

Water-Based Microwave-Assisted Extraction of Pigments from Madder Optimized by a Box–Behnken Design


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material, Standards, and Chemicals
2.2. Extraction
2.2.1. Microwave-Assisted Extraction
2.2.2. Reference Extractions
2.3. UV Spectroscopy
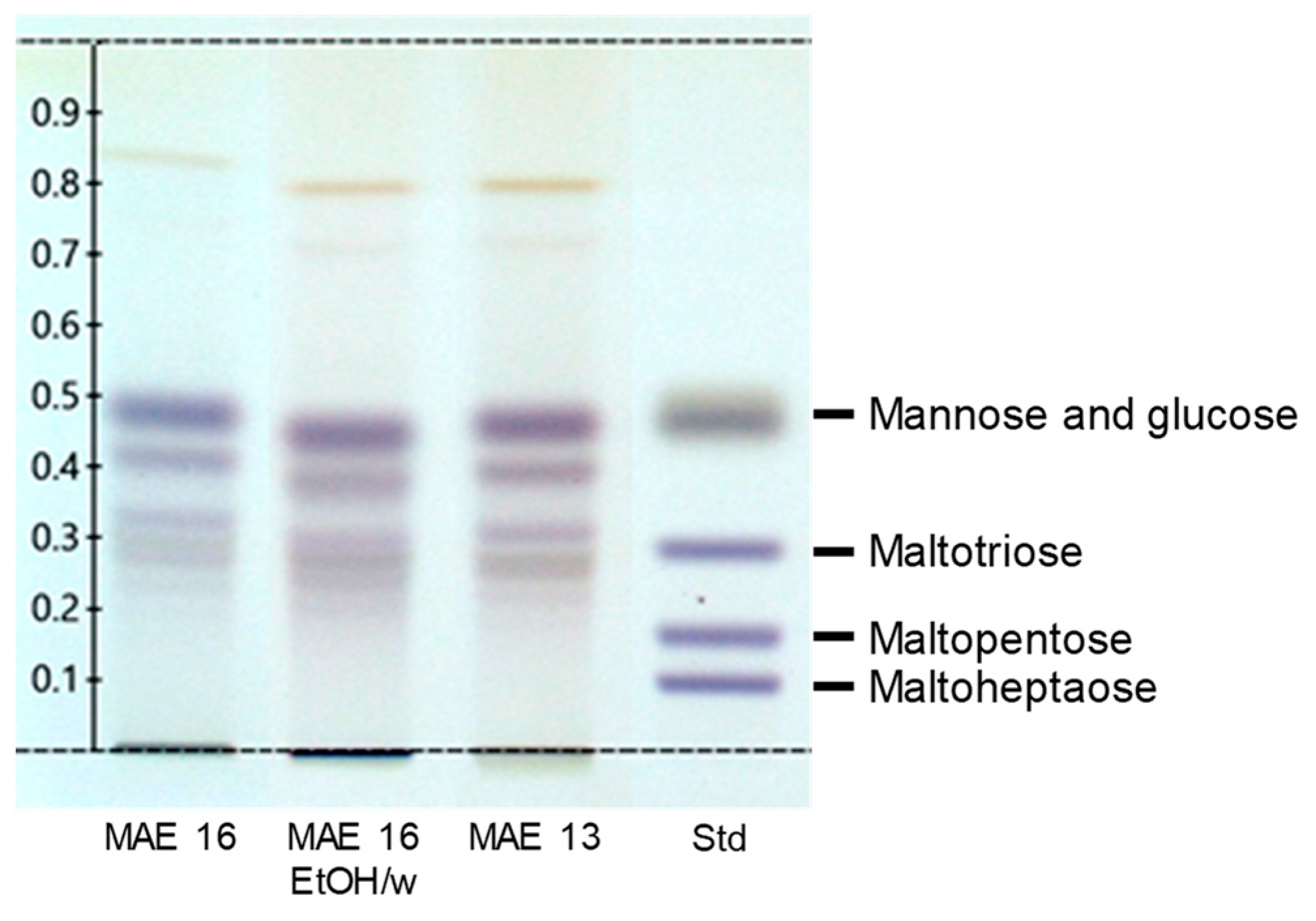
2.4. Estimation of Carbohydrate Content
2.5. Quantification of Total Phenolic Content and Anthraquinones
2.5.1. Total Phenolic Content Determination
2.5.2. Total Anthraquinone Determination
2.6. UHPLC-ESI-QTOF Analysis
3. Results and Discussion
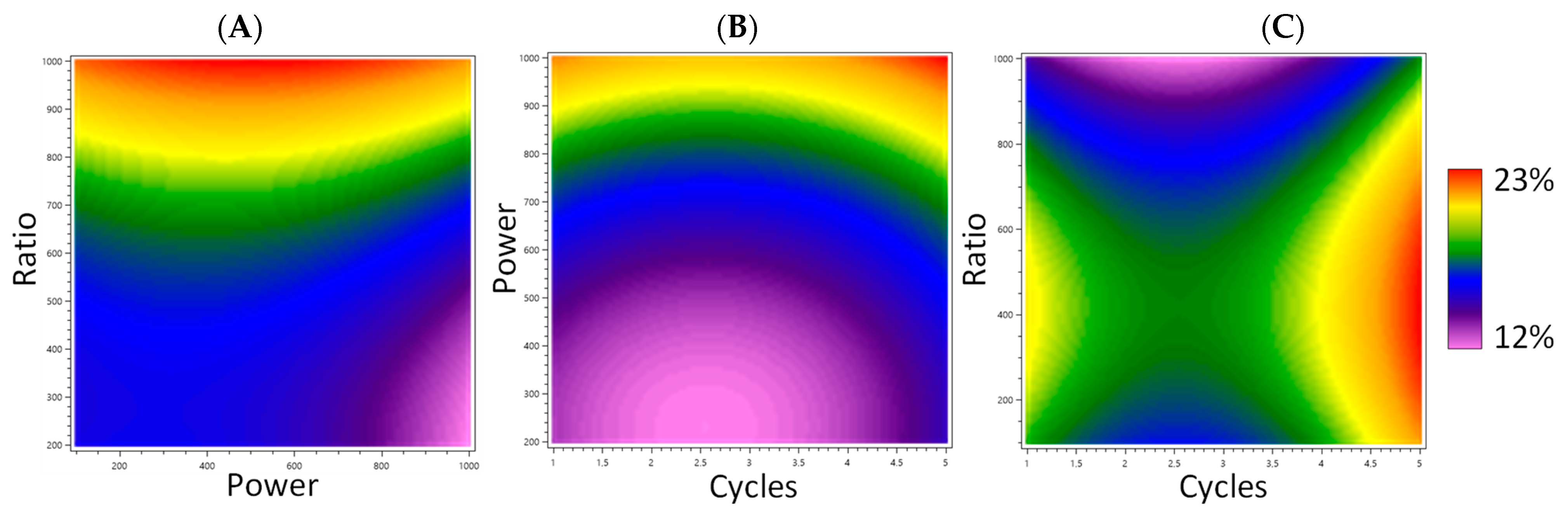
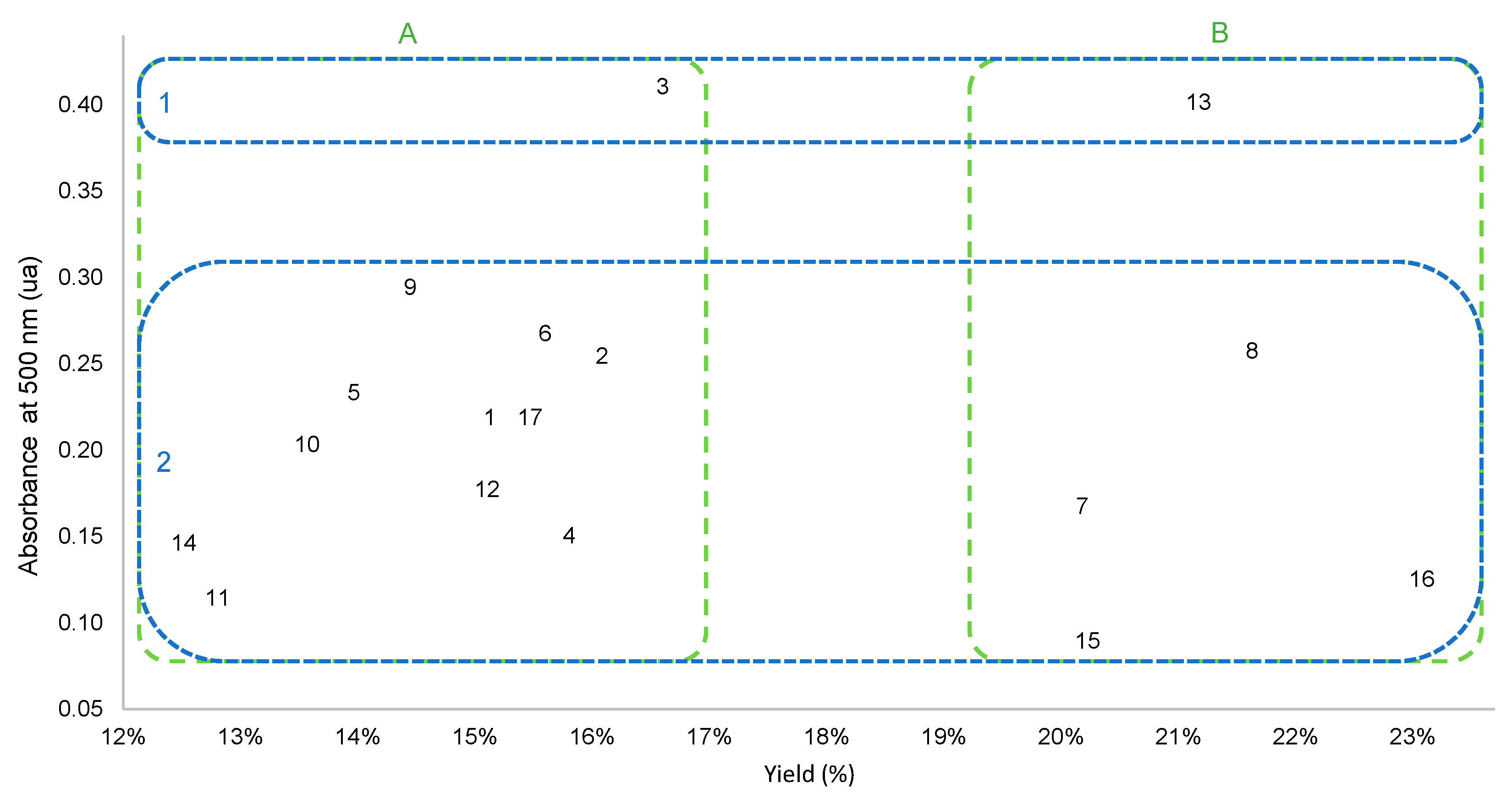
3.1. Optimization of Extraction Parameters
3.2. Influence of Extraction Parameters on the Color of Extracts
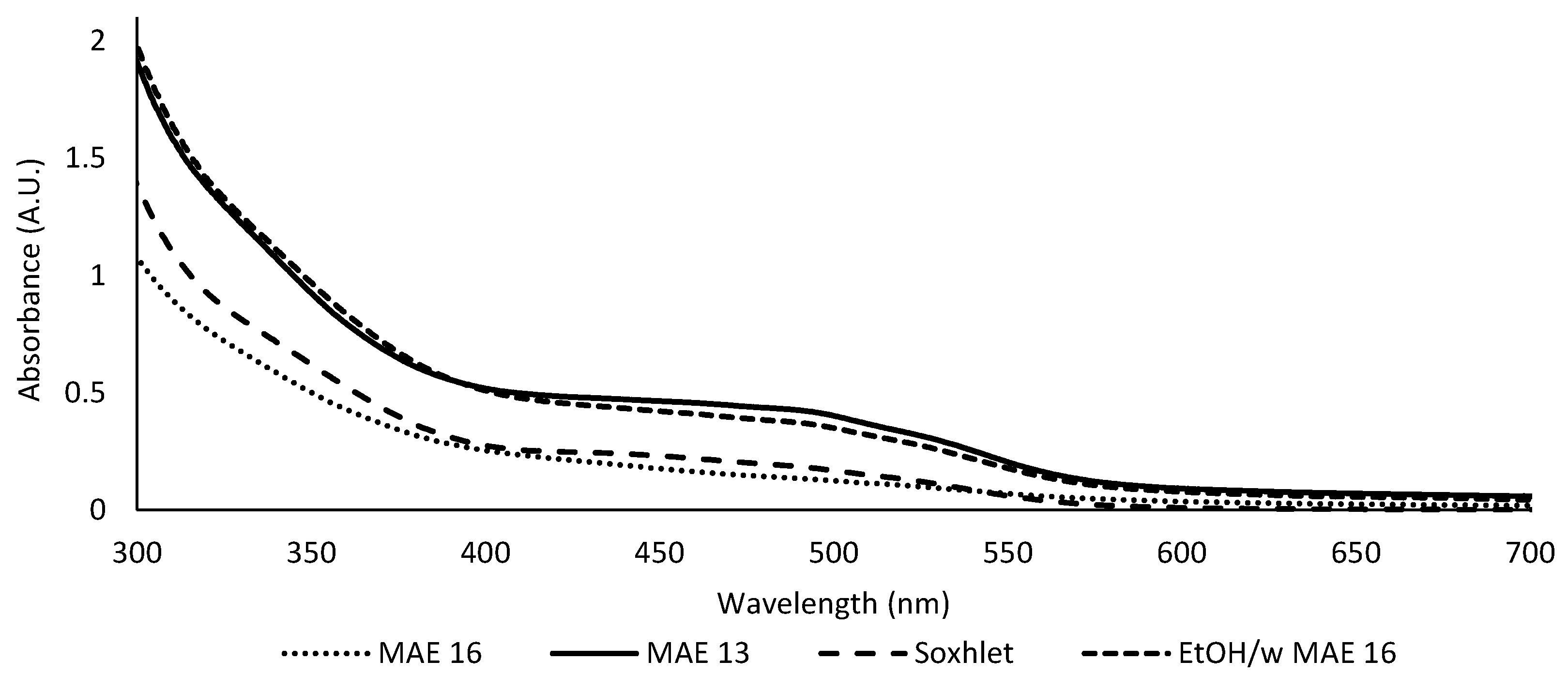
3.3. Comparison with Hydroalcoholic Reference Extractions
3.4. Detection of Metabolites
3.5. Characterization of the Pigments
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aggarwal, S. Indian Dye Yielding Plants: Efforts and Opportunities. Nat. Resour. Forum 2021, 45, 63–86. [Google Scholar] [CrossRef]
- Franca, C.C.V.; Ueno, H.M. Green Cosmetics: Perspectives and Challenges in the Context of Green Chemistry. Desenvolv. Meio Ambiente 2020, 53, 133–150. [Google Scholar] [CrossRef]
- Roohinejad, S.; Nikmaram, N.; Brahim, M.; Koubaa, M.; Khelfa, A.; Greiner, R. Potential of Novel Technologies for Aqueous Extraction of Plant Bioactives. In Water Extraction of Bioactive Compounds; Elsevier: Amsterdam, The Netherlands, 2017; pp. 399–419. ISBN 978-0-12-809380-1. [Google Scholar]
- Chemat, F.; Abert Vian, M.A.; Ravi, H.K.; Khadhraoui, B.; Hilali, S.; Perino, S.; Fabiano Tixier, A.-S. Review of Alternative Solvents for Green Extraction of Food and Natural Products: Panorama, Principles, Applications and Prospects. Molecules 2019, 24, 3007. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Hu, D.; Chen, Q.; Shi, C.; Ye, J.; Dai, Z.; Lu, Y. Water-Based Green and Sustainable Extraction Protocols for Value-Added Compounds from Natural Resources. Curr. Opin. Green Sustain. Chem. 2023, 40, 100757. [Google Scholar] [CrossRef]
- Derksen, G.C.H.; Van Beek, T.A. Rubia tinctorum L. Stud. Nat. Prod. Chem. 2002, 26, 629–684. [Google Scholar] [CrossRef]
- Wijnsma, R.; Verpoorte, R. Anthraquinones in the Rubiaceae. In Fortschritte der Chemie Organischer Naturstoffe/Progress in the Chemistry of Organic Natural Products; Herz, W., Grisebach, H., Kirby, G.W., Tamm, C., Eds.; Springer: Vienna, Austria, 1986; Volume 49, pp. 79–149. ISBN 978-3-7091-8848-4. [Google Scholar]
- Mouri, C.; Laursen, R. Identification of Anthraquinone Markers for Distinguishing Rubia Species in Madder-Dyed Textiles by HPLC. Microchim. Acta 2012, 179, 105–113. [Google Scholar] [CrossRef]
- Willemen, H.; van den Meijdenberg, G.J.P.; van Beek, T.A.; Derksen, G.C.H. Comparison of Madder (Rubia tinctorum L.) and Weld (Reseda Luteola L.) Total Extracts and Their Individual Dye Compounds with Regard to Their Dyeing Behaviour, Colour, and Stability towards Light. Color. Technol. 2019, 135, 40–47. [Google Scholar] [CrossRef]
- Dulo, B.; Phan, K.; Githaiga, J.; Raes, K.; De Meester, S. Natural Quinone Dyes: A Review on Structure, Extraction Techniques, Analysis and Application Potential. Waste Biomass Valorization 2021, 12, 6339–6374. [Google Scholar] [CrossRef]
- Duval, J.; Pecher, V.; Poujol, M.; Lesellier, E. Research Advances for the Extraction, Analysis and Uses of Anthraquinones: A Review. Ind. Crops Prod. 2016, 94, 812–833. [Google Scholar] [CrossRef]
- Derksen, G.C.H.; van Holthoon, F.L.; Willemen, H.M.; Krul, C.A.M.; Franssen, M.C.R.; van Beek, T.A. Development of a Process for Obtaining Non-Mutagenic Madder Root (Rubia tinctorum) Extract for Textile Dyeing. Ind. Crops Prod. 2021, 164, 113344. [Google Scholar] [CrossRef]
- Chemat, F.; Abert-Vian, M.; Fabiano-Tixier, A.S.; Strube, J.; Uhlenbrock, L.; Gunjevic, V.; Cravotto, G. Green Extraction of Natural Products. Origins, Current Status, and Future Challenges. TrAC Trends Anal. Chem. 2019, 118, 248–263. [Google Scholar] [CrossRef]
- Barrera Vázquez, M.F.; Comini, L.R.; Milanesio, J.M.; Montoya, S.C.N.; Cabrera, J.L.; Bottini, S.; Martini, R.E. Pressurized Hot Water Extraction of Anthraquinones from Heterophyllaea pustulata Hook f. (Rubiaceae). J. Supercrit. Fluids 2015, 101, 170–175. [Google Scholar] [CrossRef]
- Destandau, E.; Michel, T. Microwave-Assisted Extraction. In Natural Product Extraction: Principles and Applications; Rostagno, M., Prado, J.M., Eds.; Green Chemistry Series; Royal Society of Chemistry: London, UK, 2022; ISBN 978-1-83916-589-4. [Google Scholar]
- Shotipruk, A.; Kiatsongserm, J.; Pavasant, P.; Goto, M.; Sasaki, M. Pressurized Hot Water Extraction of Anthraquinones from the Roots of Morinda citrifolia. Biotechnol. Prog. 2004, 20, 1872–1875. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.L.C.; Bruns, R.E.; Ferreira, H.S.; Matos, G.D.; David, J.M.; Brandão, G.C.; da Silva, E.G.P.; Portugal, L.A.; dos Reis, P.S.; Souza, A.S.; et al. Box-Behnken Design: An Alternative for the Optimization of Analytical Methods. Anal. Chim. Acta 2007, 597, 179–186. [Google Scholar] [CrossRef]
- Grigoras, C.G.; Destandau, E.; Fougère, L.; Elfakir, C. Evaluation of Apple Pomace Extracts as a Source of Bioactive Compounds. Ind. Crops Prod. 2013, 49, 794–804. [Google Scholar] [CrossRef]
- Dabiri, M.; Salimi, S.; Ghassempour, A.; Rassouli, A.; Talebi, M. Optimization of Microwave-Assisted Extraction for Alizarin and Purpurin in Rubiaceae Plants and Its Comparison with Conventional Extraction Methods. J. Sep. Sci. 2005, 28, 387–396. [Google Scholar] [CrossRef]
- Hemwimon, S.; Pavasant, P.; Shotipruk, A. Microwave-Assisted Extraction of Antioxidative Anthraquinones from Roots of Morinda Citrifolia. Sep. Purif. Technol. 2007, 54, 44–50. [Google Scholar] [CrossRef]
- Sharif, S.; Nabais, P.; Melo, M.J.; Oliveira, M.C. Traditional Yellow Dyes Used in the 21st Century in Central Iran: The Knowledge of Master Dyers Revealed by HPLC-DAD and UHPLC-HRMS/MS. Molecules 2020, 25, 908. [Google Scholar] [CrossRef]
- Singh, R.; Geetanjali; Chauhan, S.M.S. 9,10-Anthraquinones and Other Biologically Active Compounds from the Genus Rubia. Chem. Biodivers. 2004, 1, 1241–1264. [Google Scholar] [CrossRef]
- Eltamany, E.E.; Nafie, M.S.; Khodeer, D.M.; El-Tanahy, A.H.H.; Abdel-Kader, M.S.; Badr, J.M.; Abdelhameed, R.F.A. Rubia tinctorum Root Extracts: Chemical Profile and Management of Type II Diabetes Mellitus. RSC Adv. 2020, 10, 24159–24168. [Google Scholar] [CrossRef]
- Çalis, I.; Heilmann, J.; Tasdemir, D.; Linden, A.; Ireland, C.M.; Sticher, O. Flavonoid, Iridoid, and Lignan Glycosides from Putoria c alabrica. J. Nat. Prod. 2001, 64, 961–964. [Google Scholar] [CrossRef]
- Lu, C.-M.; Yang, J.-J.; Wang, P.-Y.; Lin, C.-C. A New Acylated Flavonol Glycoside and Antioxidant Effects of Hedyotis diffusa. Planta Med. 2000, 66, 374–377. [Google Scholar] [CrossRef]
- Michel, T. Nouvelles Méthodologies D’extraction, de Fractionnement et D’identification: Application aux Molécules Bioactives de L’argousier (Hippophae rhamnoides). Ph.D. Thesis, Université d’Orléans, Orléans, France, 2011. [Google Scholar]
- Fabre, N.; Rustan, I.; de Hoffmann, E.; Quetin-Leclercq, J. Determination of Flavone, Flavonol, and Flavanone Aglycones by Negative Ion Liquid Chromatography Electrospray Ion Trap Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2001, 12, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Rafaelly, L.; Heron, S.; Nowik, W.; Tchapla, A. Optimisation of ESI-MS Detection for the HPLC of Anthraquinone Dyes. Dye. Pigment. 2008, 77, 191–203. [Google Scholar] [CrossRef]
- Derksen, G.C.H.; Niederländer, H.A.G.; van Beek, T.A. Analysis of Anthraquinones in Rubia tinctorum L. by Liquid Chromatography Coupled with Diode-Array UV and Mass. J. Chromatogr. A 2002, 978, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.; Xiong, A.; Shen, D.; Yang, L.; Wang, Z. Characterization of the Principal Constituents of Danning Tablets, a Chinese Formula Consisting of Seven Herbs, by an UPLC-DAD-MS/MS Approach. Molecules 2016, 21, 631. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| X1 (Cycles) | X2 (Power in W) | X3 (Ratio in mg/20 mL) | Coded Variables |
|---|---|---|---|
| 1 | 200 | 100 | −1 |
| 3 | 600 | 550 | 0 |
| 5 | 1000 | 1000 | 1 |
| Run Order | X1 (Cycles) | X2 (Power in W) | X3 (Ratio in mg/20 mL) |
|---|---|---|---|
| 1 | 3 | 600 | 550 |
| 2 | 1 | 600 | 550 |
| 3 | 5 | 200 | 100 |
| 4 | 5 | 600 | 1000 |
| 5 | 1 | 200 | 100 |
| 6 | 3 | 600 | 550 |
| 7 | 1 | 1000 | 1000 |
| 8 | 5 | 1000 | 100 |
| 9 | 3 | 600 | 100 |
| 10 | 3 | 200 | 550 |
| 11 | 1 | 200 | 1000 |
| 12 | 3 | 600 | 550 |
| 13 | 1 | 1000 | 100 |
| 14 | 5 | 200 | 1000 |
| 15 | 3 | 1000 | 1000 |
| 16 | 5 | 1000 | 550 |
| 17 | 3 | 600 | 550 |
| Model Term | Sum of Squares | Degrees of Freedom | Mean Square | F-Value | p-Value | Significant |
|---|---|---|---|---|---|---|
| Regression | 0.0170 | 9 | 1.89 × 10−03 | 45.1054 | 0.0000 a | Yes |
| Number of cycles (X1) | 0.0003 | 1 | 2.86 × 10−04 | 6.8016 | 0.0350 a | Yes |
| Microwave power (X2) | 0.0136 | 1 | 2.22 × 10−04 | 5.2905 | 0.0550 | |
| Plant/water ratio (X3) | 0.0004 | 1 | 8.30 × 10−05 | 1.9776 | 0.2025 | |
| X1X1 | 0.0012 | 1 | 5.16 × 10−04 | 12.2858 | 0.0099 a | Yes |
| X2X2 | 0.0010 | 1 | 1.01 × 10−03 | 24.1367 | 0.0017 a | Yes |
| X3X3 | 0.0003 | 1 | 3.25 × 10−04 | 7.7453 | 0.0272 a | Yes |
| X1X2 | 0.0000 | 1 | 1.47 × 10−06 | 0.0351 | 0.8567 | |
| X1X3 | 0.0001 | 1 | 2.79 × 10−05 | 0.6654 | 0.4415 | |
| X2X3 | 0.0002 | 1 | 2.45 × 10−04 | 5.8341 | 0.0464 a | Yes |
| Residual | 0.0003 | 7 | 4.20 × 10−05 | |||
| Total | 0.0173 | 16 |
| Extract | 13 | 16 | 16 EtOH/w | Soxhlet |
|---|---|---|---|---|
| n = 3 | n = 3 | n = 3 | n = 1 | |
| Yield (%) | 21.5 ± 0.5% | 21.6 ± 1.1% | 20.7 ± 0.4% | 14.1 |
| Abs at 500 nm (ua) | 0.35 ± 0.05 | 0.16 ± 0.01 | 0.31 ± 0.09 | 0.17 |
| Anthraquinones | Total Phenolic Content | |
|---|---|---|
| (mg alizarin eq/g) | (mg gallic acid eq/g) | |
| n = 3 * | n = 3 * | |
| 13 | 49.4 ± 9.3 a | 21 ± 4 a |
| 16 | 25.2 ± 1.7 b | 15 ± 1 b |
| 16 EtOH/w | 48.0 ± 11.5 a | 21 ± 2 a |
| # | Rt (min) | Meas. m/z ([M+H]+) | Error (ppm) | Meas. m/z ([M-H]−) | Error (ppm) | Molecular Formula | MS/MS+ (Intensity) | MS/MS− (Intensity) | ʎmax (nm) | Putative Identification |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 4.55 | 355.0989 | 0 | C16H18O9 | 163.0385 (6081) | 277, 330 | Chlorogenic acid | |||
| 145.0260 (1207) | ||||||||||
| 135.0442 (1017) | ||||||||||
| 117.0353 (740) | ||||||||||
| 107.0488 (408) | ||||||||||
| 2 | 5.01 | N.D. | - | 593.1135 | 2.3 | C26H26O16 | - | 549.1213 (3075) | 252, 284, 432 | Galiosin |
| 255.0224 (1874) | ||||||||||
| 3 | 5.41 | 743.2041 | −1.1 | 741.1845 | −2.7 | C32H38O20 | 611.1614 (1054) | - | 270, 353 | Quercetin xyloside (or arabinoside) rhamnoside glucoside |
| 465.1093 (308) | ||||||||||
| 303.0526 (377) | ||||||||||
| 4 | 5.65 | 597.1459 | 0 | 595.133 | 1.6 | C26H28O16 | 465.0962 (1235) | - | 260, 355 | Quercetin xylosyl glucoside |
| 303.0471 (391) | ||||||||||
| 5 | 5.86 | 611.1608 | 0.5 | 609.1472 | −1.9 | C27H31O16 | 465.1042 (3613) | 301.0331 (668) | 257, 350 | Rutin |
| 303.0529 (559) | ||||||||||
| 6 | 6.13 | 465.1019 | 1.7 | 463.0874 | 1.8 | C21H20O12 | 303.0511 (3050) | 300.0225 (2713) | 264, 350 | Hyperoside |
| 7 | 6.78 | N.D. | 563.1394 | 2.2 | C26H28O14 | - | 269.0399 (1740) | 280, 330 (br) | Apigenin xyloside glucoside | |
| 251.0335 (334) | ||||||||||
| 8 | 7.32 | 565.1553 | −0.2 | 563.1414 | −1.3 | C26H28O14 | - | 269.0432 (1382) | 276, 305 (sh), 336, 416 | Lucidin primveroside |
| 9 | 7.39 | 579.1695 | 2.3 | 577.1564 | −0.2 | C27H30O14 | - | 269.0496 (3421) | 276, 305 (sh), 345, 416 (br) | Lucidin rhamnoside glucoside |
| 10 | 8.03 | 303.0503 | −1 | 301.0332 | 8.2 | C15H10O7 | 257.0429 (752) | 273.0346 (207) | 272, 368 | Quercetin |
| 247.0569 (309) | 257.0308 (104) | |||||||||
| 239.0195 (185) | 245.0413 (237) | |||||||||
| 229.0508 (957) | 178.9980 (470) | |||||||||
| 201.0561 (770) | 164.0093 (270) | |||||||||
| 165.0176 (947) | 151.0027 (1325) | |||||||||
| 153.0175 (1632) | 121.0285 (278) | |||||||||
| 149.0237 (376) | ||||||||||
| 137.0228 (1222) | ||||||||||
| 121.0287 (692) | ||||||||||
| 111.0079 (326) | ||||||||||
| 11 | 8.25 | 301.0347 | −1.4 | 299.0173 | 5.6 | C15H8O7 | 283.0233 (4995) | 255.0273 (56,787) | 256, 284 (sh), 328 (sh), 492 (br) | Pseudopurpurin |
| 273.0419 (1928) | 227.0347 (26,091) | |||||||||
| 245.0412 (3174) | 199.0372 (520) | |||||||||
| 231.0264 (3092) | 183.0431 (2059) | |||||||||
| 227.0300 (12,919) | 171.0444 (2032) | |||||||||
| 213.0215 (7041) | 129.0353 (812) | |||||||||
| 199.0369 (3634) | ||||||||||
| 187.0374 (4122) | ||||||||||
| 181.0328 (2458) | ||||||||||
| 175.0374 (976) | ||||||||||
| 171.0453 (1000) | ||||||||||
| 157.0259 (4415) | ||||||||||
| 143.0472 (507) | ||||||||||
| 129.0334 (1361) | ||||||||||
| 12 | 8.32 | 285.0394 | 0 | 283.0229 | 6.9 | C15H8O6 | 267.0304 (20,184) | 265.0066 (2300) | 248, 288, 432 (br) | Munjistin |
| 249.0198 (2216) | 239.0350 (66,054) | |||||||||
| 227.0337 (1135) | 211.0353 (24,985) | |||||||||
| 213.0210 (1019) | 195.0451 (21,570) | |||||||||
| 211.0387 (1316) | 167.0481 (2117) | |||||||||
| 13 | 8.95 | 269.0445 | −0.2 | 267.0283 | 5.9 | C15H8O5 | 223.0390 (211) | 223.0373 (10,478) | 276, 341, 452 (br) | Nordamnacanthal |
| 195.0357 (634) | 195.0411 (2660) | |||||||||
| 167.0558 (540) | ||||||||||
| 139.0512 (385) | ||||||||||
| 120.0807 (205) | ||||||||||
| 105.0343 (329) | ||||||||||
| 14 | 9.52 | 271.0597 | 1.6 | 269.0436 | 7.3 | C15H10O5 | - | 251.0295 (3615) | 276, 305 (sh), 340, 416 (br) | Lucidin |
| 239.0309 (265) | ||||||||||
| 223.0418 (233) | ||||||||||
| 195.0414 (384) | ||||||||||
| 179.0508 (314) | ||||||||||
| 15 | 10.63 | 271.0608 | - | 269.0438 | 6.5 | C15H10O5 | 228.0415 (151) | 254.0176 (477) | 261, 350 (br) | Apigenin |
| 215.0408 (225) | 241.0456 (512) | |||||||||
| 226.0231 (425) | ||||||||||
| 223.0381 (733) | ||||||||||
| 213.0187 (411) | ||||||||||
| 211.0360 (290) | ||||||||||
| 199.0376 (255) | ||||||||||
| 195.0418 (432) | ||||||||||
| 182.0366 (327) | ||||||||||
| 16 | 10.83 | 241.0496 | −0.1 | 239.0338 | 4.9 | C14H8O4 | - | 211.0349 (819) | 281, 339, 425 | Dihydroxyanthraquinone |
| 195.0486 (353) | ||||||||||
| 183.0426 (106) | ||||||||||
| 158.9252 (112) | ||||||||||
| 155.0532 (87) | ||||||||||
| 135.5830 (51) | ||||||||||
| 17 | 10.94 | 257.0444 | −0.2 | 255.0277 | 8.8 | C14H8O5 | 169.0335 (546) | 227.0297 (4771) | 253, 288, 480 (br), 515 | Purpurin |
| 18 | 11.35 | 287.0548 | 0.6 | N.D. | - | C15H10O6 | 255.0265 (994) | - | 255, 287, 476 (br) | Trihydroxy-hydroxymethylanthraquinone |
| 237.0150 (1104) | ||||||||||
| 227.0339 (411) | ||||||||||
| 199.0405 (811) | ||||||||||
| 181.0287 (405) | ||||||||||
| 155.0500 (427) | ||||||||||
| 115.0542 (351) | ||||||||||
| 19 | 12.07 | 255.0652 | 0.1 | 253.0495 | 4.7 | C15H10O4 | - | 225.0537 (1675) | 276, 408 (br) | Rubiadin |
| 209.0619 (424) | ||||||||||
| 181.0658 (101) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chambaud, M.; Colas, C.; Destandau, E. Water-Based Microwave-Assisted Extraction of Pigments from Madder Optimized by a Box–Behnken Design. Separations 2023, 10, 433. https://doi.org/10.3390/separations10080433
Chambaud M, Colas C, Destandau E. Water-Based Microwave-Assisted Extraction of Pigments from Madder Optimized by a Box–Behnken Design. Separations. 2023; 10(8):433. https://doi.org/10.3390/separations10080433
Chicago/Turabian StyleChambaud, Marine, Cyril Colas, and Emilie Destandau. 2023. "Water-Based Microwave-Assisted Extraction of Pigments from Madder Optimized by a Box–Behnken Design" Separations 10, no. 8: 433. https://doi.org/10.3390/separations10080433
APA StyleChambaud, M., Colas, C., & Destandau, E. (2023). Water-Based Microwave-Assisted Extraction of Pigments from Madder Optimized by a Box–Behnken Design. Separations, 10(8), 433. https://doi.org/10.3390/separations10080433