
Development and Application of Molecularly Imprinted Polymers for the Selective Extraction of Chlordecone from Bovine Serum

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Instrumentation
2.2. Instrumentation and Analytical Conditions
2.3. Condition of Synthesis of Molecularly Imprinted Polymers
2.4. Preliminary Evaluation of the MIP/NIPs by Solid-Phase Extraction in Pure Organic Media
2.5. Characterization of the Most Promising MIP/NIP (MIP/NIP 2) in Pure Organic Media
2.6. Extraction Procedure Applied to Bovine Serum
2.7. Evaluation of the Matrix Effects during LC/MS-MS Analysis
3. Results and Discussion
3.1. Screening of the Synthesis Conditions
3.1.1. Choice of the Synthesis Conditions
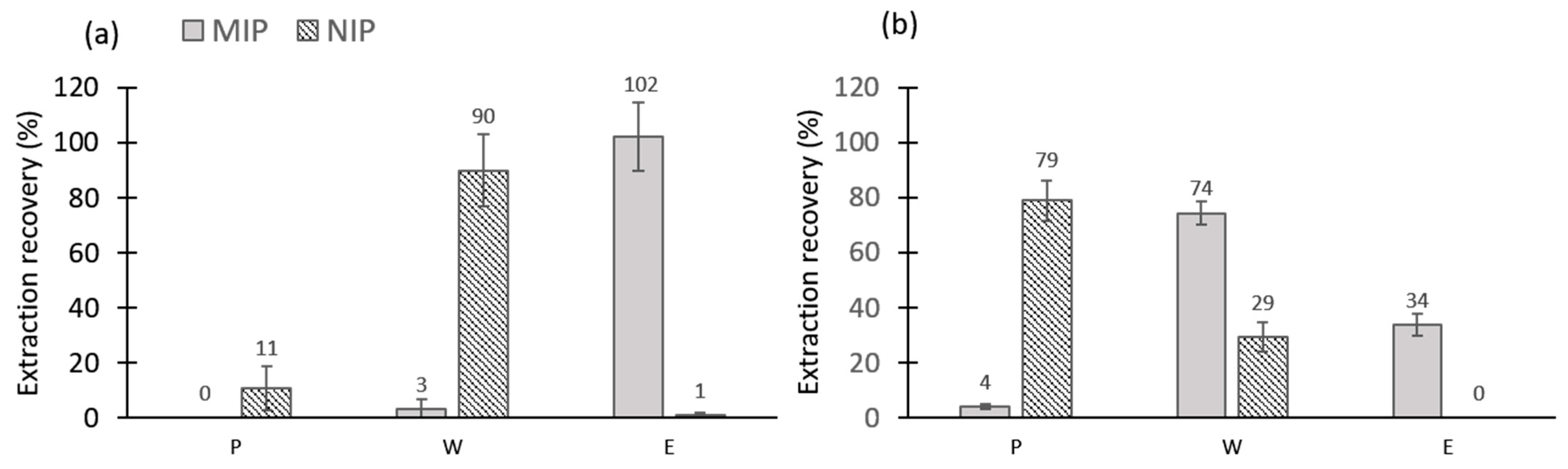
3.1.2. Evaluation of the MIP/NIPs by Solid-Phase Extraction in Pure Organic Solvents
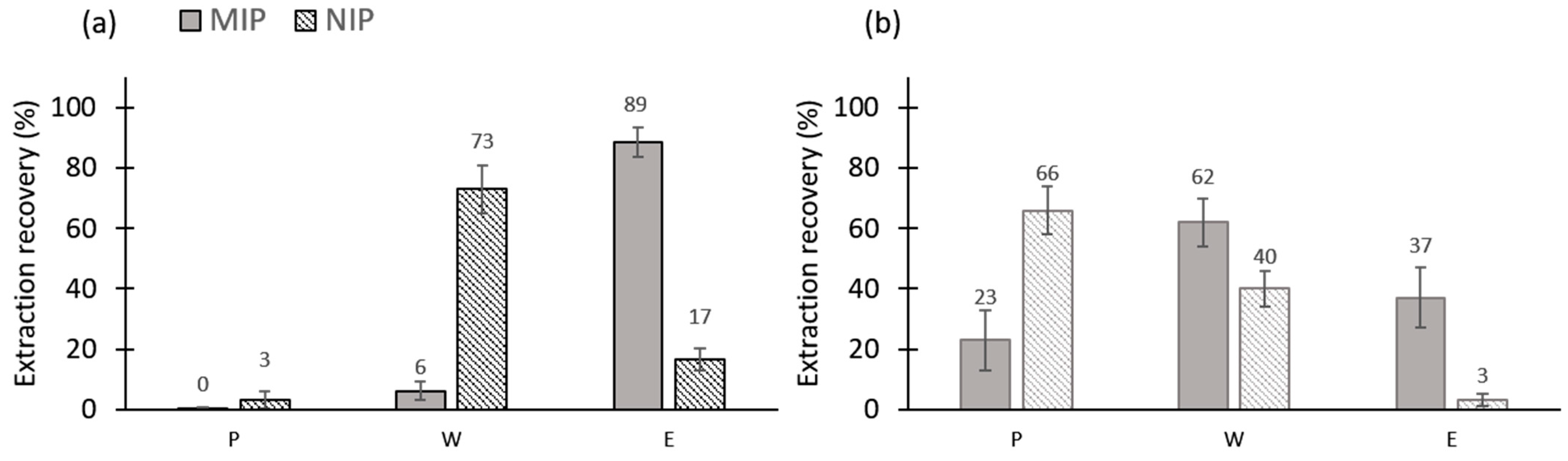
3.2. Characterization of the Most Promising MIP/NIPs (MIP/NIP 2) in Pure Organic Media
3.2.1. Repeatability of the Extraction Procedure
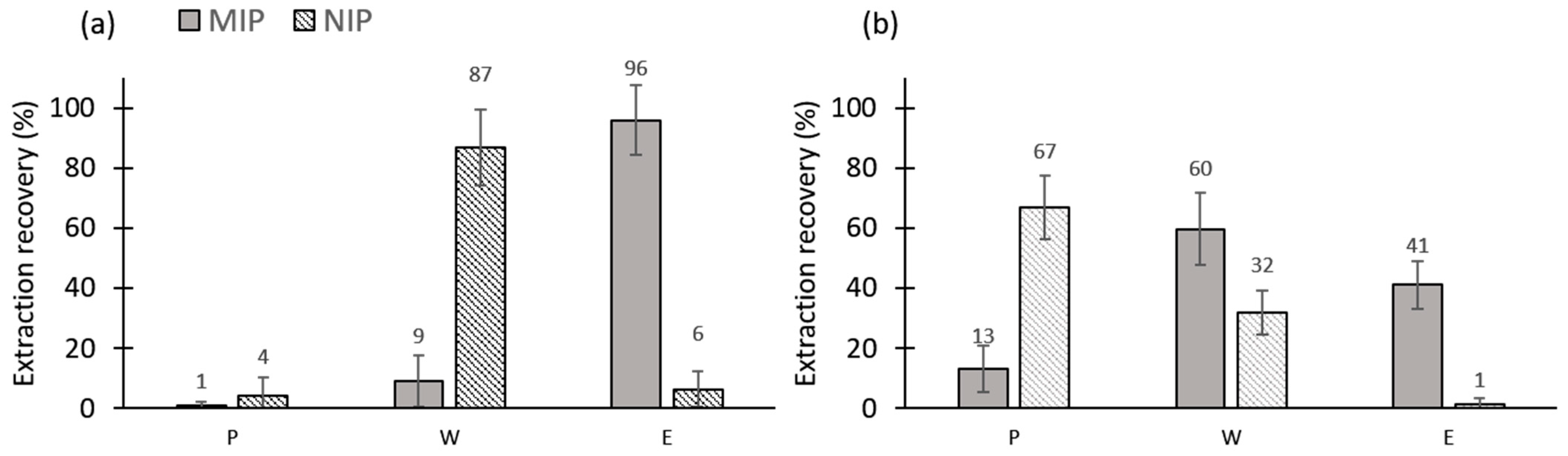
3.2.2. Repeatability of the Cartridge Filling
3.2.3. Reproducibility of the MIP/NIP 2 Synthesis
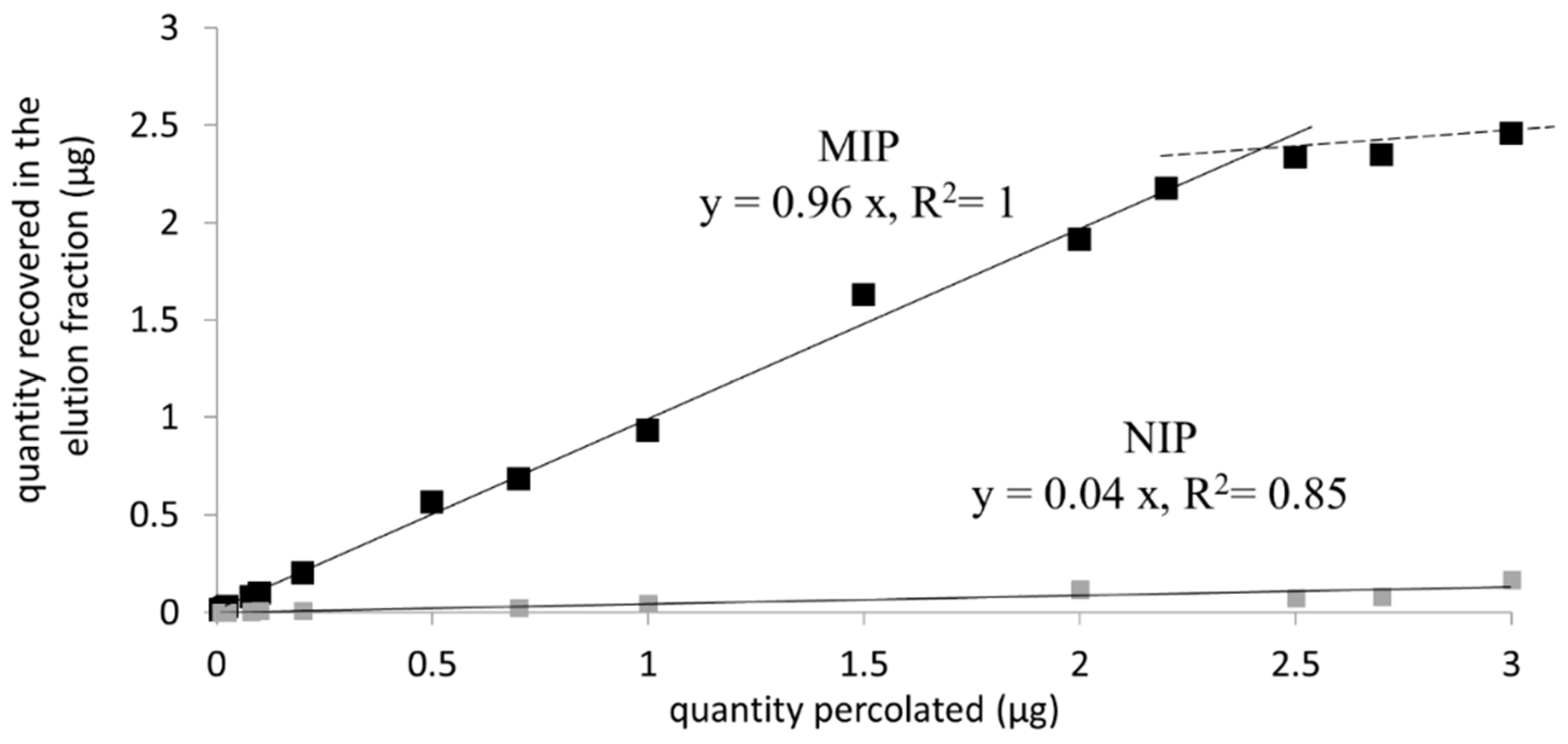
3.2.4. Capacity of the MIP 2
3.3. Optimized Extraction Procedure Applied to Bovine Serum
3.3.1. Retention of CLD in Bovine’s Serum
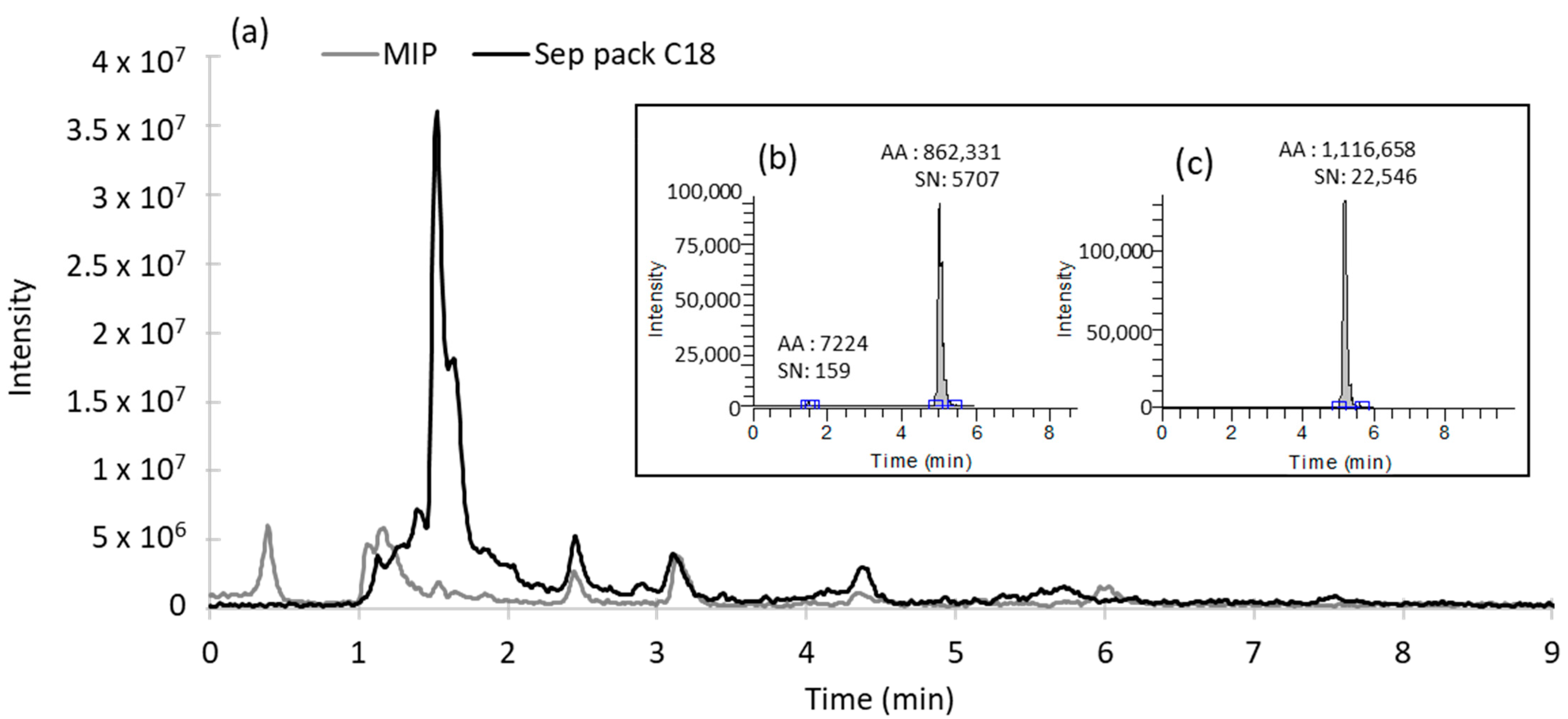
3.3.2. Comparison of Performances with a Conventional Sorbent and Evaluation of Matrix Effects
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jondreville, C.; Lavigne, A.; Jurjanz, S.; Dalibard, C.; Liabeuf, J.-M.; Clostre, F.; Lesueur-Jannoyer, M. Contamination of Free-Range Ducks by Chlordecone in Martinique (French West Indies): A Field Study. Sci. Total Environ. 2014, 493, 336–341. [Google Scholar] [CrossRef]
- Boucher, O.; Simard, M.-N.; Muckle, G.; Rouget, F.; Kadhel, P.; Bataille, H.; Chajès, V.; Dallaire, R.; Monfort, C.; Thomé, J.-P.; et al. Exposure to an Organochlorine Pesticide (Chlordecone) and Development of 18-Month-Old Infants. Neurotoxicology 2013, 35, 162–168. [Google Scholar] [CrossRef]
- Cordier, S.; Bouquet, E.; Warembourg, C.; Massart, C.; Rouget, F.; Kadhel, P.; Bataille, H.; Monfort, C.; Boucher, O.; Muckle, G.; et al. Perinatal Exposure to Chlordecone, Thyroid Hormone Status and Neurodevelopment in Infants: The Timoun Cohort Study in Guadeloupe (French West Indies). Environ. Res. 2015, 138, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Hervé, D.; Costet, N.; Kadhel, P.; Rouget, F.; Monfort, C.; Thomé, J.-P.; Multigner, L.; Cordier, S. Prenatal Exposure to Chlordecone, Gestational Weight Gain, and Birth Weight in a Guadeloupean Birth Cohort. Environ. Res. 2016, 151, 436–444. [Google Scholar] [CrossRef]
- Fournier, A.; Feidt, C.; Lastel, M.-L.; Archimede, H.; Thome, J.-P.; Mahieu, M.; Rychen, G. Toxicokinetics of Chlordecone in Goats: Implications for Risk Management in French West Indies. Chemosphere 2017, 171, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Fourcot, A.; Feidt, C.; Bousquet-Mélou, A.; Ferran, A.A.; Gourdine, J.L.; Bructer, M.; Joaquim-Justo, C.; Rychen, G.; Fournier, A. Modeling Chlordecone Toxicokinetics Data in Growing Pigs Using a Nonlinear Mixed-Effects Approach. Chemosphere 2020, 250, 126151. [Google Scholar] [CrossRef]
- EURL | Residues of Pesticides | Pesticide EU-MRLs Database. Available online: https://www.eurl-pesticides.eu/docs/public/tmplt_article.asp?LabID=100&CntID=649&Theme_ID=1&Pdf=False&Lang=EN (accessed on 26 October 2021).
- NOTE ANSES on the Establishment of a Maximum Residue Limit for Chlordecone in Fat for Meat Products. Saisine n°2018-SA-0202. Available online: https://www.anses.fr/fr/system/files/ERCA2018SA0265.pdf (accessed on 1 December 2021).
- Meijs, A.W.H.M.; Ernst, G.F. Rapid Gas Chromatographic Method for the Determination of Kepone in the Eel. J. Chromatogr. A 1979, 171, 486–489. [Google Scholar] [CrossRef]
- Bordet, F.; Thieffinne, A.; Mallet, J.; Heraud, F.; Blateau, A.; Inthavong, D. In-House Validation for Analytical Methods and Quality Control for Risk Evaluation of Chlordecone in Food. Null 2007, 87, 985–998. [Google Scholar] [CrossRef]
- Saint-Hilaire, M.; Bertin, T.; Inthavong, C.; Lavison-Bompard, G.; Guérin, T.; Fournier, A.; Feidt, C.; Rychen, G.; Parinet, J. Validation of Analytical Methods for Chlordecone and Its Metabolites in the Urine and Feces of Ewes. J. Chromatogr. B 2018, 1093–1094, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Saint-Hilaire, M.; Inthavong, C.; Bertin, T.; Lavison-Bompard, G.; Guérin, T.; Fournier, A.; Feidt, C.; Rychen, G.; Parinet, J. Development and Validation of an HPLC-MS/MS Method with QuEChERS Extraction Using Isotopic Dilution to Simultaneously Analyze Chlordecone and Chlordecol in Animal Livers. Food Chem. 2018, 252, 147–153. [Google Scholar] [CrossRef]
- Bichon, E.; Guiffard, I.; Vénisseau, A.; Marchand, P.; Antignac, J.-P.; Le Bizec, B. Ultra-Trace Quantification Method for Chlordecone in Human Fluids and Tissues. J. Chromatogr. A 2015, 1408, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Woignier, T.; Fernandes, P.; Jannoyer-Lesueur, M.; Soler, A. Sequestration of Chlordecone in the Porous Structure of an Andosol and Effects of Added Organic Matter: An Alternative to Decontamination. Eur. J. Soil Sci. 2012, 63, 717–723. [Google Scholar] [CrossRef]
- Debier, C.; Pomeroy, P.P.; Dupont, C.; Joiris, C.; Comblin, V.; Boulengé, E.L.; Larondelle, Y.; Thomé, J.-P. Dynamics of PCB Transfer from Mother to Pup during Lactation in UK Grey Seals Halichoerus Grypus: Differences in PCB Profile between Compartments of Transfer and Changes during the Lactation Period. Mar. Ecol. Prog. Ser. 2003, 247, 249–256. [Google Scholar] [CrossRef]
- Multigner, L.; Ndong, J.R.; Giusti, A.; Romana, M.; Delacroix-Maillard, H.; Cordier, S.; Jégou, B.; Thome, J.P.; Blanchet, P. Chlordecone Exposure and Risk of Prostate Cancer. JCO 2010, 28, 3457–3462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichon, V.; Delaunay, N.; Combès, A. Sample Preparation Using Molecularly Imprinted Polymers. Anal. Chem. 2020, 92, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Lin, S.; Gan, N.; Chen, X.; Cao, Y.; Li, T.; Zhan, P. Multi-Walled Carbon Nanotube Modified Dummy-Template Magnetic Molecularly Imprinted Microspheres as Solid-Phase Extraction Material for the Determination of Polychlorinated Biphenyls in Fish. J. Sep. Sci. 2014, 37, 1591–1600. [Google Scholar] [CrossRef]
- Xu, X.; Liang, S. Molecularly Imprinted Solid-Phase Extraction Method for the Gas Chromatographic Analysis of Organochlorine Fungicides in Ginseng. J. Sep. Sci. 2019, 42, 1393–1403. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yan, H.; Qiu, M.; Qiao, J.; Yang, G. Determination of Dicofol in Aquatic Products Using Molecularly Imprinted Solid-Phase Extraction Coupled with GC-ECD Detection. Talanta 2011, 85, 2100–2105. [Google Scholar] [CrossRef]
- Yan, H.; Sun, N.; Han, Y.; Yang, C.; Wang, M.; Wu, R. Ionic Liquid-Mediated Molecularly Imprinted Solid-Phase Extraction Coupled with Gas Chromatography-Electron Capture Detector for Rapid Screening of Dicofol in Vegetables. J. Chromatogr. A 2013, 1307, 21–26. [Google Scholar] [CrossRef]
- Yan, H.; Yang, C.; Sun, Y.; Row, K.H. Ionic Liquid Molecularly Imprinted Polymers for Application in Pipette-Tip Solid-Phase Extraction Coupled with Gas Chromatography for Rapid Screening of Dicofol in Celery. J. Chromatogr. A 2014, 1361, 53–59. [Google Scholar] [CrossRef]
- Qiao, F.; Gao, M.; Yan, H. Molecularly Imprinted Ionic Liquid Magnetic Microspheres for the Rapid Isolation of Organochlorine Pesticides in Environmental Water. J. Sep. Sci. 2016, 39, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Lv, T.; Yan, H.; Wu, G.; Li, H. Glyoxal–Urea–Formaldehyde Molecularly Imprinted Resin as Pipette Tip Solid-Phase Extraction Adsorbent for Selective Screening of Organochlorine Pesticides in Spinach. J. Agric. Food Chem. 2015, 63, 9650–9656. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Pan, M.; Fang, G.; Jing, W.; He, S.; Wang, S. An Ionic Liquid Modified Dummy Molecularly Imprinted Polymer as a Solid-Phase Extraction Material for the Simultaneous Determination of Nine Organochlorine Pesticides in Environmental and Food Samples. Anal. Methods 2013, 5, 6128–6134. [Google Scholar] [CrossRef]
- Shaikh, H.; Memon, N.; Bhanger, M.I.; Nizamani, S.M.; Denizli, A. Core–Shell Molecularly Imprinted Polymer-Based Solid-Phase Microextraction Fiber for Ultra Trace Analysis of Endosulfan I and II in Real Aqueous Matrix through Gas Chromatography–Micro Electron Capture Detector. J. Chromatogr. A 2014, 1337, 179–187. [Google Scholar] [CrossRef]
- Zuo, H.G.; Yang, H.; Zhu, J.X.; Ding, Y. Preparation of a Novel RAM-MIP for Selective Solid-Phase Extraction and Gas Chromatography Determination of Heptachlor, Endosulfan and Their Metabolite Residues in Pork. Anal. Methods 2017, 9, 6009–6018. [Google Scholar] [CrossRef]
- Silverio, O.V.; So, R.C.; Elnar, K.J.S.; Malapit, C.A.; Nepomuceno, M.C.M. Development of Dieldrin, Endosulfan, and Hexachlorobenzene-Imprinted Polymers for Dye-Displacement Array Sensing. J. Appl. Polym. Sci. 2017, 134. [Google Scholar] [CrossRef]
- Singh, K.; Balasubramanian, S.; Rani, B.E.A. Computational and Experimental Studies of Molecularly Imprinted Polymers for Organochlorine Pesticides Heptachlor and DDT. Curr. Anal. Chem. 2012, 8, 562–568. [Google Scholar] [CrossRef]
- Singh, K.; Pasha, A.; Rani, B.A. Preparation of Molecularly Imprinted Polymers for Heptachlor: An Organochlorine Pesticide. Chron. Young Sci. 2013, 4, 46. [Google Scholar]
- Anirudhan, T.S.; Alexander, S. Design and Fabrication of Molecularly Imprinted Polymer-Based Potentiometric Sensor from the Surface Modified Multiwalled Carbon Nanotube for the Determination of Lindane (γ-Hexachlorocyclohexane), an Organochlorine Pesticide. Biosens. Bioelectron. 2015, 64, 586–593. [Google Scholar] [CrossRef]
- Pelin Böke, C.; Karaman, O.; Medetalibeyoglu, H.; Karaman, C.; Atar, N.; Lütfi Yola, M. A New Approach for Electrochemical Detection of Organochlorine Compound Lindane: Development of Molecular Imprinting Polymer with Polyoxometalate/Carbon Nitride Nanotubes Composite and Validation. Microchem. J. 2020, 157, 105012. [Google Scholar] [CrossRef]
- Pichon, V. Selective Sample Treatment Using Molecularly Imprinted Polymers. J. Chromatogr A 2007, 1152, 41–53. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Xia, B.; Liu, J.; Ding, L.; Zhou, Y. Determination of Kepone and Its Metabolite in Water and Soil by High-Performance Liquid Chromatography–Mass Spectrometry. Anal. Lett. 2015, 48, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Thibert, V.; Legeay, P.; Chapuis-Hugon, F.; Pichon, V. Synthesis and Characterization of Molecularly Imprinted Polymers for the Selective Extraction of Cocaine and Its Metabolite Benzoylecgonine from Hair Extract before LC–MS Analysis. Talanta 2012, 88, 412–419. [Google Scholar] [CrossRef]
- Boulanouar, S.; Combès, A.; Mezzache, S.; Pichon, V. Synthesis and Application of Molecularly Imprinted Polymers for the Selective Extraction of Organophosphorus Pesticides from Vegetable Oils. J. Chromatogr. A 2017, 1513, 59–68. [Google Scholar] [CrossRef]
- Varenne, F.; Kadhirvel, P.; Bosman, P.; Renault, L.; Combès, A.; Pichon, V. Synthesis and Characterization of Molecularly Imprinted Polymers for the Selective Extraction of Oxazepam from Complex Environmental and Biological Samples. Anal. Bioanal. Chem. 2021. [Google Scholar] [CrossRef]
- Kadhirvel, P.; Combès, A.; Bordron, L.; Pichon, V. Development and Application of Water-Compatible Molecularly Imprinted Polymers for the Selective Extraction of Carbamazepine from Environmental Waters. Anal. Bioanal Chem 2019, 411, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Pichon, V.; Haupt, K. Affinity Separations on Molecularly Imprinted Polymers with Special Emphasis on Solid-Phase Extraction. J. Liq. Chromatogr. Relat. Technol. 2006, 29, 989–1023. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Template (for MIPs Only) | Monomer | Porogen |
|---|---|---|---|
| MIP/NIP 1 | CLD-OH | MAA | Chloroform |
| MIP/NIP 2 | CLD-OH | 4-VP | ACN |
| MIP/NIP 3 | CLD-OH | 4-VP | Toluene |
| MIP/NIP 4 | CLD-OH | 4-VP | Chloroform |
| Washing Fraction | Elution Fraction | |
|---|---|---|
| MIP 2 | 18 ± 7 | 87 ± 6 |
| NIP 2 | 58 ± 8 | 21 ± 5 |
| Sep-Pack C18 | 0 | 76 ± 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bosman, P.; Combès, A.; Lambert, M.; Lavison-Bompard, G.; Pichon, V. Development and Application of Molecularly Imprinted Polymers for the Selective Extraction of Chlordecone from Bovine Serum. Separations 2021, 8, 237. https://doi.org/10.3390/separations8120237
Bosman P, Combès A, Lambert M, Lavison-Bompard G, Pichon V. Development and Application of Molecularly Imprinted Polymers for the Selective Extraction of Chlordecone from Bovine Serum. Separations. 2021; 8(12):237. https://doi.org/10.3390/separations8120237
Chicago/Turabian StyleBosman, Pauline, Audrey Combès, Marine Lambert, Gwenaëlle Lavison-Bompard, and Valérie Pichon. 2021. "Development and Application of Molecularly Imprinted Polymers for the Selective Extraction of Chlordecone from Bovine Serum" Separations 8, no. 12: 237. https://doi.org/10.3390/separations8120237
APA StyleBosman, P., Combès, A., Lambert, M., Lavison-Bompard, G., & Pichon, V. (2021). Development and Application of Molecularly Imprinted Polymers for the Selective Extraction of Chlordecone from Bovine Serum. Separations, 8(12), 237. https://doi.org/10.3390/separations8120237