2053.31 | 8.80 | ||||||||
| 6.99 | 0.12 | 1.73 | 4357.33 | 432.77 | 9.93 | 7.09 | 0.11 | 1.50 | 5364.67 | 264.97 | 4.94 | ||
| 3184.67 | 401.22 | 12.60 | 3720.00 | 380.23 | 10.22 | ||||||||
| 2276.67 | 361.60 | 15.88 | 2194.00 | 96.32 | 4.39 | ||||||||
| 1047.00 | 142.20 | 13.58 | 1120.00 | 91.70 | 8.19 | ||||||||
| NM | NM | ||||||||||||
| 16,061.33 | 653.48 | 4.07 | 18,309.00 | 622.21 | 3.40 | ||||||||
| 8001.00 | 873.09 | 10.91 | 8225.33 | 511.64 | 6.22 | ||||||||
| 8.21 | 0.10 | 1.19 | 1541.00 | 200.16 | 12.99 | 8.03 | 0.06 | 0.71 | 1658.67 | 167.13 | 10.08 | ||
| 1251.67 | 45.89 | 3.67 | 1247.33 | 12.28 | 0.98 | ||||||||
| 764.67 | 52.22 | 6.83 | 914.33 | 35.11 | 3.84 | ||||||||
| 360.33 | 34.92 | 9.69 | 473.67 | 36.81 | 7.77 | ||||||||
| M | M | ||||||||||||
| 25,352.33 | 1608.50 | 6.34 | 25,782.00 | 1047.61 | 4.06 | ||||||||
| 12,222.00 | 1597.23 | 13.07 | 13,023.67 | 1133.56 | 8.70 | ||||||||
| 9.28 | 0.11 | 1.20 | 2206.00 | 363.67 | 16.49 | 9.02 | 0.07 | 0.75 | 2688.33 | 251.22 | 9.34 | ||
| 1714.33 | 147.75 | 8.62 | 2088.67 | 132.63 | 6.35 | ||||||||
| 1072.33 | 109.40 | 10.20 | 1538.00 | 85.21 | 5.54 | ||||||||
| 503.67 | 43.86 | 8.71 | 621.33 | 45.18 | 7.27 | ||||||||
| Parameters | VMA | HVA | NM | M |
|---|---|---|---|---|
| Linearity [µg/mL] | 0.25–10 | 0.25–10 | 0.25–10 | 0.25–10 |
| LOQ [µg/mL] | 0.25 | 0.25 | 0.25 | 0.25 |
| LOD [µg/mL] | 0.08 | 0.08 | 0.08 | 0.08 |
| R2 | 0.9963 | 0.9995 | 0.9982 | 0.9997 |
| Regression equation | y = 5113.5x + 1123.1 | y = 4637.5x + 80.25 | y = 1782.6x − 120.04 | y = 2664.2 + 3.03 |
| AR [%] | 91.7–116.39 | 101.30–117.97 | 112.61–120.00 | 91.83–113.76 |
| Analytes | Nominal Concentration µg/mL | Intra-Day Assay | Inter-Day Assay | ||
|---|---|---|---|---|---|
| Measured Concentration µg/mL | CV (%) | Measured Concentration µg/mL | CV (%) | ||
| VMA | 10 | 9.78 | 1.74 | 10.09 | 1.66 |
| 1 | 1.00 | 4.01 | 1/00 | 3.12 | |
| 0.25 | 0.22 | 12.57 | 0.24 | 8.98 | |
| HVA | 10 | 10.12 | 3.30 | 10.20 | 2.34 |
| 1 | 1.07 | 4.83 | 1.06 | 5.72 | |
| 0.25 | 0.22 | 8.82 | 0.23 | 6.93 | |
| NM | 10 | 10.35 | 3.20 | 10.34 | 3.29 |
| 1 | 1.01 | 8.04 | 1.02 | 7.00 | |
| 0.25 | 0.25 | 8.24 | 0.24 | 5.04 | |
| M | 10 | 10.05 | 7.58 | 10.26 | 5.20 |
| 1 | 1.01 | 9.35 | 1.04 | 6.76 | |
| 0.25 | 0.24 | 5.10 | 0.25 | 3.22 | |
| No | Diagnosis | Concentration Found in Urine Sample (µg/mL) | ||||
|---|---|---|---|---|---|---|
| pH 5.5 | pH 9.0 | |||||
| VMA | HVA | VMA/HVA Ratio | NM | M | ||
| 1 | Neuroendocrine tumors, NBL | 1.89 | 1.48 | 1.27 | 4.45 | 12.27 * |
| 2 | n.m | n.m | n.m. | n.m | n.m | |
| 3 | 6.81 | 12.41 * | 0.54 | 5.20 | n.m | |
| 4 | 3.50 | 10.47 | 0.33 | 0.49 | 0.16 | |
| 5 | 11.22 * | 13.77 * | 0.81 | n.o | n.m | |
| 6 | 3.95 | 7.00 | 0.56 | 3.27 | 0.87 | |
| 7 | n.m | 16.27 * | n.m. | 9.08 | 8.47 | |
| 8 | n.m | n.m | n.m. | 1.40 | 0.66 | |
| Median | 3.95 | 11.44 | 0.56 | 3.86 | 0.87 | |
| Min | 1.89 | 1.48 | 0.33 | 0.49 | 0.16 | |
| Max | 11.22 | 16.27 | 1.27 | 9.08 | 12.27 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaczmarczyk, N.; Treder, N.; Kowalski, P.; Plenis, A.; Roszkowska, A.; Bączek, T.; Olędzka, I. Investigation of Imidazolium-Based Ionic Liquids as Additives for the Separation of Urinary Biogenic Amines via Capillary Electrophoresis. Separations 2023, 10, 116. https://doi.org/10.3390/separations10020116
Kaczmarczyk N, Treder N, Kowalski P, Plenis A, Roszkowska A, Bączek T, Olędzka I. Investigation of Imidazolium-Based Ionic Liquids as Additives for the Separation of Urinary Biogenic Amines via Capillary Electrophoresis. Separations. 2023; 10(2):116. https://doi.org/10.3390/separations10020116
Chicago/Turabian StyleKaczmarczyk, Natalia, Natalia Treder, Piotr Kowalski, Alina Plenis, Anna Roszkowska, Tomasz Bączek, and Ilona Olędzka. 2023. "Investigation of Imidazolium-Based Ionic Liquids as Additives for the Separation of Urinary Biogenic Amines via Capillary Electrophoresis" Separations 10, no. 2: 116. https://doi.org/10.3390/separations10020116
APA StyleKaczmarczyk, N., Treder, N., Kowalski, P., Plenis, A., Roszkowska, A., Bączek, T., & Olędzka, I. (2023). Investigation of Imidazolium-Based Ionic Liquids as Additives for the Separation of Urinary Biogenic Amines via Capillary Electrophoresis. Separations, 10(2), 116. https://doi.org/10.3390/separations10020116