Estimating Detection Limits in Chromatography from Calibration Data: Ordinary Least Squares Regression vs. Weighted Least Squares


Abstract
:1. Introduction
2. Experimental and Statistical Calculations
3. Statistical Considerations
4. Results
5. Discussion
6. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Currie, L.A. Nomenclature in evaluation of analytical methods including detection and quantification capabilities. Pure Appl. Chem. 1995, 67, 1699–1723. [Google Scholar] [CrossRef]
- International Union of Pure and Applied Chemistry. IUPAC Compendium of Chemical Terminology—The Gold Book; IUPAC. Available online: https://goldbook.iupac.org (accessed on 8 March 2018).
- Heyden, Y.V.; Boqué, R. The limit of detection. LCGC Eur. 2009, 22, 82–85. [Google Scholar]
- International Conference on Harmonisation (ICH). Q2(R1): Validation of Analytical Procedures—Text and Methodology. ICH Harmonised Tripartite Guideline. 1994/1996. Available online: http://www.ich.org/products/guidelines/quality/quality-single/article/validation-of-analytical-procedures-text-and-methodology.html (accessed on 31 July 2018).
- Shabir, G.A. Validation of high-performance liquid chromatography methods for pharmaceutical analysis: Understanding the differences and similarities between validation requirements of the US Food and Drug Administration, the US Pharmacopeia and the International Conference on Harmonization. J. Chromatogr. A 2003, 987, 57–66. [Google Scholar] [CrossRef] [PubMed]
- US-EPA. 40 CFR Appendix B to Part 136—Definition and Procedure for the Determination of the Method Detection Limit; Revision 2, EPA 821-R-16-006; US Environmental Protection Agency: Washington, DC, USA, 2016. Available online: https://www.gpo.gov/fdsys/granule/CFR-2011-title40-vol23/CFR-2011-title40-vol23-part136-appB/content-detail.html (accessed on 31 July 2018).
- Wenzl, T.; Haedrich, J.; Schaechtele, A.; Robouch, P.; Stroka, J. Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Feed and Food; EUR 28099; Publications Office of the European Union: Luxembourg, 2016; ISBN 978-92-79-61768-3. [Google Scholar]
- Vial, J.; Jardy, A. Experimental comparison of the different approaches to estimate LOD and LOQ of an HPLC method. Anal. Chem. 1999, 71, 2672–2677. [Google Scholar] [CrossRef]
- Shrivastava, A.; Gupta, V.B. Methods for the determination of limit of detection and limit of quantification of the analytical methods. Chron. Young Sci. 2011, 2, 21–25. [Google Scholar] [CrossRef]
- Araujo, P. Key aspects of analytical method validation and linearity evaluation. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Currie, L.A. Limits for qualitative detection and quantitative determination: Application to radiochemistry. Anal. Chem. 1968, 40, 586–593. [Google Scholar] [CrossRef]
- Long, G.L.; Winefordner, J.D. Limit of detection: A closer look at the IUPAC definition. Anal. Chem. 1983, 55, 712A–724A. [Google Scholar] [CrossRef]
- Bernal, E. Limit of detection and limit of quantification: Determination in gas chromatograpy. In Advances in Gas Chromatography; Guo, X., Ed.; IntechOpen: Rijeka, Croatia, 2014; pp. 57–81. ISBN 978-953-51-1227-3. Available online: https://www.intechopen.com/books/advances-in-gas-chromatography (accessed on 8 March 2018).
- Desimoni, E.; Brunetti, B. About estimating the limit of detection by the signal to noise approach. Pharm. Anal. Acta 2015, 6, 1000355. [Google Scholar] [CrossRef]
- International Organization for Standardization (ISO). Capability of Detection, Part 1: Terms and Definitions; ISO 11843-1; ISO: Geneve, Switzerland, 1997. [Google Scholar]
- Magnusson, B.; Ornemark, U. (Eds.) EURACHEM Guide: The Fitness for Purpose of Analytical Methods—A Laboratory Guide to Method Validation and Related Topics, 2nd ed.; LCG: Teddington, UK, 2014; ISBN 978-91-87461-59-0. [Google Scholar]
- Hubaux, A.; Vos, G. Decision and detection limits for linear calibration curves. Anal. Chem. 1970, 42, 849–855. [Google Scholar] [CrossRef]
- International Organization for Standardization (ISO). Capability of Detection. Part 2: Methodology in the Linear Calibration Case; ISO 11843-2; ISO: Geneve, Switzerland, 2000. [Google Scholar]
- Massart, D.L.; Vandeginste, B.G.M.; Buydens, L.M.C.; De Jong, S.; Lewi, P.H.; Smeyers-Verbeke, J. Handbook of Chemometrics and Qualimetrics: Part A; Elsevier: Amsterdam, The Netherlands, 1997; ISBN 0-444-89724-0. [Google Scholar]
- Mandel, J. The Statistical Analysis of Experimental Data; Dover Pub. Inc.: New York, NY, USA, 1964; ISBN 978-0486646664. [Google Scholar]
- Levene, H. Robust test for equality of variances. In Contributions to Probability and Statistics: Essays in Honor of Harold Hotelling; Olkin, I., Ghurye, S.G., Hoedffding, W., Madow, W.G., Mann, H.B., Eds.; Stanford University Press: Palo Alto, CA, USA, 1960; pp. 278–292. ISBN 978-0804705967. [Google Scholar]
- Brown, M.B.; Forsythe, A.B. Robust test for the equality of variances. J. Am. Stat. Assoc. 1974, 69, 364–367. [Google Scholar] [CrossRef]
- Jacquez, J.A.; Norusis, M. Sampling experiments on the estimation of parameters in heteroscedastic linear regression. Biometrics 1973, 29, 771–780. [Google Scholar] [CrossRef]
- Miller, J.N.; Miller, J.C. Statistics and Chemometrics for Analytical Chemistry, 6th ed.; Prentice Hall: Harlow, UK, 2010; ISBN 978-0273730422. [Google Scholar]
- Voigtman, E. Limits of Detection in Chemical Analysis; Wiley: Hoboken, NJ, USA, 2017; ISBN 978-1119188971. [Google Scholar]
- Oppenheimer, L.; Capizzi, T.P.; Weppelman, R.M.; Mehta, H. Determining the lowest limit of reliable assay measurement. Anal. Chem. 1983, 55, 638–643. [Google Scholar] [CrossRef]
- Zorn, M.E.; Gibbons, R.D.; Sonzogni, W.C. Weigthed least-squares approach to calculating limits of detection and quantification by modeling variability as a function of concentration. Anal. Chem. 1997, 69, 3069–3075. [Google Scholar] [CrossRef] [PubMed]
- Sanagi, M.M.; Ling, S.L.; Nasir, Z.; Wan Ibrahim, W.A.; Naim, A.A. Comparison of signal-to-noise, blank determination, and linear regression methods for the estimation of detection and quantification limits of volatile organic compounds by gas chromatography. J. AOAC Int. 2009, 92, 1833–1838. [Google Scholar] [PubMed]
- Ismail, R.; Lee, H.Y.; Mahyudin, N.A.; Bakar, F.A. Linearity study on detection and quantification limits for the determination of avermectins using linear regression. J. Food Drug Anal. 2014, 22, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Zorn, M.E.; Gibbons, R.D.; Sonzogni, W.C. Evaluation of approximate methods for calculating the limit of detection and quantification. Environ. Sci. Technol. 1999, 33, 2291–2295. [Google Scholar] [CrossRef]
- Desimoni, E.; Brunetti, B. About estimating the limit of detection of heteroscedastic analytical systems. Anal. Chim. Acta 2009, 655, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.M. Ordinary least squares with laboratory calibrations: A practical way to show students that this fitting model may easily yield biased results when used indiscriminately. World J. Anal. Chem. 2017, 5, 1–8. [Google Scholar] [CrossRef]
- Raposo, F. Evaluation of analytical calibration based on least-square linear regression for instrumental techniques: A tutorial review. TRAC Trends Anal. Chem. 2016, 77, 167–185. [Google Scholar] [CrossRef]
- Uhrovcik, J. Strategy for determination of LOD and LOQ values—Some basic aspects. Talanta 2014, 119, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.M.; Castel-Branco, M.M.; Falcao, A.C. Linear regression for calibration revisited: Weighting schemes for bioanalytical methods. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2002, 774, 215–222. [Google Scholar] [CrossRef]
- Tellinghuisen, J. Weighted least-squares in calibration: What difference does it make? Analyst 2007, 132, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.C.; Zhang, E.; Dong, H.; Tellinghuisen, J. Weighted least squares in calibration: Estimating data variance functions in high-performance liquid chromatography. J. Chromatogr. A 2008, 1206, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Liu, G.; Wang, J.; Aubry, A.F.; Arnold, M.E. Selecting the correct weighting factor for linear and quadratic calibration curves with least-square regression algorithm in bioanalytical LC-MS/MS assays and impacts of using incorrect weighting factors on curve stability, data quality, and assay performance. Anal. Chem. 2014, 86, 8959–8966. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, M.; Hibbert, D.B. Linearity and the limitations of least squares calibration. J. Chromatogr. A 1997, 762, 73–82. [Google Scholar] [CrossRef]
- Vanatta, L.E.; Coleman, D.E. Calculation of detection limits for a single-laboratory ion-chromatographic method to determine parts-per-trillion ions in ultrapure water. J. Chromatogr. A 1997, 770, 105–114. [Google Scholar] [CrossRef]
- Luo, W.; Li, H.; Zhang, Y.; Ang, C.Y.W. Determination of formaldehyde in blood plasma by high-performance liquid chromatography with fluorescence detection. J. Chromatogr. B Biomed. Sci. Appl. 2001, 753, 253–257. [Google Scholar] [CrossRef]
- Ribani, M.; Collins, C.H.; Bottoli, C.B.G. Validation of chromatographic methods: Evaluation of detection and quantification limits in the determination of impurities in omeprazol. J. Chromatogr. A 2007, 1156, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Proestos, C.; Sereli, D.; Komaitis, M. Determination of phenolic compounds in aromatic plants by RP-HPLC and GC-MS. Food Chem. 2006, 95, 44–52. [Google Scholar] [CrossRef]
- Muranaka, L.S.; Giorgiano, T.E.; Takita, M.A.; Forim, M.R.; Silva, L.F.C.; Coletta-Filho, H.D.; Machado, M.A.; de Souza, A.A. N-Acetylcysteine in agriculture, a novel use for an old molecule: Focus on controlling the plant-pathogen Xylella fastidiosa. PLoS ONE 2013, 8, e72937. [Google Scholar] [CrossRef] [PubMed]
- Zabell, A.P.R.; Lytle, F.E.; Julina, R.K. A proposal to improve calibration and outlier detection in high-throughput mass spectrometry. Clin. Mass Spectrom. 2016, 2, 25–33. [Google Scholar] [CrossRef]
- Meites, L.; Smit, H.C.; Kateman, G. The effects of errors in measuring the independent variable in least-squares regression analysis. Anal. Chim. Acta 1984, 164, 287–291. [Google Scholar] [CrossRef]
- Gibbons, R.D.; Jarke, F.H.; Stoub, K.P. Detection limits for linear calibration curves with increasing variance and multiple future detection decisions. In Waste Testing and Quality Assurance: Third Volume; STP 1075; ASTM Publication Code Number 04-010750-56; Friedman, D., Ed.; ASTM: Philadelphia, PA, USA, 1991; pp. 377–390. ISBN 978-0-8031-1294-0. [Google Scholar]
- Kiser, M.M.; Dolan, J.W. Selecting the best curve fit. LCGC Eur. 2004, 17, 138–143. [Google Scholar]



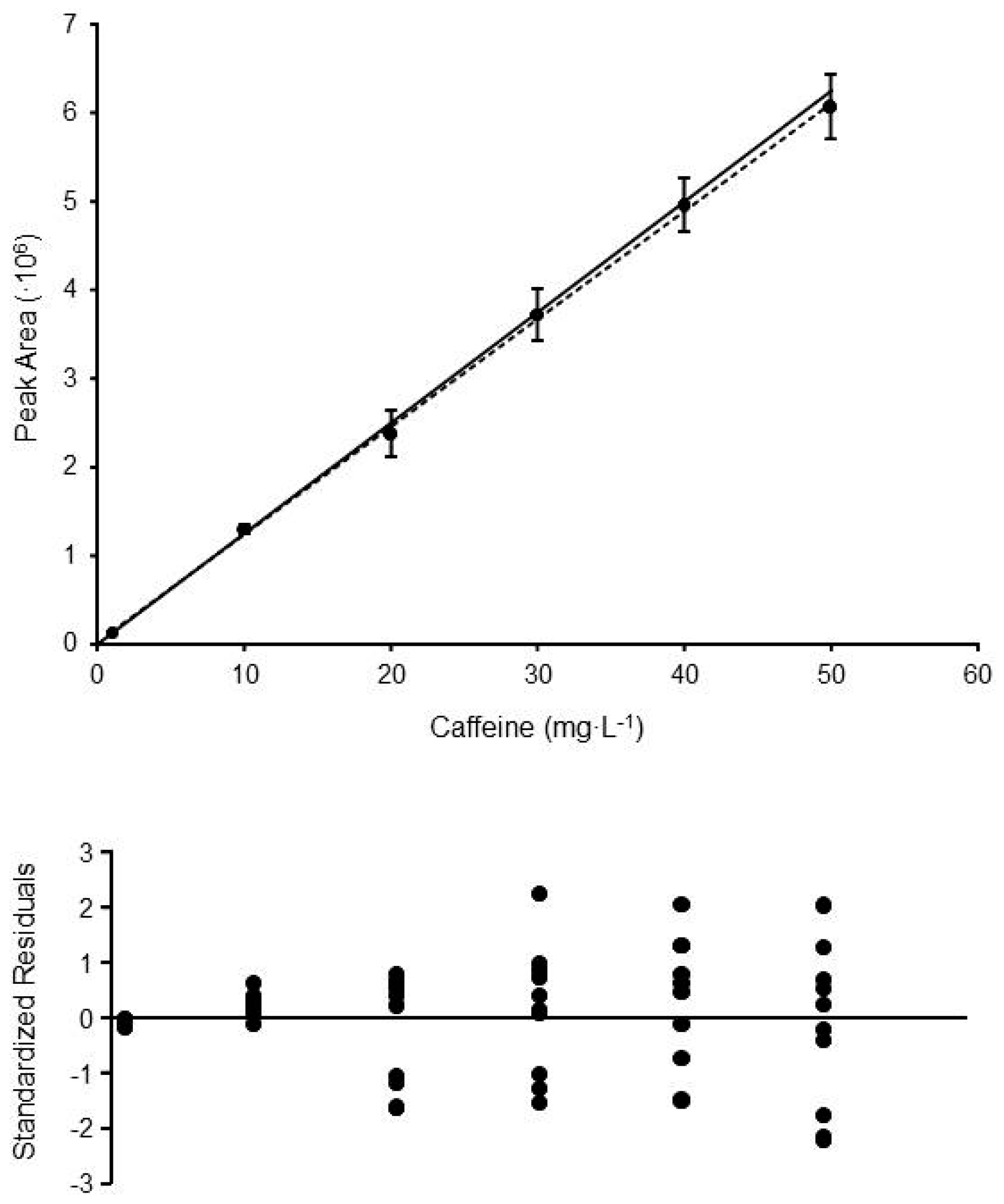{kind=link}
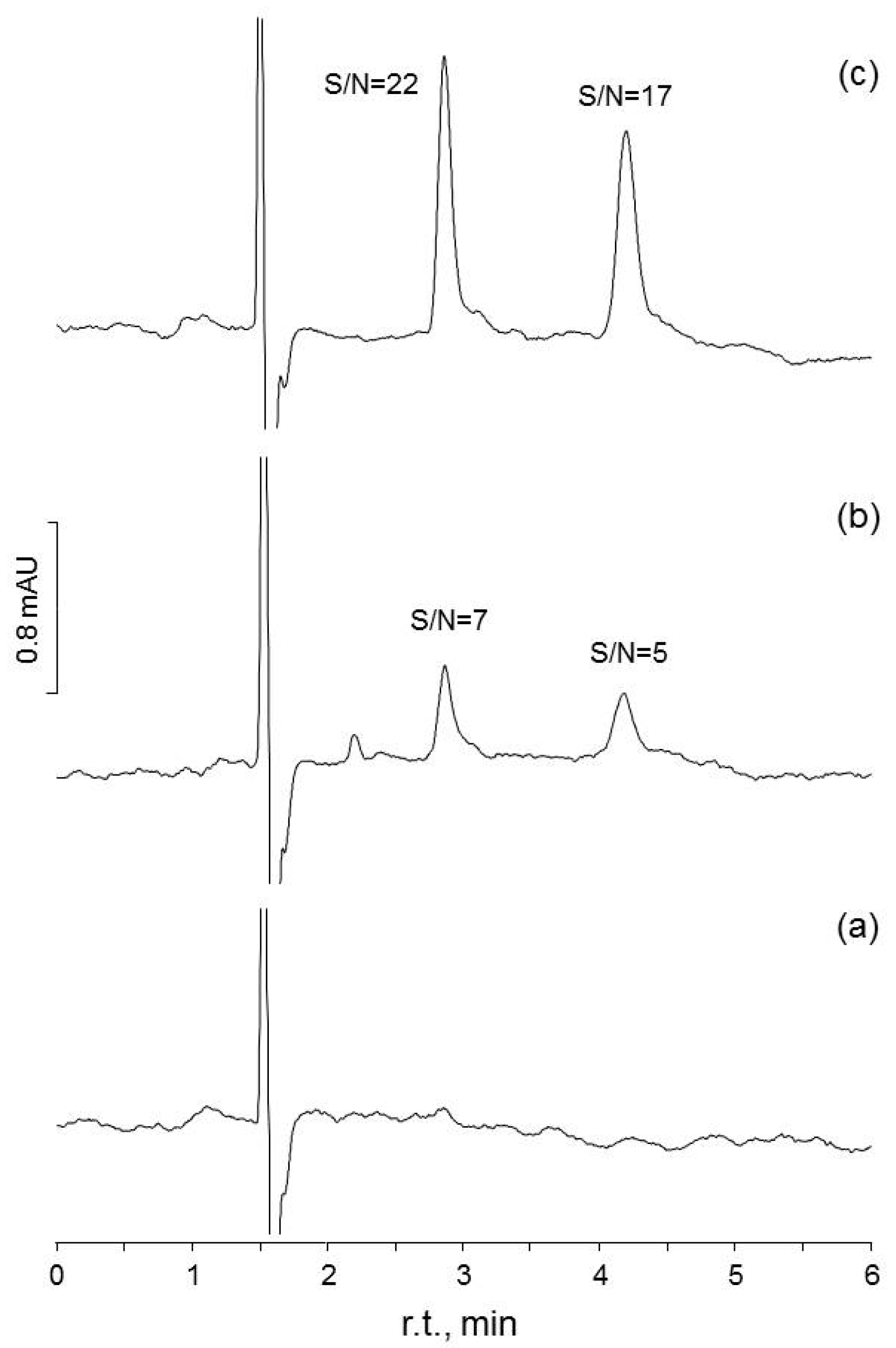{kind=link}
| Method | LOF | Levene | Model | sres | b0 | b1 | b0 = 0 | b1 = 0 |
|---|---|---|---|---|---|---|---|---|
| (p-Value) | (p-Value) | (sb0) | (sb1) | (p-Value) | (p-Value) | |||
| #1 | 0.419 | 0.001 | OLS | 0.0043 | 0.0029 | 6.73·10−4 | 0.981 | <0.001 |
| (0.0029) | (6·10−6) | |||||||
| WLS | 0.4294 | 0.0053 | 6.64·10−4 | 0.004 | <0.001 | |||
| (0.0009) | (6·10−6) | |||||||
| Blank | sbl= | 0.0012 | ||||||
| #2 | 0.952 | 0.008 | OLS | 0.0263 | 0.0023 | 1.73·10−3 | 0.901 | <0.001 |
| (0.0177) | (4·10−5) | |||||||
| WLS | 1.5490 | 0.0147 | 1.67·10−3 | 0.079 | <0.001 | |||
| (0.0063) | (3·10−5) | |||||||
| Blank | sbl= | 0.0015 | ||||||
| #3 | 0.107 | <0.001 | OLS | 0.01367 | 0.0096 | 1.69·10−3 | 0355 | <0.001 |
| (0.0092) | (2·10−5) | |||||||
| WLS | 0.9568 | 0.0154 | 1.67·10−3 | 0.016 | <0.001 | |||
| (0.0039) | (2·10−5) | |||||||
| Blank | sbl= | 0.0045 | ||||||
| #4 | 0.362 | <0.001 | OLS | 0.0143 | 0.0163 | 1.72·10−3 | 0.166 | <0.001 |
| (0.0096) | (2·10−5) | |||||||
| WLS | 0.6781 | 0.0147 | 1.72·10−3 | 0.005 | <0.001 | |||
| (0.0027) | (2·10−5) | |||||||
| Blank | sbl= | 0.0043 | ||||||
| #5 | 0.168 | <0.001 | OLS | 0.0088 | 0.0125 | 1.72·10−3 | 0.098 | <0.001 |
| (0.0058) | (3·10−5) | |||||||
| WLS | 0.4307 | 0.0150 | 1.70·10−3 | 0.0004 | <0.001 | |||
| (0.0013) | (2·10−5) | |||||||
| Blank | sbl= | 0.0025 | ||||||
| #6 | 0.745 | 0.002 | OLS | 5.78·10-3 | 8·10−3 | 3.94·10−3 | 0.158 | <0.001 |
| (5·10−3) | (4·10−5) | |||||||
| WLS | 0.3604 | 6·10−3 | 3.91·10−3 | 0.022 | <0.001 | |||
| (2·10−3) | (4·10−5) | |||||||
| Blank | sbl= | 3·10−3 |
| Method | LOF | Levene | Model | sres | b0 | b1 | b0 = 0 | b1 = 0 |
|---|---|---|---|---|---|---|---|---|
| (p-Value) | (p-Value) | (sb0) | (sb1) | (p-Value) | (p-Value) | |||
| #1 | 0.473 | <0.001 | OLS | 233,473 | 249,259 | 860,379 | 0.152 | <0.001 |
| (140,875) | (13,251) | |||||||
| WLS | 0.6620 | 27,495 | 887,238 | 0.515 | <0.001 | |||
| (38,502) | (24,696) | |||||||
| Blank | sbl= | 33,580 | ||||||
| #2 | 0.372 | <0.001 | OLS | 93,959 | 41,563 | 149,078 | 0.505 | <0.001 |
| (56,865) | (2679) | |||||||
| WLS | 0.7872 | 53,521 | 148,848 | 0.030 | <0.001 | |||
| (16,267) | (4955) | |||||||
| Blank | sbl= | 22,673 | ||||||
| #3 | 0.188 | <0.001 | OLS | 72,065 | −50,424 | 179,903 | 0.312 | <0.001 |
| (43,615) | (3131) | |||||||
| WLS | 1.3649 | −25,615 | 174,245 | 0.133 | <0.001 | |||
| (13,614) | (8048) | |||||||
| Blank | sbl= | 9628 |
| Method | LOF | Levene | Model | sres | b0 | b1 | b0 = 0 | b1 = 0 |
|---|---|---|---|---|---|---|---|---|
| (p-Value) | (p-Value) | (sb0) | (sb1) | (p-Value) | (p-Value) | |||
| #1 | 0.378 | <0.001 | OLS | 52,459 | 57,359 | 385,884 | 0.083 | <0.001 |
| (27,656) | (1804) | |||||||
| WLS | 2.2863 | −2136 | 393,588 | 0.302 | <0.001 | |||
| (1894) | (8524) | |||||||
| Blank | sbl= | 840 | ||||||
| #2 | 0.147 | <0.001 | OLS | 62,530 | 47,160 | 400,992 | 0.194 | <0.001 |
| (32,266) | (3074) | |||||||
| WLS | 0.8077 | 5079 | 407,693 | 0.012 | <0.001 | |||
| (1431) | (4671) | |||||||
| Blank | sbl= | 2458 | ||||||
| #3 | 0.621 | <0.001 | OLS | 68,178 | 24,242 | 121,810 | 0.653 | <0.001 |
| (50,056) | (1653) | |||||||
| WLS | 0.5428 | 1678 | 124,904 | 0.845 | <0.001 | |||
| (8024) | (2013) | |||||||
| Blank | sbl= | 13,719 | ||||||
| #4 | 0.537 | <0.001 | OLS | 77,517 | 113,661 | 129,902 | 0.117 | <0.001 |
| (56,913) | (1879) | |||||||
| WLS | 0.7255 | 13,710 | 134,518 | 0.574 | <0.001 | |||
| (23,893) | (2889) | |||||||
| Blank | sbl= | 32,336 | ||||||
| #5 | 0.761 | <0.001 | OLS | 46,038 | −19,495 | 11,310 | 0.615 | <0.001 |
| (35,823) | (118) | |||||||
| WLS | 0.2274 | −50,238 | 11,444 | 0.004 | <0.001 | |||
| (8227) | (87) | |||||||
| Blank | sbl= | 7538 | ||||||
| #6 | 0.741 | 0.004 | OLS | 48,296 | −101,407 | 10,799 | 0.054 | <0.001 |
| (37,580) | (124) | |||||||
| WLS | 0.2353 | −73,067 | 10,682 | 0.002 | <0.001 | |||
| (9659) | (85) | |||||||
| Blank | sbl= | 11,925 | ||||||
| #7 | 0.600 | 0.002 | OLS | 68,095 | −93,167 | 10,995 | 0.154 | <0.001 |
| (52,987) | (175) | |||||||
| WLS | 0.7003 | −43,109 | 10,628 | 0.195 | <0.001 | |||
| (27,742) | (229) | |||||||
| Blank | sbl= | 31,162 | ||||||
| #8 | 0.774 | 0.007 | OLS | 104,152 | −163,207 | 18,540 | 0.114 | <0.001 |
| (81,043) | (267) | |||||||
| WLS | 0.8237 | −65,272 | 17,849 | 0.234 | <0.001 | |||
| (46,590) | (383) | |||||||
| Blank | sbl= | 44,535 |
| Method | LOF | Levene | Model | sres | b0 | b1 | b0 = 0 | b1 = 0 |
|---|---|---|---|---|---|---|---|---|
| (p-Value) | (p-Value) | (sb0) | (sb1) | (p-Value) | (p-Value) | |||
| #1 | 0.868 | <0.001 | OLS | 205 | 105 | 692 | 0.338 | <0.001 |
| (99) | (6) | |||||||
| WLS | 0.4109 | 89 | 709 | 0.008 | <0.001 | |||
| (21) | (10) | |||||||
| Blank | sbl= | 48 | ||||||
| #2 | 0.486 | <0.001 | OLS | 321 | 3 | 1031 | 0.986 | <0.001 |
| (155) | (9) | |||||||
| WLS | 1.2439 | −140 | 1049 | 0.112 | <0.001 | |||
| (72) | (24) | |||||||
| Blank | sbl= | 66 | ||||||
| #3 | 0.534 | <0.001 | OLS | 135 | 140 | 509 | 0.084 | <0.001 |
| (65) | (4) | |||||||
| WLS | 1.0296 | 44 | 536 | 0.166 | <0.001 | |||
| (27) | (8) | |||||||
| Blank | sbl= | 29 |
| Method | Blank | OLS | WLS (sb0) | ||
|---|---|---|---|---|---|
| (sbl) | (sb0) | wi = 1/si2 | wi = 1/xi2 | wi = 1/yi2 | |
| GC-FID #1 | 6 mg·L−1 | 14 mg·L−1 | 12 mg·L−1 | 10 mg·L−1 | 10 mg·L−1 |
| GC-FID #2 | 9 mg·L−1 | 34 mg·L−1 | 13 mg·L−1 | 10 mg·L−1 | 11 mg·L−1 |
| GC-FID #3 | 9 mg·L−1 | 18 mg·L−1 | 17 mg·L−1 | 10 mg·L−1 | 11 mg·L−1 |
| GC-FID #4 | 8 mg·L−1 | 19 mg·L−1 | 14 mg·L−1 | 8 mg·L−1 | 11 mg·L−1 |
| GC-FID #5 | 5 mg·L−1 | 11 mg·L−1 | 11 mg·L−1 | 10 mg·L−1 | 10 mg·L−1 |
| GC-FID #6 | 3 mg·L−1 | 4 mg·L−1 | 3 mg·L−1 | 3 mg·L−1 | 3 mg·L−1 |
| GC-MS #1 | 0.2 ppbv | 0.5 ppbv | 0.1 ppbv | 0.1 ppbv | 0.1 ppbv |
| GC-MS #2 | 0.5 ppbv | 1.3 ppbv | 0.7 ppbv | 0.6 ppbv | 0.6 ppbv |
| GC-MS #3 | 0.2 ppbv | 0.8 ppbv | 0.3 ppbv | 0.2 ppbv | 0.2 ppbv |
| HPLC-UV #1 | 0.01 mg·L−1 | 0.24 mg·L−1 | 0.02 mg·L−1 | 0.02 mg·L−1 | 0.02 mg·L−1 |
| HPLC-UV #2 | 0.02 mg·L−1 | 0.27 mg·L−1 | 0.02 mg·L−1 | 0.02 mg·L−1 | 0.02 mg·L−1 |
| HPLC-UV #3 | 0.4 mg·L−1 | 1.4 mg·L−1 | 0.2 mg·L−1 | 0.1 mg·L−1 | 0.1 mg·L−1 |
| HPLC-UV #4 | 0.8 mg·L−1 | 1.4 mg·L−1 | 0.6 mg·L−1 | 0.2 mg·L−1 | 0.2 mg·L−1 |
| HPLC-UV #5 | 2 μM | 10 μM | 2 μM | 3 μM | 3 μM |
| HPLC-UV #6 | 4 μM | 11 μM | 3 μM | 3 μM | 3 μM |
| HPLC-UV #7 | 10 μM | 16 μM | 9 μM | 5 μM | 6 μM |
| HPLC-UV #8 | 8 μM | 14 μM | 9 μM | 8 μM | 8 μM |
| CZE-UV #1 | 0.2 mg·L−1 | 0.5 mg·L−1 | 0.2 mg·L−1 | 0.2 mg·L−1 | 0.2 mg·L−1 |
| CZE-UV #2 | 0.2 mg·L−1 | 0.5 mg·L−1 | 0.2 mg·L−1 | 0.1 mg·L−1 | 0.1 mg·L−1 |
| CZE-UV #3 | 0.2 mg·L−1 | 0.8 mg·L−1 | 0.2 mg·L−1 | 0.1 mg·L−1 | 0.1 mg·L−1 |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanchez, J.M. Estimating Detection Limits in Chromatography from Calibration Data: Ordinary Least Squares Regression vs. Weighted Least Squares. Separations 2018, 5, 49. https://doi.org/10.3390/separations5040049
Sanchez JM. Estimating Detection Limits in Chromatography from Calibration Data: Ordinary Least Squares Regression vs. Weighted Least Squares. Separations. 2018; 5(4):49. https://doi.org/10.3390/separations5040049
Chicago/Turabian StyleSanchez, Juan M. 2018. "Estimating Detection Limits in Chromatography from Calibration Data: Ordinary Least Squares Regression vs. Weighted Least Squares" Separations 5, no. 4: 49. https://doi.org/10.3390/separations5040049
APA StyleSanchez, J. M. (2018). Estimating Detection Limits in Chromatography from Calibration Data: Ordinary Least Squares Regression vs. Weighted Least Squares. Separations, 5(4), 49. https://doi.org/10.3390/separations5040049