
An Optimised MS-Based Versatile Untargeted Metabolomics Protocol


Abstract
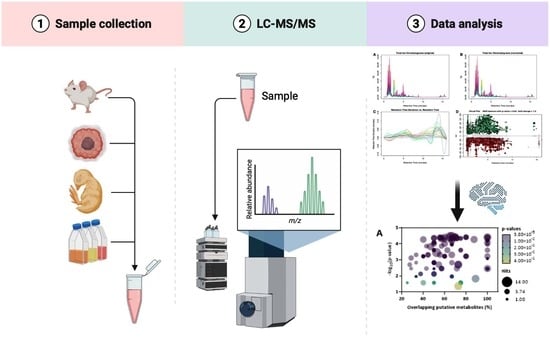
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Metabolomics by Mass Spectrometry
1.2. Development of the Protocol
1.3. Advantages of the Method
1.4. Current Limitations
2. Experimental Design
2.1. Bacteria

2.2. Eukaryotic Cells
2.3. Metabolite Extraction from Suspension Cells Using Freeze–Thaw Cycles

2.4. Metabolite Extraction from Adherent Cells by Cell Scraping

2.5. Mouse Samples

2.6. Mouse Tissue Sample Preparation
2.7. Mouse Plasma Sample Preparation
2.8. Mouse Urine Sample Preparation
2.9. Mouse Faecal Matter Samples Preparation
2.10. Human Samples

2.11. Controls
2.12. Data Processing
3. Materials and Equipment
3.1. Reagents
3.2. Experimental Models
3.3. Equipment
- UHPLC system composed of an Elute UHPLC HPG 1300 pump with two pairs of serial-coupled, individually controlled linear drive pump heads, an Elute autosampler and an Elute CSV column oven preheater, equipped with C18 reverse phase and a HILIC column, with pre-columns, and coupled to an Impact II QqTOF mass spectrometer with an electrospray ion source (UHR-ESI-QqTOF, Bruker Daltonics GmbH & Co., Bremmen, Germany). Further details are available and discussed in the Supplementary Materials;
- Vials with caps and inserts;
- Polypropilene microcentrifuge tubes, 1.5 mL and 2 mL;
- 0.22 μm polyethersulfone (PES) membrane (Branchia, cat. # SFPE-22E-050);
- Ultrasonic bath (Bandelin Electronic, RK 52);
- Potter–Elvehjem homogenizer (Sigma (St. Louis, MO, USA), cat. # P7734);
- Micro-pestles for 1.5 and 2.0 mL tubes (Carl Roth, cat. # CXH7.1);
- SPECTROstar BMG Labtech equipped with a multiwell plate reader and a low-volume microspot plate (BMG Labtech, SpectrostarNano).
3.4. Software
- Data Analysis v4.1-4.5 (Bruker(Billerica, MA, USA));
- MARS software v3.32 (BMG Labtech);
- ProteoWizard MSConvert v3.0 (https://proteowizard.sourceforge.io/, accessed on 3 February 2022) [40];
- XCMS v3.7.1 (https://xcmsonline.scripps.edu/, accessed on 1 October 2021) [32];
3.5. Reagent and Sample Setup
3.5.1. Samples
3.5.2. Quality Control (QC)
3.5.3. Mobile Phase Solutions
- Mobile phase A1 is 0.1% (vol/vol) formic acid in water; this is prepared by adding 1.0 mL of formic acid to 1 L of LC-MS-grade water and mixing thoroughly.
- Mobile phase B1 is 0.1% (vol/vol) formic acid in acetonitrile; this is prepared by adding 1.0 mL of formic acid to 1 L of LC-MS-grade acetonitrile and mixing thoroughly.
- Mobile phase A2 is 10 mM ammonium acetate in water, with 0.1% (vol/vol) acetic acid. This is prepared by dissolving 0.7708 g of ammonium acetate in 1 L of LC-MS-grade water and adding 1.0 mL of acetic acid.
- Mobile phase B2 is 10 mM ammonium acetate in acetonitrile containing 2% (vol/vol) water and 0.1% (vol/vol) acetic acid). This is prepared by (i) dissolving 0.7708 g of ammonium acetate in 20 mL of LC-MS grade water, (ii) diluting this with 980 mL of warm LC-MS-grade acetonitrile, and (iii) adding 1 mL of acetic acid. Note: Mobile phase B2 must be prepared by adding warm acetonitrile (ca. 37 °C) or ammonium acetate will precipitate. Upon proper dissolution, no temperature-dependent precipitation has been observed.
3.5.4. Sodium Formate/Acetate Calibrant Solution
3.6. Equipment Setup
3.6.1. UHPLC Instrument Setup
3.6.2. Mass Spectrometer Setup
4. Expected Results and Data Analysis

5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Everett, J.R.; Loo, R.L.; Pullen, F.S. Pharmacometabonomics and personalized medicine. Ann. Clin. Biochem. Int. J. Biochem. Lab. Med. 2013, 50, 523–545. [Google Scholar] [CrossRef] [PubMed]
- Kaddurah-Daouk, R.; Weinshilboum, R.M. Pharmacometabolomics: Implications for Clinical Pharmacology and Systems Pharmacology. Clin. Pharmacol. Ther. 2014, 95, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.F.; Marques, M.M. Pharmacometabolomics in Drug Discovery and Development. In Systems Medicine; Wolken-hauer, O., Ed.; Academic Press: Oxford, UK, 2021; pp. 480–500. [Google Scholar] [CrossRef]
- Tounta, V.; Liu, Y.; Cheyne, A.; Larrouy-Maumus, G. Metabolomics in infectious diseases and drug discovery. Mol. Omics 2021, 17, 376–393. [Google Scholar] [CrossRef]
- Aderemi, A.; Ayeleso, A.; Oyedapo, O.; Mukwevho, E. Metabolomics: A Scoping Review of Its Role as a Tool for Disease Biomarker Discovery in Selected Non-Communicable Diseases. Metabolites 2021, 11, 418. [Google Scholar] [CrossRef]
- Gertsman, I.; Barshop, B.A. Promises and pitfalls of untargeted metabolomics. J. Inherit. Metab. Dis. 2018, 41, 355–366. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.; Atherton, H.J.; Goodacre, R.; Griffin, J.L. Systems level studies of mammalian metabolomes: The roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem. Soc. Rev. 2011, 40, 387–426. [Google Scholar] [CrossRef]
- Everett, J.R. From Metabonomics to Pharmacometabonomics: The Role of Metabolic Profiling in Personalized Medicine. Front. Pharmacol. 2016, 7, 297. [Google Scholar] [CrossRef]
- Kaddurah-Daouk, R.; Kristal, B.S.; Weinshilboum, R.M. Metabolomics: A Global Biochemical Approach to Drug Response and Disease. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 653–683. [Google Scholar] [CrossRef]
- Patejko, M.; Jacyna, J.; Markuszewski, M.J. Sample preparation procedures utilized in microbial metabolomics: An overview. J. Chromatogr. B 2017, 1043, 150–157. [Google Scholar] [CrossRef]
- Kamal, K.M.; Maifiah, M.H.M.; Rahim, N.A.; Hashim, Y.Z.H.-Y.; Sani, M.S.A.; Azizan, K.A. Bacterial Metabolomics: Sample Preparation Methods. Biochem. Res. Int. 2022, 2022, 9186536. [Google Scholar] [CrossRef]
- Pinu, F.R.; Villas-Boas, S.G.; Aggio, R. Analysis of Intracellular Metabolites from Microorganisms: Quenching and Extraction Protocols. Metabolites 2017, 7, 53. [Google Scholar] [CrossRef]
- Wang, T.; Wang, X.; Zhuang, Y.; Wang, G. A systematic evaluation of quenching and extraction procedures for quantitative metabolome profiling of HeLa carcinoma cell under 2D and 3D cell culture conditions. Biotechnol. J. 2023; early view. [Google Scholar] [CrossRef]
- Mushtaq, M.Y.; Choi, Y.H.; Verpoorte, R.; Wilson, E.G. Extraction for Metabolomics: Access to The Metabolome. Phytochem. Anal. 2014, 25, 291–306. [Google Scholar] [CrossRef]
- Villas-Boas, S.; Højer-Pedersen, J.; Åkesson, M.; Smedsgaard, J.; Nielsen, J. Global metabolite analysis of yeast: Evaluation of sample preparation methods. Yeast 2005, 22, 1155–1169. [Google Scholar] [CrossRef]
- Dettmer, K.; Nürnberger, N.; Kaspar, H.; Gruber, M.A.; Almstetter, M.F.; Oefner, P.J. Metabolite extraction from adherently growing mammalian cells for metabolomics studies: Optimization of harvesting and extraction protocols. Anal. Bioanal. Chem. 2011, 399, 1127–1139. [Google Scholar] [CrossRef]
- Muschet, C.; Möller, G.; Prehn, C.; de Angelis, M.H.; Adamski, J.; Tokarz, J. Removing the bottlenecks of cell culture metabolomics: Fast normalization procedure, correlation of metabolites to cell number, and impact of the cell harvesting method. Metabolomics 2016, 12, 151. [Google Scholar] [CrossRef]
- Marques, C.F.; Marques, M.M.; Justino, G.C. The mechanisms underlying montelukast’s neuropsychiatric effects—New insights from a combined metabolic and multiomics approach. Life Sci. 2022, 310, 121056. [Google Scholar] [CrossRef]
- Overmyer, K.A.; Thonusin, C.; Qi, N.R.; Burant, C.F.; Evans, C.R. Impact of Anesthesia and Euthanasia on Metabolomics of Mammalian Tissues: Studies in a C57BL/6J Mouse Model. PLoS ONE 2015, 10, e0117232. [Google Scholar] [CrossRef]
- Want, E.J.; Masson, P.; Michopoulos, F.; Wilson, I.D.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Loftus, N.; Holmes, E.; Nicholson, J. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat. Protoc. 2013, 8, 17–32. [Google Scholar] [CrossRef]
- Gagné, F. Tissue Preparation and Subcellular Fractionation Techniques. In Biochemical Ecotoxicology: Principles and Methods; Elsevier: Amsterdam, The Netherlands, 2014; pp. 21–31. [Google Scholar] [CrossRef]
- Kaluarachchi, M.; Boulangé, C.L.; Karaman, I.; Lindon, J.C.; Ebbels, T.M.D.; Elliott, P.; Tracy, R.P.; Olson, N.C. A comparison of human serum and plasma metabolites using untargeted 1H NMR spectroscopy and UPLC-MS. Metabolomics 2018, 14, 32. [Google Scholar] [CrossRef]
- Liu, X.; Hoene, M.; Wang, X.; Yin, P.; Häring, H.-U.; Xu, G.; Lehmann, R. Serum or plasma, what is the difference? Investigations to facilitate the sample material selection decision making process for metabolomics studies and beyond. Anal. Chim. Acta 2018, 1037, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Yin, P.; Peter, A.; Franken, H.; Zhao, X.; Neukamm, S.S.; Rosenbaum, L.; Lucio, M.; Zell, A.; Häring, H.-U.; Xu, G.; et al. Preanalytical Aspects and Sample Quality Assessment in Metabolomics Studies of Human Blood. Clin. Chem. 2013, 59, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Paglia, G.; Del Greco, F.M.; Sigurdsson, B.B.; Rainer, J.; Volani, C.; Hicks, A.A.; Pramstaller, P.P.; Smarason, S.V. Influence of collection tubes during quantitative targeted metabolomics studies in human blood samples. Clin. Chim. Acta 2018, 486, 320–328. [Google Scholar] [CrossRef]
- Want, E.J. LC-MS Untargeted Analysis. Methods Mol. Biol. 2018, 1738, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Deda, O.; Gika, H.G.; Theodoridis, G.A. Rat Fecal Metabolomics-Based Analysis. Methods Mol. Biol. 2018, 1738, 149–157. [Google Scholar] [CrossRef]
- Su, X.; Klein, M.S.; Lewis, I.A.; Fiehn, O.; Rabinowitz, J.D. Metabolite Measurement: Pitfalls to Avoid and Practices to Follow. Annu. Rev. Biochem. 2017, 86, 277–304. [Google Scholar] [CrossRef]
- Katajamaa, M.; Miettinen, J.; Orešič, M. MZmine: Toolbox for processing and visualization of mass spectrometry based molecular profile data. Bioinformatics 2006, 22, 634–636. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef]
- Tautenhahn, R.; Patti, G.J.; Rinehart, D.; Siuzdak, G. XCMS Online: A Web-Based Platform to Process Untargeted Metabolomic Data. Anal. Chem. 2012, 84, 5035–5039. [Google Scholar] [CrossRef]
- Forsberg, E.M.; Huan, T.; Rinehart, D.; Benton, H.P.; Warth, B.; Hilmers, B.; Siuzdak, G. Data processing, multi-omic pathway mapping, and metabolite activity analysis using XCMS Online. Nat. Protoc. 2018, 13, 633–651. [Google Scholar] [CrossRef]
- Benton, H.P.; Ivanisevic, J.; Mahieu, N.G.; Kurczy, M.E.; Johnson, C.H.; Franco, L.; Rinehart, D.; Valentine, E.; Gowda, H.; Ubhi, B.K.; et al. Autonomous Metabolomics for Rapid Metabolite Identification in Global Profiling. Anal. Chem. 2015, 87, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Zhou, G.; Ewald, J.; Chang, L.; Hacariz, O.; Basu, N.; Xia, J. Using MetaboAnalyst 5.0 for LC–HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat. Protoc. 2022, 17, 1735–1761. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: A web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S.; Valencia, A. MetPA: A web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 2010, 26, 2342–2344. [Google Scholar] [CrossRef] [PubMed]
- HUPO Proteomics Standards Initiative (PSI). HUPO-PSI Working Groups and Outputs. Available online: http://psidev.info/index.php?q=node/80#mzdata (accessed on 10 September 2022).
- Pedrioli, P.G.A.; Eng, J.; Hubley, R.; Vogelzang, M.; Deutsch, E.; Raught, B.; Pratt, B.; Nilsson, E.; Angeletti, R.H.; Apweiler, R.; et al. A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol. 2004, 22, 1459–1466. [Google Scholar] [CrossRef]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Marques, C.F.; Pinheiro, P.F.; Justino, G.C. Optimized protocol for obtaining and characterizing primary neuron-enriched cultures from embryonic chicken brains. STAR Protoc. 2022, 3, 101753. [Google Scholar] [CrossRef]
- Du Sert, N.P.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. Br. J. Pharmacol. 2020, 177, 3617–3624. [Google Scholar] [CrossRef]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef]
- Vinaixa, M.; Samino, S.; Saez, I.; Duran, J.; Guinovart, J.J.; Yanes, O. A Guideline to Univariate Statistical Analysis for LC/MS-Based Untargeted Metabolomics-Derived Data. Metabolites 2012, 2, 775–795. [Google Scholar] [CrossRef]
- Marques, C.F.; Pinheiro, P.F.; Justino, G.C. Protocol to study in vitro drug metabolism and identify montelukast metabolites from purified enzymes and primary cell cultures by mass spectrometry. STAR Protoc. 2023, 4, 102086. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marques, C.F.; Justino, G.C. An Optimised MS-Based Versatile Untargeted Metabolomics Protocol. Separations 2023, 10, 314. https://doi.org/10.3390/separations10050314
Marques CF, Justino GC. An Optimised MS-Based Versatile Untargeted Metabolomics Protocol. Separations. 2023; 10(5):314. https://doi.org/10.3390/separations10050314
Chicago/Turabian StyleMarques, Cátia F., and Gonçalo C. Justino. 2023. "An Optimised MS-Based Versatile Untargeted Metabolomics Protocol" Separations 10, no. 5: 314. https://doi.org/10.3390/separations10050314
APA StyleMarques, C. F., & Justino, G. C. (2023). An Optimised MS-Based Versatile Untargeted Metabolomics Protocol. Separations, 10(5), 314. https://doi.org/10.3390/separations10050314