
MALDI-TOF MS: A Promising Analytical Approach to Cancer Diagnostics and Monitoring

 , and
, and 
Abstract
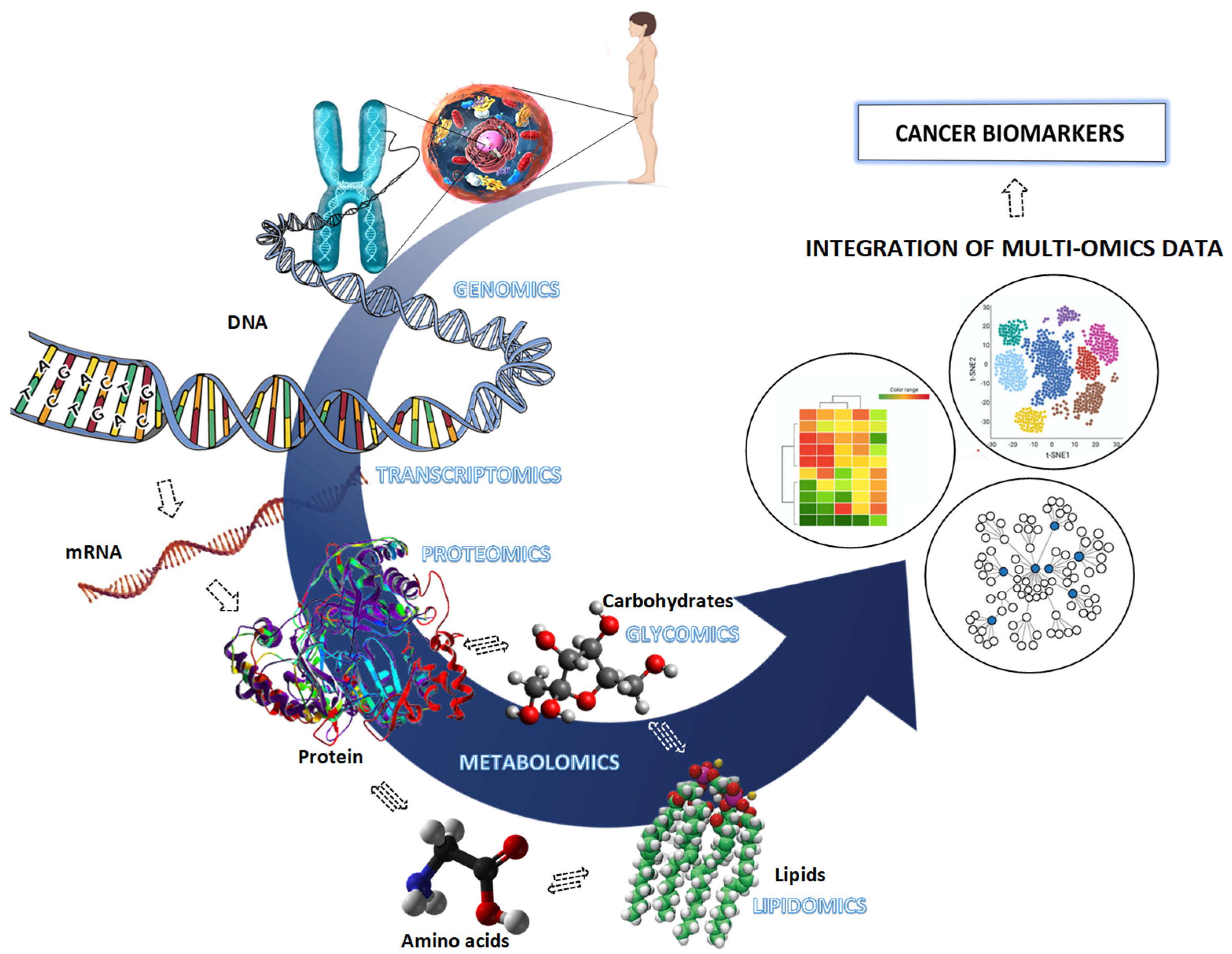
:1. Introduction
2. MALDI-TOF MS Proteome/Peptidome Profile
2.1. Sampling and Sample Preparation Approaches in MALDI-TOF MS Analysis
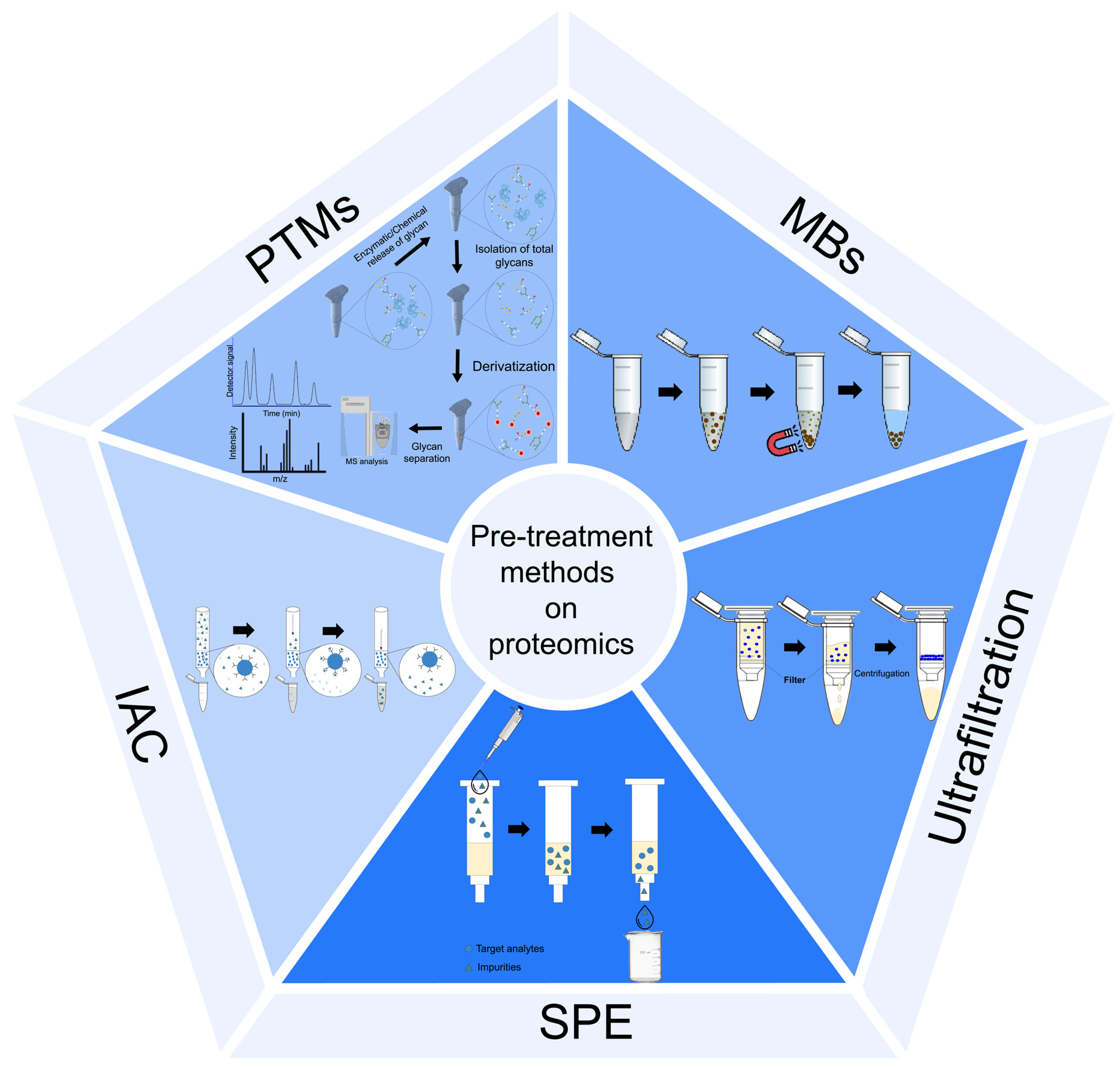
2.2. Sample Pre-Treatment Procedures
2.2.1. Ultrafiltration
2.2.2. Solid-Phase Extraction
2.2.3. Magnetic Beads
2.2.4. Immunoaffinity Chromatography
2.2.5. Glycoproteome and Phosphoproteome Enrichment
2.3. MALDI Target Preparation
2.4. Statistical Analysis
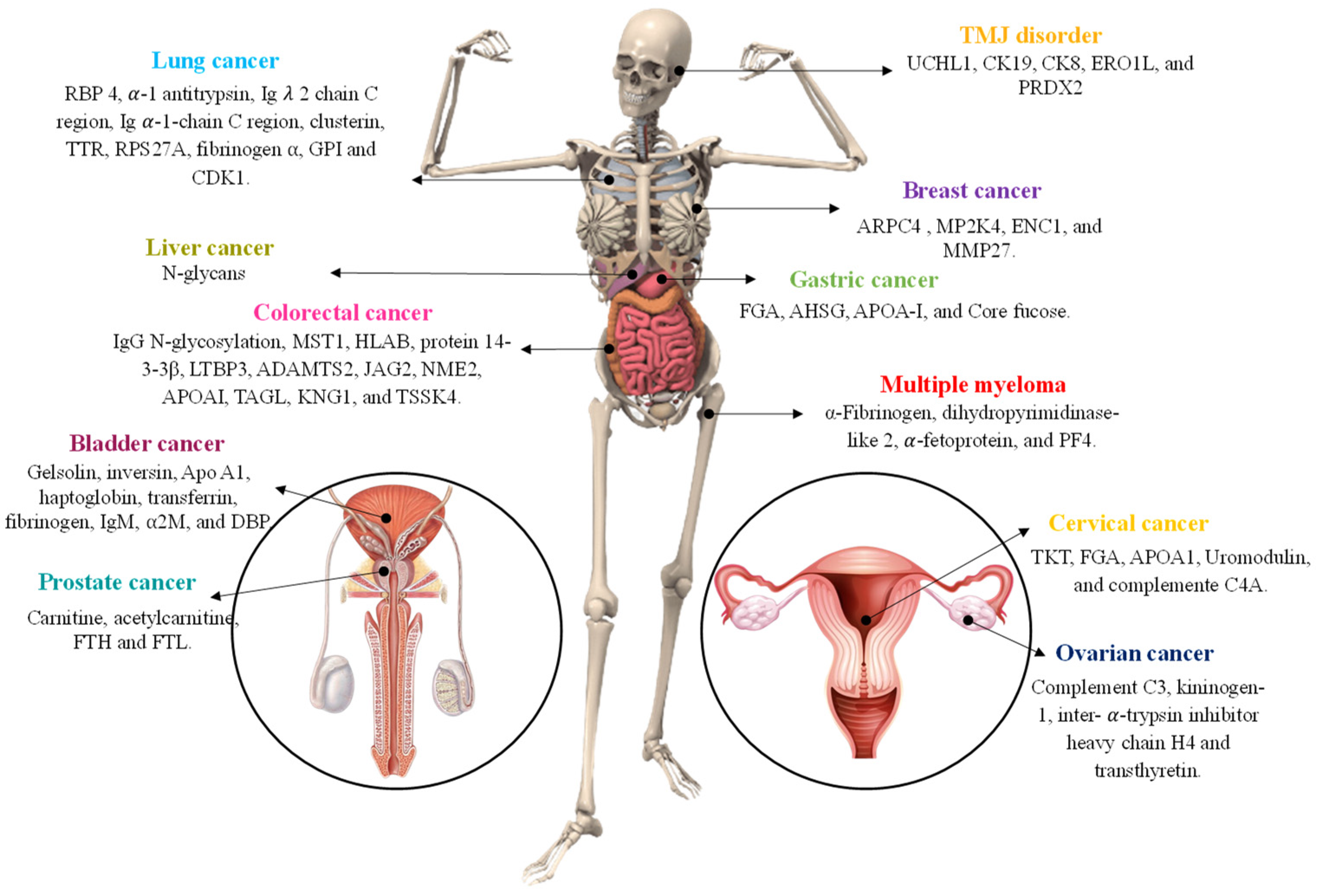
3. MALDI-TOF MS Applications in Cancer Diagnosis
3.1. Bladder Cancer
3.2. Breast Cancer
3.3. Cervical Cancer
3.4. Colorectal Cancer
3.5. Gastric Cancer
3.6. Liver Cancer
3.7. Lung Cancer
3.8. Ovarian Cancer
3.9. Prostate Cancer
3.10. Other Cancers
4. Mass Spectrometry-Based Proteome
5. Future Perspective
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Yabroff, K.R.; Wu, X.-C.; Negoita, S.; Stevens, J.; Coyle, L.; Zhao, J.; Mumphrey, B.J.; Jemal, A.; Ward, K.C. Association of the COVID-19 Pandemic with Patterns of Statewide Cancer Services. J. Natl. Cancer Inst. 2022, 114, 907–909. [Google Scholar] [CrossRef]
- Silva, C.; Perestrelo, R.; Silva, P.; Capelinha, F.; Tomás, H.; Câmara, J.S. Volatomic Pattern of Breast Cancer and Cancer-Free Tissues as a Powerful Strategy to Identify Potential Biomarkers. Analyst 2019, 144, 4153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, C.; Qiu, L.; Li, J.; Fu, T.; Zhang, X.; Tan, W. Cancer Biomarker Discovery Using DNA Aptamers. Analyst 2016, 141, 461–466. [Google Scholar] [CrossRef] [Green Version]
- Tanase, C.; Albulescu, R.; Neagu, M. Proteomic Approaches for Biomarker Panels in Cancer. J. Immunoass. Immunochem. 2016, 37, 1–15. [Google Scholar] [CrossRef]
- Pin, E.; Fredolini, C.; Petricoin, E.F. The Role of Proteomics in Prostate Cancer Research: Biomarker Discovery and Validation. Clin. Biochem. 2013, 46, 524–538. [Google Scholar] [CrossRef]
- Tanase, C.P.; Codrici, E.; Popescu, I.D.; Mihai, S.; Enciu, A.-M.; Necula, L.G.; Preda, A.; Ismail, G.; Albulescu, R.; Tanase, C.P.; et al. Prostate Cancer Proteomics: Current Trends and Future Perspectives for Biomarker Discovery. Oncotarget 2017, 8, 18497–18512. [Google Scholar] [CrossRef] [Green Version]
- Zografos, E.; Anagnostopoulos, A.K.; Papadopoulou, A.; Legaki, E.; Zagouri, F.; Marinos, E.; Tsangaris, G.T.; Gazouli, M. Serum Proteomic Signatures of Male Breast Cancer. Cancer Genom. Proteom. 2019, 16, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Duong, V.A.; Park, J.M.; Lim, H.J.; Lee, H. Proteomics in Forensic Analysis: Applications for Human Samples. Appl. Sci. 2021, 11, 3393. [Google Scholar] [CrossRef]
- Hameed, S.; Fatima, Z. Integrated Omics Approaches to Infectious Diseases; Springer: Singapore, 2021; ISBN 978-981-16-0690-8. [Google Scholar]
- Dupree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A Critical Review of Bottom-Up Proteomics: The Good, the Bad, and the Future of This Field. Proteomes 2020, 8, 14. [Google Scholar] [CrossRef]
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: Technologies and Their Applications. J. Chromatogr. Sci. 2017, 55, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Greco, V.; Piras, C.; Pieroni, L.; Ronci, M.; Putignani, L.; Roncada, P.; Urbani, A. Applications of MALDI-TOF Mass Spectrometry in Clinical Proteomics. Expert. Rev. Proteom. 2018, 15, 683–696. [Google Scholar] [CrossRef]
- Hosseini, S.; Martinez-Chapa, S.O. Fundamentals of MALDI-ToF-MS Analysis; SpringerBriefs in Applied Sciences and Technology; Springer: Singapore, 2017; ISBN 978-981-10-2355-2. [Google Scholar]
- Rizk, M.M.; Sharaki, O.A.; Meleis, M.E.; Younan, D.N.; Elkayal, A.A.; Moez, P. Detection of Epithelial Ovarian Cancer Using C8Magnetic Bead Separation and MALDI-TOF Plasma Proteome Profiling in Egyptian Females. Asian Pac. J. Cancer Prev. 2019, 20, 3603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiatly, A.; Horala, A.; Hajduk, J.; Matysiak, J.; Nowak-Markwitz, E.; Kokot, Z.J. MALDI-TOF-MS Analysis in Discovery and Identification of Serum Proteomic Patterns of Ovarian Cancer. BMC Cancer 2017, 17, 472. [Google Scholar]
- Bai, J.; Yang, Y.; Wang, J.; Zhang, L.; Wang, F.; He, A. Variability of Serum Novel Serum Peptide Biomarkers Correlates with the Disease States of Multiple Myeloma. Clin. Proteom. 2019, 16, 17. [Google Scholar] [CrossRef] [Green Version]
- Kirana, C.; Peng, L.; Miller, R.; Keating, J.P.; Glenn, C.; Shi, H.; Jordan, T.W.; Maddern, G.J.; Stubbs, R.S. Combination of Laser Microdissection, 2D-DIGE and MALDI-TOF MS to Identify Protein Biomarkers to Predict Colorectal Cancer Spread. Clin. Proteom. 2019, 16, 3. [Google Scholar]
- Nedjadi, T.; Albarakati, N.; Benabdelkamel, H.; Masood, A.; Alfadda, A.A.; Al-Maghrabi, J. Proteomic Profiling of Plasma-Derived Biomarkers in Patients with Bladder Cancer: A Step towards Clinical Translation. Life 2021, 11, 1294. [Google Scholar] [PubMed]
- Serafim, V.; Shah, A.; Puiu, M.; Andreescu, N.; Coricovac, D.; Nosyrev, A.E.; Spandidos, D.A.; Tsatsakis, A.M.; Dehelean, C.; Pinzaru, I. Classification of Cancer Cell Lines Using Matrix-Assisted Laser Desorption/Ionization Time-of-flight Mass Spectrometry and Statistical Analysis. Int. J. Mol. Med. 2017, 40, 1096–1104. [Google Scholar] [PubMed] [Green Version]
- Padoan, A.; Basso, D.; Zambon, C.F.; Prayer-Galetti, T.; Arrigoni, G.; Bozzato, D.; Moz, S.; Zattoni, F.; Bellocco, R.; Plebani, M. MALDI-TOF Peptidomic Analysis of Serum and Post-Prostatic Massage Urine Specimens to Identify Prostate Cancer Biomarkers. Clin. Proteom. 2018, 15, 23. [Google Scholar]
- Xu, J.; Xu, B.; Tang, C.; Li, X.; Qin, H.; Wang, W.; Wang, H.; Wang, Z.; Li, L.; Li, Z.; et al. The Exploration of Peptide Biomarkers in Malignant Pleural Effusion of Lung Cancer Using Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry. Dis. Markers 2017, 2017, 3160426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Tang, C.; Li, X.; Wang, J.W.; Qin, H.; Xu, B.; Chen, J.; Gao, H.; He, K.; Liu, X. Detection and Significance of Small-Cell Lung Cancer Serum Protein Markers Using MALDI-TOF-MS. Int. J. Clin. Exp. Med. 2017, 10, 929–936. [Google Scholar]
- Yu, Z.; Zhao, C.; Hu, S.; Zhang, H.; Li, W.; Zhang, R.; Luo, Q.; Yang, H. MALDI-MS-Based Biomarker Analysis of Extracellular Vesicles from Human Lung Carcinoma Cells. RSC Adv. 2021, 11, 25375–25380. [Google Scholar] [CrossRef] [PubMed]
- Sargazi, M.; Hashemi, S.H.; Kaykhaii, M. Modern Sample Preparation Techniques: A Brief Introduction. In Sample Preparation Techniques for Chemical Analysis; Kaykhaii, M., Ed.; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Zacharis, K.; Câmara, J.S.; Perestrelo, R.; Berenguer, C.V.; Andrade, C.F.P.; Gomes, T.M.; Olayanju, B.; Kabir, A.; Rocha, C.M.R.; Teixeira, J.A.; et al. Green Extraction Techniques as Advanced Sample Preparation Approaches in Biological, Food, and Environmental Matrices: A Review. Molecules 2022, 27, 2953. [Google Scholar] [CrossRef]
- Hajduk, J.; Matysiak, J.; Kokot, Z.J. Challenges in Biomarker Discovery with MALDI-TOF MS. Clin. Chim. Acta 2016, 458, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Petre, G.; Durand, H.; Pelletier, L.; Poulenard, M.; Nugue, G.; Ray, P.F.; Rendu, J.; Coutton, C.; Berger, F.; Bidart, M. Rapid Proteomic Profiling by MALDI-TOF Mass Spectrometry for Better Brain Tumor Classification. Proteom. Clin. Appl. 2020, 14, e1900116. [Google Scholar] [CrossRef]
- LaputkovÃi, G.; Talian, I.; SchwartzovÃi, V.; SchwartzovÃi, Z. MALDI-TOF MS Profiling in the Discovery and Identification of Salivary Proteomic Patterns of Temporomandibular Joint Disorders. Open Chem. 2020, 18, 1173–1180. [Google Scholar] [CrossRef]
- Lemańska-Perek, A.; Lis-Kuberka, J.; Lepczyński, A.; Dratwa-Chałupnik, A.; Tupikowski, K.; Kątnik-Prastowska, I.; Ożgo, M. Potential Plasma Biomarkers of Bladder Cancer Identified by Proteomic Analysis: A Pilot Study. Adv. Clin. Exp. Med. 2019, 28, 339–346. [Google Scholar] [CrossRef]
- Ding, D.; Chen, M.; Xiao, X.; Cao, P.; Li, S. Novel Serum Peptide Model Revealed by MALDI-TOF-MS and Its Diagnostic Value in Early Bladder Cancer. Int. J. Biol. Markers 2020, 35, 59–66. [Google Scholar] [CrossRef]
- Azevedo, R.; Soares, J.; Gaiteiro, C.; Peixoto, A.; Lima, L.; Ferreira, D.; Relvas-Santos, M.; Fernandes, E.; Tavares, A.; Cotton, S.; et al. Glycan Affinity Magnetic Nanoplatforms for Urinary Glycobiomarkers Discovery in Bladder Cancer. Talanta 2018, 184, 347–355. [Google Scholar] [CrossRef]
- Zaki, G.; Shoeib, T. Concentrations of Several Phthalates Contaminants in Egyptian Bottled Water: Effects of Storage Conditions and Estimate of Human Exposure. Sci. Total Environ. 2018, 618, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Bose, S.; Ahn, S.H.; Son, B.H.; Ko, B.S.; Kim, H.J.; Chung, I.Y.; Kim, J.; Lee, W.; Ko, M.S.; et al. Breast Cancer Diagnosis by Analysis of Serum N-Glycans Using MALDI-TOF Mass Spectroscopy. PLoS ONE 2020, 15, e0231004. [Google Scholar] [CrossRef] [Green Version]
- Sousa, P.; Camacho, I.; Câmara, J.S.; Perestrelo, R. Urinary Proteomic/Peptidomic Biosignature of Breast Cancer Patients Using 1D SDS-PAGE Combined with Matrix-Assisted Laser Desorption/Ionization-Time of Flight Mass Spectrometry. Separations 2023, 10, 291. [Google Scholar] [CrossRef]
- Kontostathi, G.; Zoidakis, J.; Makridakis, M.; Lygirou, V.; Mermelekas, G.; Papadopoulos, T.; Vougas, K.; Vlamis-Gardikas, A.; Drakakis, P.; Loutradis, D.; et al. Cervical Cancer Cell Line Secretome Highlights the Roles of Transforming Growth Factor-Beta-Induced Protein Ig-H3, Peroxiredoxin-2, and NRF2 on Cervical Carcinogenesis. Biomed. Res. Int. 2017, 2017, 4180703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, C.C.; Chou, H.C.; Chen, Y.J.; Kuo, W.H.; Chan, C.H.; Lin, Y.C.; Liao, E.C.; Chang, S.J.; Chan, H.L. Role of PGRMC1 in Cell Physiology of Cervical Cancer. Life Sci. 2019, 231, 116541. [Google Scholar] [CrossRef]
- Chen, Y.; Xiong, X.; Wang, Y.; Zhao, J.; Shi, H.; Zhang, H.; Wang, Y.; Wei, Y.; Xue, W.; Zhang, J. Proteomic Screening for Serum Biomarkers for Cervical Cancer and Their Clinical Significance. Med. Sci. Monit. 2019, 25, 288. [Google Scholar] [CrossRef]
- Liu, S.; Cheng, L.; Fu, Y.; Liu, B.F.; Liu, X. Characterization of IgG N-Glycome Profile in Colorectal Cancer Progression by MALDI-TOF-MS. J. Proteom. 2018, 181, 225–237. [Google Scholar] [CrossRef]
- Yu, J.; Zhai, X.; Li, X.; Zhong, C.; Guo, C.; Yang, F.; Yuan, Y.; Zheng, S. Identification of MST1 as a Potential Early Detection Biomarker for Colorectal Cancer through a Proteomic Approach. Sci. Rep. 2017, 7, 14265. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Huang, Y.; Lin, C.; Li, X.; Fang, X.; Zhong, C.; Yuan, Y.; Zheng, S. Identification of Kininogen 1 as a Serum Protein Marker of Colorectal Adenoma in Patients with a Family History of Colorectal Cancer. J. Cancer 2018, 9, 540–547. [Google Scholar] [CrossRef] [Green Version]
- Buttacavoli, M.; Albanese, N.N.; Roz, E.; Pucci-Minafra, I.; Feo, S.; Cancemi, P. Proteomic Profiling of Colon Cancer Tissues: Discovery of New Candidate Biomarkers. Int. J. Mol. Sci. 2020, 21, 3096. [Google Scholar] [CrossRef]
- Lee, H.J.; Saralamma, V.V.G.; Kim, S.M.; Ha, S.E.; Vetrivel, P.; Kim, E.H.; Lee, S.J.; Heo, J.D.; Rampogu, S.; Lee, K.W.; et al. Comparative Proteomic Profiling of Tumor-Associated Proteins in Human Gastric Cancer Cells Treated with Pectolinarigenin. Nutrients 2018, 10, 1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, F.; Wu, H.; Qu, K.; Sun, Q.; Li, F.; Shi, C.; Li, Y.; Xiong, X.; Qin, Q.; Yu, T.; et al. Identification of Serum Proteins AHSG, FGA and APOA-I as Diagnostic Biomarkers for Gastric Cancer. Clin. Proteom. 2018, 15, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, R.; Zhao, J.; Qin, W.; Zhang, Z.; Zhao, R.; Han, J.; Yang, Y.; Li, L.; Wang, X.; Ren, S.; et al. Discovery of Non-Invasive Glycan Biomarkers for Detection and Surveillance of Gastric Cancer. J. Cancer 2017, 8, 1908. [Google Scholar] [CrossRef]
- Qin, Y.; Zhong, Y.; Ma, T.; Zhang, J.; Yang, G.; Guan, F.; Li, Z.; Li, B. A Pilot Study of Salivary N-Glycome in HBV-Induced Chronic Hepatitis, Cirrhosis, and Hepatocellular Carcinoma. Glycoconj. J. 2017, 34, 523–535. [Google Scholar] [CrossRef]
- Park, H.G.; Jang, K.S.; Park, H.M.; Song, W.S.; Jeong, Y.Y.; Ahn, D.H.; Kim, S.M.; Yang, Y.H.; Kim, Y.G. MALDI-TOF MS-Based Total Serum Protein Fingerprinting for Liver Cancer Diagnosis. Analyst 2019, 144, 2231–2238. [Google Scholar] [CrossRef]
- Sun, F.; Wang, G.; Zhang, J.; Su, J. Application Value of MALDI-TOF-MS in Proteomics of Hepatitis B Virus-Related Liver Cancer. Chin. J. Exp. Clin. Virol. 2019, 6, 95–98. [Google Scholar]
- Li, B.; Li, B.; Guo, T.; Sun, Z.; Li, X.; Li, X.; Wang, H.; Chen, W.; Chen, P.; Qiao, M.; et al. Application Value of Mass Spectrometry in the Differentiation of Benign and Malignant Liver Tumors. Med. Sci. Monit. 2017, 23, 1636. [Google Scholar] [CrossRef] [Green Version]
- Mahalingam, D.; Chelis, L.; Nizamuddin, I.; Lee, S.S.; Kakolyris, S.; Halff, G.; Washburn, K.; Attwood, K.; Fahad, I.; Grigorieva, J.; et al. Detection of Hepatocellular Carcinoma in a High-Risk Population by a Mass Spectrometry-Based Test. Cancers 2021, 13, 3109. [Google Scholar] [CrossRef]
- Hou, W.L.; Chang, M.; Liu, X.F.; Hu, L.S.; Hua, S.C. Proteomic and Ultrastructural Analysis of Clara Cell and Type II Alveolar Epithelial Cell-Type Lung Cancer Cells. Transl. Cancer Res. 2020, 9, 565–576. [Google Scholar] [CrossRef]
- Saleem, M.; Raza, S.K.; G Musharraf, S. A Comparative Protein Analysis of Lung Cancer, along with Three Controls Using a Multidimensional Proteomic Approach. Exp. Biol. Med. 2019, 244, 36–41. [Google Scholar] [CrossRef]
- Song, Y.; Xu, X.; Wang, N.; Zhang, T.; Hu, C. MALDI-TOF-MS Analysis in Low Molecular Weight Serum Peptidome Biomarkers for NSCLC. J. Clin. Lab. Anal. 2022, 36, e24254. [Google Scholar] [CrossRef]
- Lv, P.; Liu, Z.; Xu, B.; Tang, C.; Li, X.; Qin, H.; Yang, S.; Gao, H.; He, K.; Liu, X. Exploratory Study on Application of MALDI-TOF-MS to Detect Serum and Urine Peptides Related to Small Cell Lung Carcinoma. Mol. Med. Rep. 2020, 21, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Pais, R.J.; Zmuidinaite, R.; Lacey, J.C.; Jardine, C.S.; Iles, R.K. A Rapid and Affordable Screening Tool for Early-Stage Ovarian Cancer Detection Based on MALDI-ToF MS of Blood Serum. Appl. Sci. 2022, 12, 3030. [Google Scholar] [CrossRef]
- Andersen, M.K.; Høiem, T.S.; Claes, B.S.R.; Balluff, B.; Martin-Lorenzo, M.; Richardsen, E.; Krossa, S.; Bertilsson, H.; Heeren, R.M.A.; Rye, M.B.; et al. Spatial Differentiation of Metabolism in Prostate Cancer Tissue by MALDI-TOF MSI. Cancer Metab. 2021, 9, 9. [Google Scholar] [CrossRef]
- Sun, J.; Yu, G.; Yang, Y.; Qiao, L.; Xu, B.; Ding, C.; Liu, Y.; Yu, S. Evaluation of Prostate Cancer Based on MALDI-TOF MS Fingerprinting of Nanoparticle-Treated Serum Proteins/Peptides. Talanta 2020, 220, 121331. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhao, X.; Lei, T.; Zhang, M. Screening, Identification of Prostate Cancer Urinary Biomarkers and Verification of Important Spots. Investig. New Drugs 2019, 37, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Zhao, J.; Wang, X.; Yan, S.; Chu, H.; Gao, M.; Zhang, X. Integrated Pipeline of Rapid Isolation and Analysis of Human Plasma Exosomes for Cancer Discrimination Based on Deep Learning of MALDI-TOF MS Fingerprints. Anal. Chem. 2022, 94, 1831–1839. [Google Scholar] [CrossRef]
- Banach, P.; Dereziński, P.; Matuszewska, E.; Matysiak, J.; Bochyński, H.; Kokot, Z.J.; Nowak-Markwitz, E. MALDI-TOF-MS Analysis in the Identification of Urine Proteomic Patterns of Gestational Trophoblastic Disease. Metabolites 2019, 9, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernokalskaya, E.; Gutierrez, S.; Pitt, A.M.; Leonard, J.T. Ultrafiltration for Proteomic Sample Preparation. Electrophoresis 2004, 25, 2461–2468. [Google Scholar] [CrossRef] [PubMed]
- Callesen, A.K.; Madsen, J.S.; Vach, W.; Kruse, T.A.; Mogensen, O.; Jensen, O.N. Serum Protein Profiling by Solid Phase Extraction and Mass Spectrometry: A Future Diagnostics Tool? Proteomics 2009, 9, 1428–1441. [Google Scholar] [CrossRef]
- Gobom, J.; Nordhoff, E.; Mirgorodskaya, E.; Ekman, R.; Roepstorff, P. Sample Purification and Preparation Technique Based on Nano-Scale Reversed-Phase Columns for the Sensitive Analysis of Complex Peptide Mixtures by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry—PubMed. J. Mass. Spectrom. 1999, 34, 105–116. [Google Scholar] [CrossRef]
- Xing, R.; Wen, Y.; He, H.; Guo, Z.; Liu, Z. Recent Progress in the Combination of Molecularly Imprinted Polymer-Based Affinity Extraction and Mass Spectrometry for Targeted Proteomic Analysis. TrAC Trends Anal. Chem. 2019, 110, 417–428. [Google Scholar] [CrossRef]
- Shi, H.; Tsal, W.B.; Garrison, M.D.; Ferrari, S.; Ratner, B.D. Template-Imprinted Nanostructured Surfaces for Protein Recognition. Nature 1999, 398, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, X.; Lu, W.; Wu, X.; Li, J. Molecular Imprinting: Perspectives and Applications. Chem. Soc. Rev. 2016, 45, 2137–2211. [Google Scholar] [CrossRef] [PubMed]
- McKitterick, N.; Bicak, T.C.; Cormack, P.A.G.; Reubsaet, L.; Halvorsen, T.G. Facilitating Serum Determination of Neuron Specific Enolase at Clinically Relevant Levels by Coupling On-Line Molecularly Imprinted Solid-Phase Extraction to LC-MS/MS. Anal. Chim. Acta 2020, 1140, 210–218. [Google Scholar] [CrossRef]
- Özdaş, S.; Baydemir Peşint, G.; Arısoy, P.; Zenger, O.; Eren, B. Neopterin-Imprinted Columns for Selective Neopterin Recognition from Serum and Urine Samples. Process Biochem. 2021, 108, 1–7. [Google Scholar] [CrossRef]
- Scorrano, S.; Longo, L.; Vasapollo, G. Molecularly Imprinted Polymers for Solid-Phase Extraction of 1-Methyladenosine from Human Urine. Anal. Chim. Acta 2010, 659, 167–171. [Google Scholar] [CrossRef]
- Mavliutova, L.; Munoz Aldeguer, B.; Wiklander, J.; Wierzbicka, C.; Huynh, C.M.; Nicholls, I.A.; Irgum, K.; Sellergren, B. Discrimination between Sialic Acid Linkage Modes Using Sialyllactose-Imprinted Polymers. RSC Adv. 2021, 11, 22409–22418. [Google Scholar] [CrossRef]
- Hajduk, J.; Matysiak, J.; Kokot, P.; Nowicki, P.; Dereziński, P.; Kokot, Z.J. The Application of Fuzzy Statistics and Linear Discriminant Analysis as Criteria for Optimizing the Preparation of Plasma for Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry Peptide Profiling. Clin. Chim. Acta 2015, 448, 174–181. [Google Scholar] [CrossRef]
- Liu, W.; Gao, X.; Cai, Q.; Li, J.; Zhu, Z.; Li, C.; Yao, X.; Yang, Q.; Xiang, M.; Yan, M.; et al. Identification of Novel Serum Biomarkers for Gastric Cancer by Magnetic Bead. Front. Biosci. (Elite Ed.) 2010, 2, 961–971. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, J.; Philip, J.; Entenberg, D.; Chaparro, C.A.; Tanwar, M.K.; Holland, E.C.; Tempst, P. Serum Peptide Profiling by Magnetic Particle-Assisted, Automated Sample Processing and MALDI-TOF Mass Spectrometry. Anal. Chem. 2004, 76, 1560–1570. [Google Scholar] [CrossRef]
- Bruegel, M.; Planert, M.; Baumann, S.; Focke, A.; Bergh, F.T.; Leichtle, A.; Ceglarek, U.; Thiery, J.; Fiedler, G.M. Standardized Peptidome Profiling of Human Cerebrospinal Fluid by Magnetic Bead Separation and Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry. J. Proteom. 2009, 72, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Gode, D.; Volmer, D.A. A Novel Magnet Focusing Plate for Matrix-Assisted Laser Desorption/Ionization Analysis of Magnetic Bead-Bound Analytes. Rapid Commun. Mass. Spectrom. 2013, 27, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Moser, A.C.; Hage, D.S. Immunoaffinity Chromatography: An Introduction to Applications and Recent Developments. Bioanalysis 2010, 2, 769. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Ludwig, S.; Muller, L.; Hong, C.S.; Kirkwood, J.M.; Ferrone, S.; Whiteside, T.L. Immunoaffinity-Based Isolation of Melanoma Cell-Derived Exosomes from Plasma of Patients with Melanoma. J. Extracell. Vesicles 2018, 7, 1435138. [Google Scholar] [CrossRef] [PubMed]
- Nicol, G.R.; Han, M.; Kim, J.; Birse, C.E.; Brand, E.; Nguyen, A.; Mesri, M.; FitzHugh, W.; Kaminker, P.; Moore, P.A.; et al. Use of an Immunoaffinity-Mass Spectrometry-Based Approach for the Quantification of Protein Biomarkers from Serum Samples of Lung Cancer Patients. Mol. Cell. Proteom. 2008, 7, 1974–1982. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.S.; Faruque, H.A.; Kim, J.H.; Kim, K.J.; Choi, J.E.; Kim, B.A.; Kim, B.; Kim, Y.J.; Woo, M.H.; Park, J.Y.; et al. CD5L as an Extracellular Vesicle-Derived Biomarker for Liquid Biopsy of Lung Cancer. Diagnostics 2021, 11, 620. [Google Scholar] [CrossRef]
- Hsiao, Y.C.; Lin, S.Y.; Chien, K.Y.; Chen, S.F.; Wu, C.C.; Chang, Y.T.; Chi, L.M.; Chu, L.J.; Chiang, W.F.; Chien, C.Y.; et al. An Immuno-MALDI Mass Spectrometry Assay for the Oral Cancer Biomarker, Matrix Metalloproteinase-1, in Dried Saliva Spot Samples. Anal. Chim. Acta 2020, 1100, 118–130. [Google Scholar] [CrossRef]
- Kirwan, A.; Utratna, M.; O’Dwyer, M.E.; Joshi, L.; Kilcoyne, M. Glycosylation-Based Serum Biomarkers for Cancer Diagnostics and Prognostics. Biomed. Res. Int. 2015, 2015, 490531. [Google Scholar] [CrossRef] [Green Version]
- Kanshin, E.; Michnick, S.; Thibault, P. Sample Preparation and Analytical Strategies for Large-Scale Phosphoproteomics Experiments. Semin. Cell Dev. Biol. 2012, 23, 843–853. [Google Scholar] [CrossRef]
- Sürmen, M.G.; Sürmen, S.; Ali, A.; Musharraf, S.G.; Emekli, N. Phosphoproteomic Strategies in Cancer Research: A Minireview. Analyst 2020, 145, 7125–7149. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.Y.; Yang, C.K.; Liu, C.P.; Liu, C.Y. Stationary Phases for the Enrichment of Glycoproteins and Glycopeptides. Electrophoresis 2014, 35, 2091–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruprecht, B.; Roesli, C.; Lemeer, S.; Kuster, B. MALDI-TOF and NESI Orbitrap MS/MS Identify Orthogonal Parts of the Phosphoproteome. Proteomics 2016, 16, 1447–1456. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.F.; Hsu, C.C.; Hung, J.N.; Wang, Y.T.; Choong, W.K.; Zeng, M.Y.; Lin, P.Y.; Hong, R.W.; Sung, T.Y.; Chen, Y.J. Sequential Phosphoproteomic Enrichment through Complementary Metal-Directed Immobilized Metal Ion Affinity Chromatography. Anal. Chem. 2014, 86, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Sun, X.; Li, Y.; Deng, C.; Duan, G. Facile Synthesis of Fe3O4@PDA Core-Shell Microspheres Functionalized with Various Metal Ions: A Systematic Comparison of Commonly-Used Metal Ions for IMAC Enrichment. Talanta 2018, 178, 600–607. [Google Scholar] [CrossRef]
- Monopoli, A.; Nacci, A.; Cataldi, T.R.I.; Calvano, C.D.; Piovesana, S.; Montonea, C.M.; Cerrato, A. Synthesis and Matrix Properties of α-Cyano-5-Phenyl-2,4-Pentadienic Acid (CPPA) for Intact Proteins Analysis by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry. Molecules 2020, 25, 6054. [Google Scholar] [CrossRef]
- Lou, X.; Van Dongen, J.L.J.; Vekemans, J.A.J.M.; Meijer, E.W. Matrix Suppression and Analyte Suppression Effects of Quaternary Ammonium Salts in Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry: An Investigation of Suppression Mechanism. Rapid Commun. Mass. Spectrom. 2009, 23, 3077–3082. [Google Scholar] [CrossRef]
- Terracciano, R.; Preianò, M.; Maggisano, G.; Pelaia, C.; Savino, R. Hexagonal Mesoporous Silica as a Rapid, Efficient and Versatile Tool for MALDI-TOF MS Sample Preparation in Clinical Peptidomics Analysis: A Pilot Study. Molecules 2019, 24, 2311. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Wu, R.; Hung, Y.L.W.; Chen, X.; Chan, T.W.D. Development of a Matrix Sublimation Device with Controllable Crystallization Temperature for MALDI Mass Spectrometry Imaging. Anal. Chem. 2021, 93, 6342–6347. [Google Scholar] [CrossRef]
- Tholey, A.; Heinzle, E. Ionic (Liquid) Matrices for Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry-Applications and Perspectives. Anal. Bioanal. Chem. 2006, 386, 24–37. [Google Scholar] [CrossRef]
- Monopoli, A.; Calvano, C.D.; Nacci, A.; Palmisano, F. Boronic Acid Chemistry in MALDI MS: A Step Forward in Designing a Reactive Matrix with Molecular Recognition Capabilities. Chem. Commun. 2014, 50, 4322–4324. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.; Svatoš, A. Proton Sponge: A Novel and Versatile MALDI Matrix for the Analysis of Metabolites Using Mass Spectrometry. Anal. Chem. 2009, 81, 7954–7959. [Google Scholar] [CrossRef] [PubMed]
- Vasil’ev, Y.V.; Khvostenko, O.G.; Streletskii, A.V.; Boltalina, O.V.; Kotsiris, S.G.; Drewello, T. Electron Transfer Reactivity in Matrix-Assisted Laser Desorption/Ionization (MALDI): Ionization Energy, Electron Affinity and Performance of the DCTB Matrix within the Thermochemical Framework. J. Phys. Chem. A 2006, 110, 5967–5972. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, Y.; Tanimura, R.; Maeda, K.; Watanabe, M.; Kawabata, S.I.; Iwamoto, S.; Izumi, S.; Tanaka, K. Alkylated Dihydroxybenzoic Acid as a MALDI Matrix Additive for Hydrophobic Peptide Analysis. Anal. Chem. 2012, 84, 4237–4243. [Google Scholar] [CrossRef] [PubMed]
- López-Fernández, H.; Santos, H.M.; Capelo, J.L.; Fdez-Riverola, F.; Glez-Peña, D.; Reboiro-Jato, M. Mass-Up: An All-in-One Open Software Application for MALDI-TOF Mass Spectrometry Knowledge Discovery. BMC Bioinform. 2015, 16, 318. [Google Scholar] [CrossRef] [Green Version]
- Ketterlinus, R.; Hsieh, S.Y.; Teng, S.H.; Lee, H.; Pusch, W. Fishing for Biomarkers: Analyzing Mass Spectrometry Data with the New ClinProTools Software. Biotechniques 2005, 38, 37–40. [Google Scholar] [CrossRef]
- Titulaer, M.K.; Siccama, I.; Dekker, L.J.; van Rijswijk, A.L.C.T.; Heeren, R.M.A.; Sillevis Smitt, P.A.; Luider, T.M. A Database Application for Pre-Processing, Storage and Comparison of Mass Spectra Derived from Patients and Controls. BMC Bioinform. 2006, 7, 403. [Google Scholar] [CrossRef] [Green Version]
- Meuleman, W.; Engwegen, J.Y.M.N.; Gast, M.C.W.; Beijnen, J.H.; Reinders, M.J.T.; Wessels, L.F.A. Comparison of Normalisation Methods for Surface-Enhanced Laser Desorption and Ionisation (SELDI) Time-of-Flight (TOF) Mass Spectrometry Data. BMC Bioinform. 2008, 9, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wei, F.; Wang, F.; Li, C.; Meng, G.; Duan, H.; Ma, Q.; Zhang, W. Serum Peptidome Profiling Analysis for the Identification of Potential Biomarkers in Cervical Intraepithelial Neoplasia Patients. Biochem. Biophys. Res. Commun. 2015, 465, 476–480. [Google Scholar] [CrossRef]
- Du, P.; Stolovitzky, G.; Horvatovich, P.; Bischoff, R.; Lim, J.; Suits, F. A Noise Model for Mass Spectrometry Based Proteomics. Bioinformatics 2008, 24, 1070–1077. [Google Scholar] [CrossRef] [Green Version]
- Datta, S.; Mertens, B.J.A. Statistical Analysis of Proteomics, Metabolomics, and Lipidomics Data Using Mass Spectrometry. In Frontiers in Probability and the Statistical Sciences; Springer International Publishing: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Labas, V.; Spina, L.; Belleannee, C.; Teixeira-Gomes, A.P.; Gargaros, A.; Dacheux, F.; Dacheux, J.L. Analysis of Epididymal Sperm Maturation by MALDI Profiling and Top-down Mass Spectrometry. J. Proteom. 2015, 113, 226–243. [Google Scholar] [CrossRef] [PubMed]
- Heather, L.C.; Wang, X.; West, J.A.; Griffin, J.L. A Practical Guide to Metabolomic Profiling as a Discovery Tool for Human Heart Disease. J. Mol. Cell Cardiol. 2013, 55, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Dong, N.; Yun, Y.; Deng, B.; Ren, D.; Liu, S.; Liang, Y. Chemometric Methods in Data Processing of Mass Spectrometry-Based Metabolomics: A Review. Anal. Chim. Acta 2016, 914, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Salem, N.; Hussein, S. Data Dimensional Reduction and Principal Components Analysis. Procedia Comput. Sci. 2019, 163, 292–299. [Google Scholar] [CrossRef]
- Silva, C.; Perestrelo, R.; Silva, P.; Tomás, H.; Câmara, J.S. Breast Cancer Metabolomics: From Analytical Platforms to Multivariate Data Analysis. A Review. Metabolites 2019, 9, 102. [Google Scholar] [CrossRef] [Green Version]
- Aminu, M.; Ahmad, N.A. Complex Chemical Data Classification and Discrimination Using Locality Preserving Partial Least Squares Discriminant Analysis. ACS Omega 2020, 5, 26601–26610. [Google Scholar] [CrossRef]
- Zaki, A.; Ramadan, R.A.; Moez, P.; Ghareeb, H.; Elkarmouty, A. Plasma Peptidome Pattern of Breast Cancer Using Magnetic Beads-Based Plasma Fractionation and MALDI-TOF MS: A Case Control Study in Egypt. Asian Pac. J. Cancer Prev. 2019, 20, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Ewing, I.; Hurley, J.J.; Josephides, E.; Millar, A. The Molecular Genetics of Colorectal Cancer. Frontline Gastroenterol. 2014, 5, 26–30. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Zheng, R.; Baade, P.D.; Zhang, S.; Zeng, H.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cancer Statistics in China, 2015. CA Cancer J Clin 2016, 66, 115–132. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.T.; Liu, F.C.; Wu, C.Y.; Kuo, C.F.; Lan, W.C.; Yu, H.P. Epidemiology and Survival Outcomes of Lung Cancer: A Population-Based Study. Biomed. Res. Int. 2019, 2019, 8148156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziaran, S.; Varchulova Novakova, Z.; Bohmer, D.; Danisovic, L. Biomarkers for Determination Prostate Cancer: Implication for Diagnosis and Prognosis. Neoplasma 2015, 62, 683–691. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-de-las-Penas, C.; Svensson, P. Myofascial Temporomandibular Disorder. Curr. Rheumatol. Rev. 2016, 12, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Slade, G.D.; Ohrbach, R.; Greenspan, J.D.; Fillingim, R.B.; Bair, E.; Sanders, A.E.; Diatchenko, L.; Meloto, C.B.; Smith, S.; Maixner, W. Painful Temporomandibular Disorder: Decade of Discovery from OPPERA Studies. J. Dent. Res. 2016, 95, 1084–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerecke, C.; Fuhrmann, S.; Strifler, S.; Schmidt-Hieber, M.; Einsele, H.; Knop, S. The Diagnosis and Treatment of Multiple Myeloma. Dtsch. Arztebl. Int. 2016, 113, 470–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luu, G.T.; Sanchez, L.M. Toward Improvement of Screening through Mass Spectrometry-Based Proteomics: Ovarian Cancer as a Case Study. Int. J. Mass. Spectrom. 2021, 469, 116679. [Google Scholar] [CrossRef]
- Angel, T.E.; Aryal, U.K.; Hengel, S.M.; Baker, E.S.; Kelly, R.T.; Robinson, E.W.; Smith, R.D. Mass Spectrometry-Based Proteomics: Existing Capabilities and Future Directions. Chem. Soc. Rev. 2012, 41, 3912–3928. [Google Scholar] [CrossRef] [Green Version]
- Tsai, T.H.; Song, E.; Zhu, R.; Di Poto, C.; Wang, M.; Luo, Y.; Varghese, R.S.; Tadesse, M.G.; Ziada, D.H.; Desai, C.S.; et al. LC-MS/MS-Based Serum Proteomics for Identification of Candidate Biomarkers for Hepatocellular Carcinoma. Proteomics 2015, 15, 2369–2381. [Google Scholar] [CrossRef] [Green Version]
- Ploypetch, S.; Jaresitthikunchai, J.; Phaonakrop, N.; Sakcamduang, W.; Manee-in, S.; Suriyaphol, P.; Roytrakul, S.; Suriyaphol, G. Utilizing MALDI-TOF MS and LC-MS/MS to Access Serum Peptidome-Based Biomarkers in Canine Oral Tumors. Sci. Rep. 2022, 12, 21641. [Google Scholar] [CrossRef]
- Darie-Ion, L.; Whitham, D.; Jayathirtha, M.; Rai, Y.; Neagu, A.N.; Darie, C.C.; Petre, B.A. Applications of MALDI-MS/MS-Based Proteomics in Biomedical Research. Molecules 2022, 27, 6196. [Google Scholar] [CrossRef] [PubMed]
- Carapito, C.; Duong, V.-A.; Lee, H. Bottom-Up Proteomics: Advancements in Sample Preparation. Int. J. Mol. Sci. 2023, 24, 5350. [Google Scholar] [CrossRef]
- Karpievitch, Y.V.; Polpitiya, A.D.; Anderson, G.A.; Smith, R.D.; Dabney, A.R. Liquid Chromatography Mass Spectrometry-Based Proteomics: Biological and Technological Aspects. Ann. Appl. Stat. 2010, 4, 1797–1823. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, L.E.; Kilpatrick, E.L. Optimizing High-Resolution Mass Spectrometry for the Identification of Low-Abundance Post-Translational Modifications of Intact Proteins. J. Proteome Res. 2017, 16, 3255–3265. [Google Scholar] [CrossRef]
- Lin, Z.; Wei, L.; Cai, W.; Zhu, Y.; Tucholski, T.; Mitchell, S.D.; Guo, W.; Ford, S.P.; Diffee, G.M.; Ge, Y. Simultaneous Quantification of Protein Expression and Modifications by Top-down Targeted Proteomics: A Case of the Sarcomeric Subproteome. Mol. Cell. Proteom. 2019, 18, 594–605. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.M.; Kelleher, N.L.; Linial, M.; Goodlett, D.; Langridge-Smith, P.; Goo, Y.A.; Safford, G.; Bonilla, L.; Kruppa, G.; Zubarev, R.; et al. Proteoform: A Single Term Describing Protein Complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Tiambeng, T.N.; Cai, W.; Chen, B.; Lin, Z.; Gregorich, Z.R.; Ge, Y. Impact of Phosphorylation on the Mass Spectrometry Quantification of Intact Phosphoproteins. Anal. Chem. 2018, 90, 4935–4939. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Gregorich, Z.R.; Valeja, S.G.; Zhang, H.; Cai, W.; Chen, Y.C.; Guner, H.; Chen, A.J.; Schwahn, D.J.; Hacker, T.A.; et al. Top-down Proteomics Reveals Concerted Reductions in Myofilament and Z-Disc Protein Phosphorylation after Acute Myocardial Infarction. Mol. Cell Proteom. 2014, 13, 2752–2764. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ge, Y. Comprehensive Analysis of Protein Modifications by Top-down Mass Spectrometry. Circ. Cardiovasc. Genet. 2011, 4, 711. [Google Scholar] [CrossRef]
- Van Belkum, A.; Welker, M.; Erhard, M.; Chatellier, S. Biomedical Mass Spectrometry in Today’s and Tomorrow’s Clinical Microbiology Laboratories. J. Clin. Microbiol. 2012, 50, 1513–1517. [Google Scholar] [CrossRef] [Green Version]
- Bang, G.; Lee, H.; Kim, H.; Han, E.H.; Park, Y.H.; Kim, J.Y. Comparison of Protein Characterization Using In Solution and S-Trap Digestion Methods for Proteomics. Biochem. Biophys. Res. Commun. 2022, 589, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, O.M.; Ziganshin, R.H.; Arapidi, G.P.; Kovalchuk, S.I.; Azarkin, I.V.; Sorokina, A.V.; Govorun, V.M.; Radzinsky, V.E.; Ivanov, V.T. Scope and Limitations of MALDI-TOF MS Blood Serum Peptide Profiling in Cancer Diagnostics. Russ. J. Bioorg Chem. 2016, 42, 497–505. [Google Scholar] [CrossRef]
- Park, J.M.; Kim, M.J.; Noh, J.Y.; Yun, T.G.; Kang, M.J.; Lee, S.G.; Yoo, B.C.; Pyun, J.C. Simultaneous Analysis of Multiple Cancer Biomarkers Using MALDI-TOF Mass Spectrometry Based on a Parylene-Matrix Chip. J. Am. Soc. Mass. Spectrom. 2020, 31, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Tarfeen, N.; Nisa, K.U.; Nisa, Q. MALDI-TOF MS: Application in Diagnosis, Dereplication, Biomolecule Profiling and Microbial Ecology. Proc. Indian Natl. Sci. Acad. 2022, 88, 277–291. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathology/Sample | Extraction Procedure | Matrix | Main Conclusions | References |
|---|---|---|---|---|
| Bladder cancer | ||||
| Plasma proteome | 2D-DIGE | HCCA |
| [20] |
| Plasma proteome | SDS-PAGE | HCCA |
| [31] |
| Serum peptidome | MB-WCX | HCCA |
| [32] |
| Urine glycoproteome | MNP@lectins | - |
| [33] |
| Breast cancer (BC) | ||||
| Plasma peptidome | MB-HIC8 | HCCA |
| [34] |
| Serum proteome | 2-DE | HCCA |
| [9] |
| Blood and serum N-glycans | SPE | DHB |
| [35] |
| Urine proteome/peptidome | SDS-PAGE | SA |
| [36] |
| Cervical cancer (CC) | ||||
| Cell lines proteome | 2DE | - |
| [37] |
| Cell lines proteome | 2D-DIGE | HCCA |
| [38] |
| Serum proteome | MB-WCX | HCCA |
| [39] |
| Colorectal cancer (CRC) | ||||
| Serum IgG N-glycome | MiniChromTM Pre-packed Columns with Eshmuno® | DHB |
| [40] |
| Serum proteome | MB-WCX | HCCA |
| [41] |
| Serum proteome | MB-WCX | HCCA |
| [42] |
| Tissue proteome | 2D-DIGE | HCCA |
| [19] |
| Tissue proteome | 2D-DIGE, µZip-TipC18 | HCCA |
| [43] |
| Gastric cancer (GC) | ||||
| Gastric cell lines proteome | 2-DE | HCCA |
| [44] |
| Serum peptidome | MB-IMAC-Cu | HCCA |
| [45] |
| Serum glycoproteome | HILIC SPE | DHB |
| [46] |
| Liver cancer | ||||
| Salivary N-glycome | SPE–C18 | DHB |
| [47] |
| Serum proteome | Samples diluted with H2O and direct application | HCCA |
| [48] |
| Serum proteome | MB-WCX | - |
| [49] |
| Serum proteome | MB-WCX | HCCA |
| [50] |
| Serum proteome | Direct application | - |
| [51] |
| Lung cancer | ||||
| Cell lines proteome | 2D-DIGE | HCCA |
| [52] |
| Cell lines proteome | Ultracentrifugation | DHB, SA |
| [25] |
| Plasma proteome | 2D-SDS-PAGE | HCCA |
| [53] |
| Pleural effusion (PE) and malignant pleural effusion (MPE) peptidome profile | MB-WCX | HCCA |
| [23] |
| Serum proteome | MB-WCX | HCCA |
| [24] |
| Serum peptidome | MB-WCX | HCCA |
| [54] |
| Serum and urine peptidome | SPE-MBs | HCCA |
| [55] |
| Ovarian cancer (OC) | ||||
| Plasma proteome | MagSi-Proteomics C8 beads | HCCA |
| [16] |
| Serum proteome | ZipTip C18 with ACN and 0.1% TFA | HCCA |
| [17] |
| Serum proteome | Samples diluted with H2O (1:80 v/v) and direct application | SA |
| [56] |
| Prostate cancer (PCa) | ||||
| Tissue proteome | Direct | DHB |
| [57] |
| Serum proteome/peptidome | HICNPs | HCCA |
| [58] |
| Urine and serum peptidome | ACN and re-suspension in 0.1% TFA | HCCA |
| [22] |
| Urine proteome | 2DE | - |
| [59] |
| Other cancers | ||||
| Cell lines proteome | Centrifugation (75% ethanol, 70% formic acid, 100% ACN) | HCCA |
| [29] |
| Cell lines proteome | Centrifugation (75% ethanol, 70% formic acid, 100% ACN) | HCCA |
| [21] |
| Plasma exosomes | SSEC (consisting of Mini-SEC and HPL-SEC) | HCCA |
| [60] |
| Urine proteome | ZipTip C18 | HCCA |
| [61] |
| Salivary proteome | Centrifugation | HCCA |
| [30] |
| Serum peptidome | MB-WCX | - |
| [18] |
| MS/MS | MALDI-TOF MS | |
|---|---|---|
| Sample | Liquid form | Solid form and/or liquid form to dry on plate |
| Sample pre-treatment | Requires more extensive sample pre-treatment/clean-up | Minimal sample pre-treatment |
| Separation/fraction | Usually required an online or offline method | Not required |
| Molecules | Peptides | Proteins, large glycopeptides, oligonucleotides |
| Ionization | Soft ionization with solvent and electronebulization | Soft ionization with matrix |
| Fragmentation | Yes | No |
| Analyze time | Minutes or hours | 20–30 s |
| Quantification | Relative and/or absolute | Only relative |
| Data | Identification and characterization of peptides | Only provides putative m/z |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sousa, P.; Silva, L.; Luís, C.; Câmara, J.S.; Perestrelo, R. MALDI-TOF MS: A Promising Analytical Approach to Cancer Diagnostics and Monitoring. Separations 2023, 10, 453. https://doi.org/10.3390/separations10080453
Sousa P, Silva L, Luís C, Câmara JS, Perestrelo R. MALDI-TOF MS: A Promising Analytical Approach to Cancer Diagnostics and Monitoring. Separations. 2023; 10(8):453. https://doi.org/10.3390/separations10080453
Chicago/Turabian StyleSousa, Patrícia, Laurentina Silva, Catarina Luís, José S. Câmara, and Rosa Perestrelo. 2023. "MALDI-TOF MS: A Promising Analytical Approach to Cancer Diagnostics and Monitoring" Separations 10, no. 8: 453. https://doi.org/10.3390/separations10080453
APA StyleSousa, P., Silva, L., Luís, C., Câmara, J. S., & Perestrelo, R. (2023). MALDI-TOF MS: A Promising Analytical Approach to Cancer Diagnostics and Monitoring. Separations, 10(8), 453. https://doi.org/10.3390/separations10080453