
Mechanistic Studies of Oxygen-Atom Transfer (OAT) in the Homogeneous Conversion of N2O by Ru Pincer Complexes




Abstract
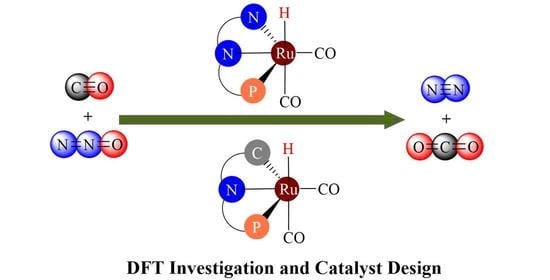
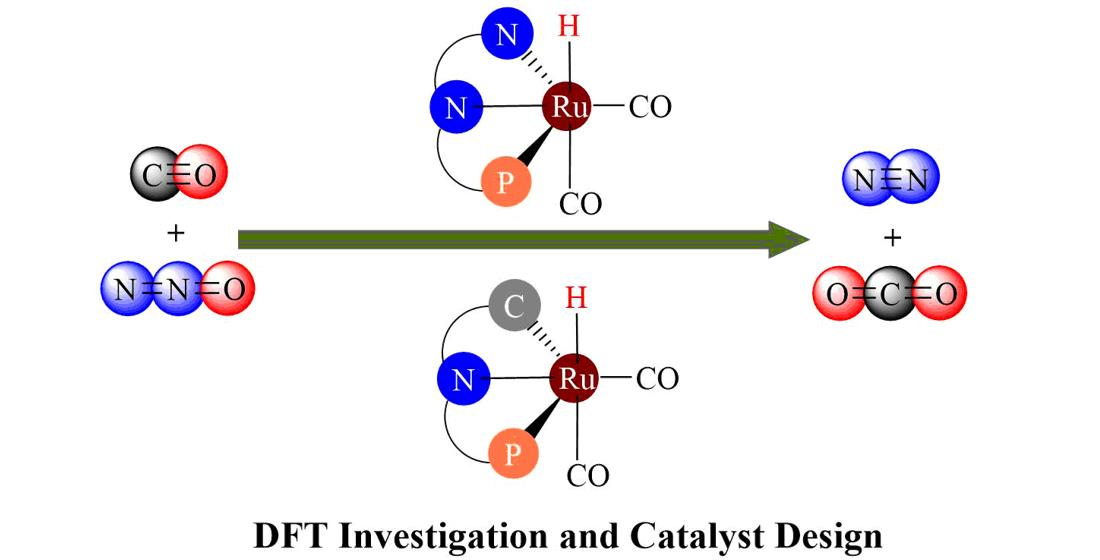
:
1. Introduction
2. Computational Methods
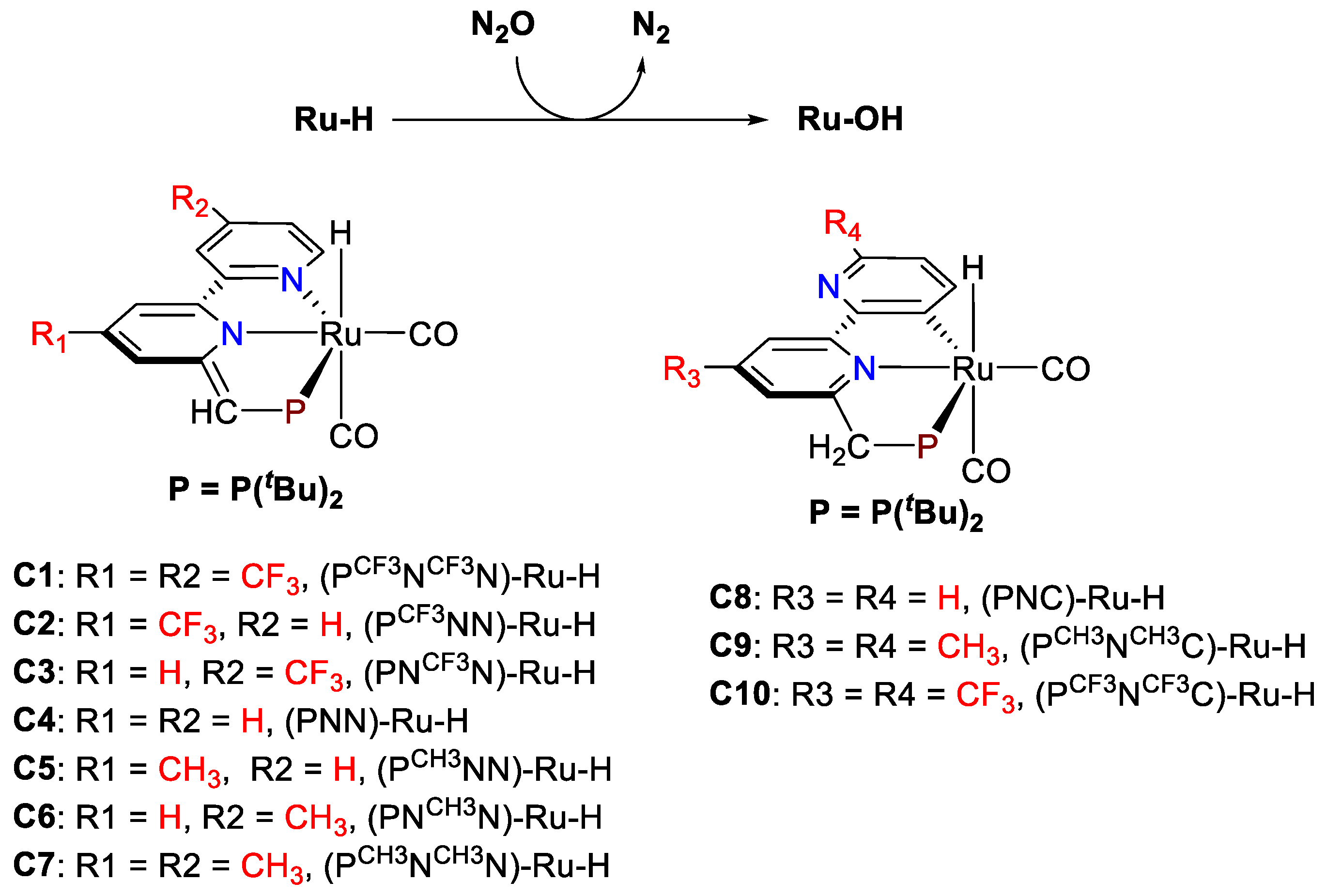
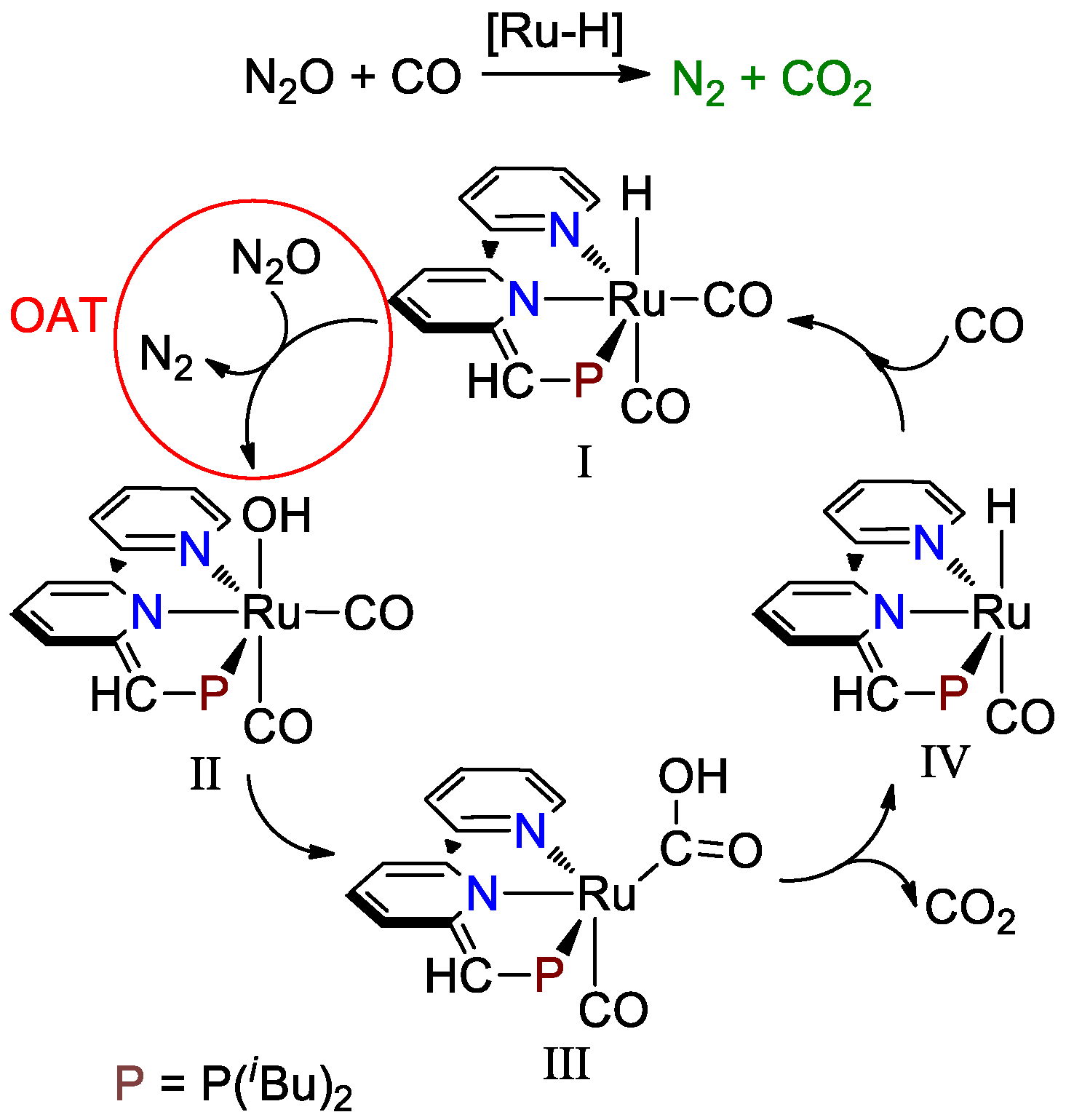
3. Results and Discussion
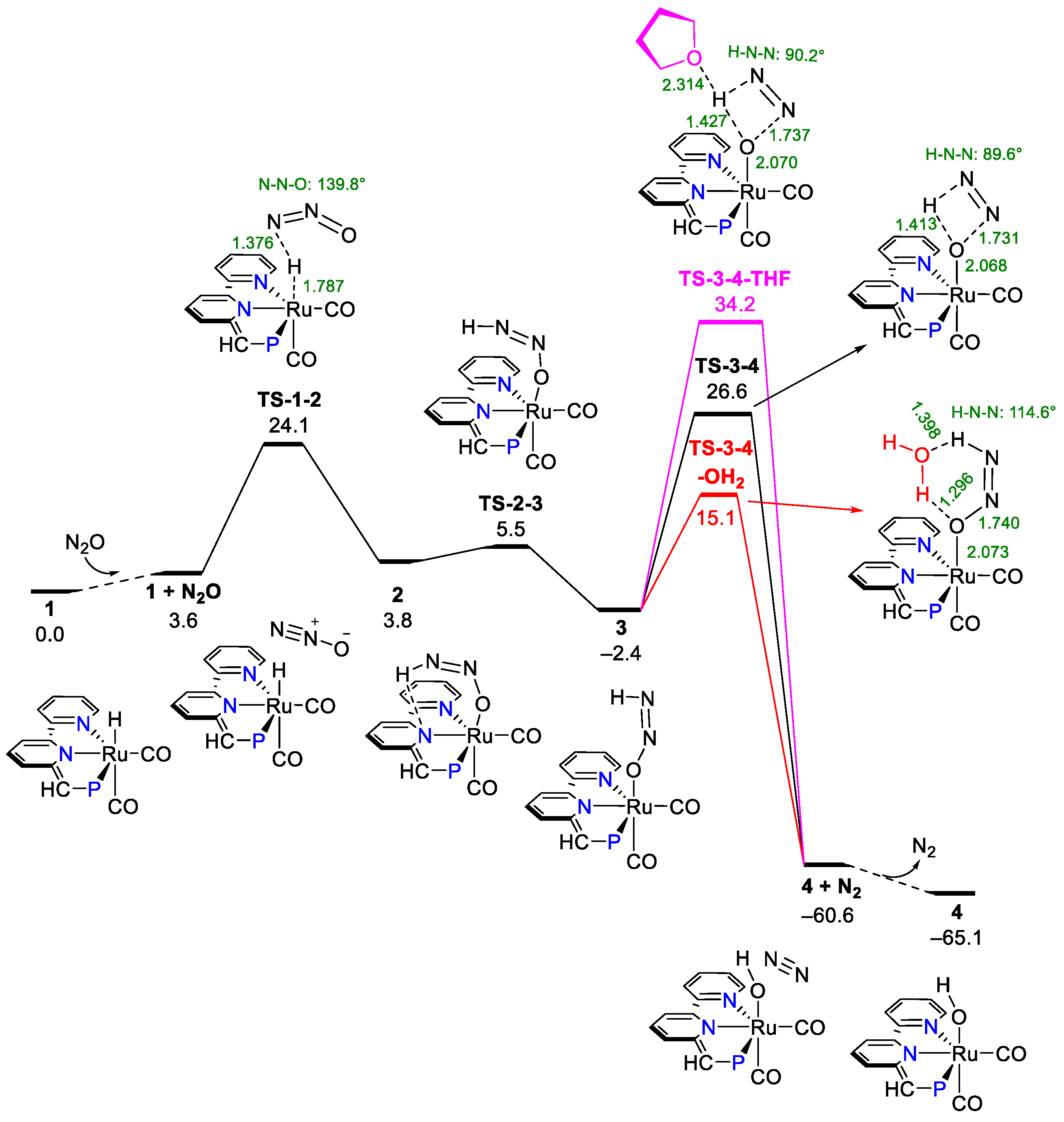
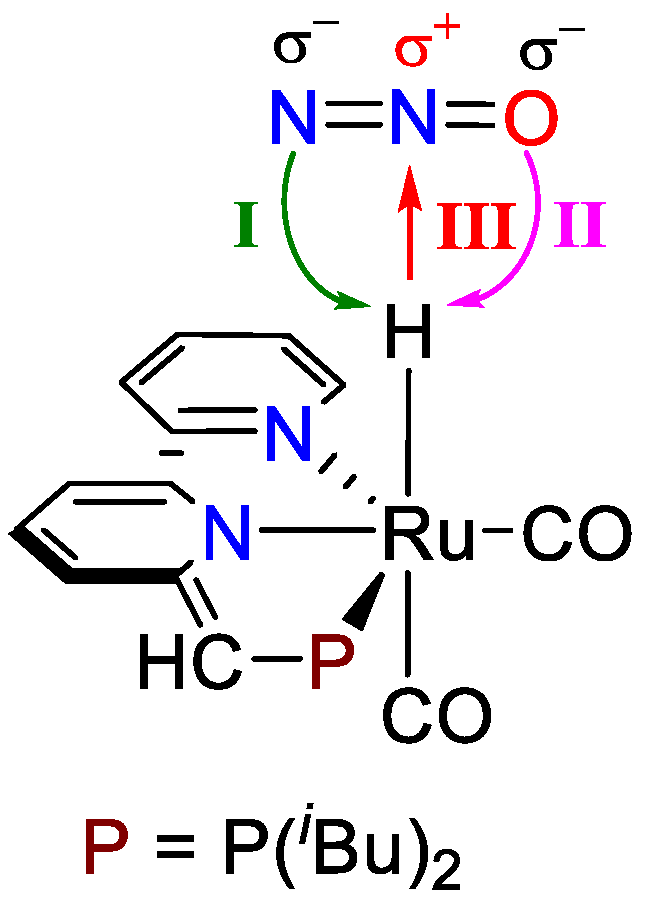
3.1. Proton Transfer from Ru-H to the Terminal N of Endo N2O
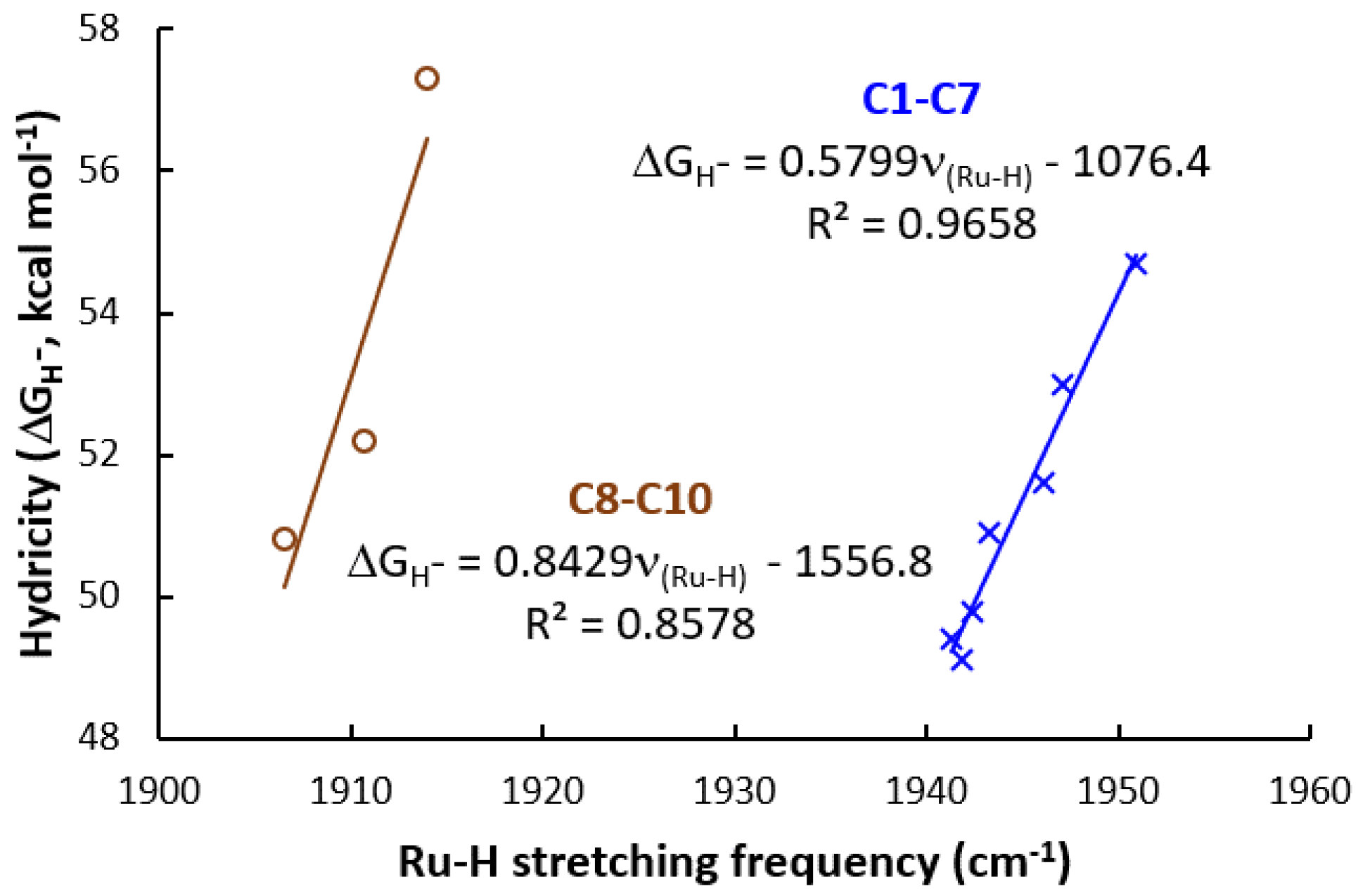
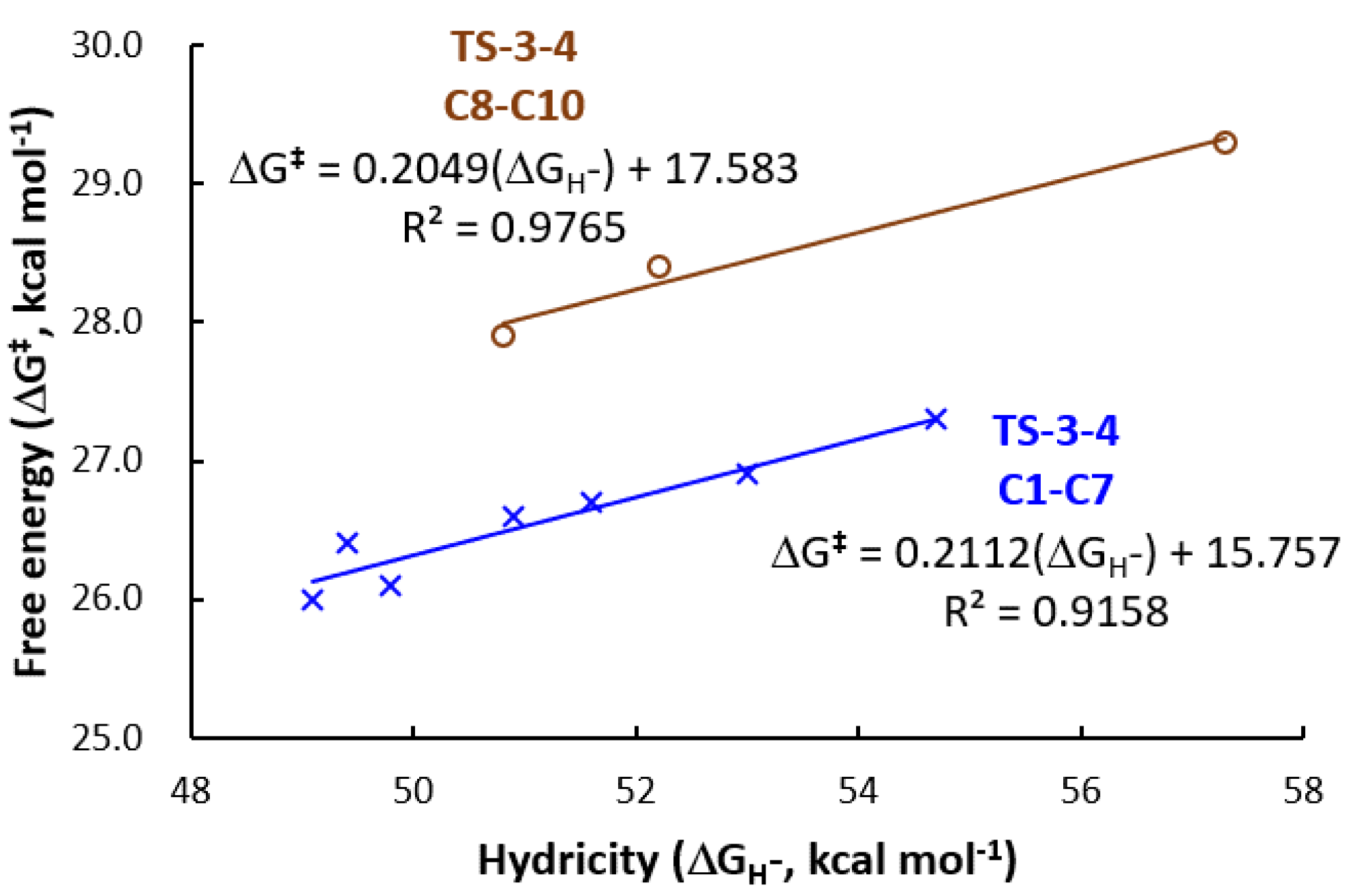
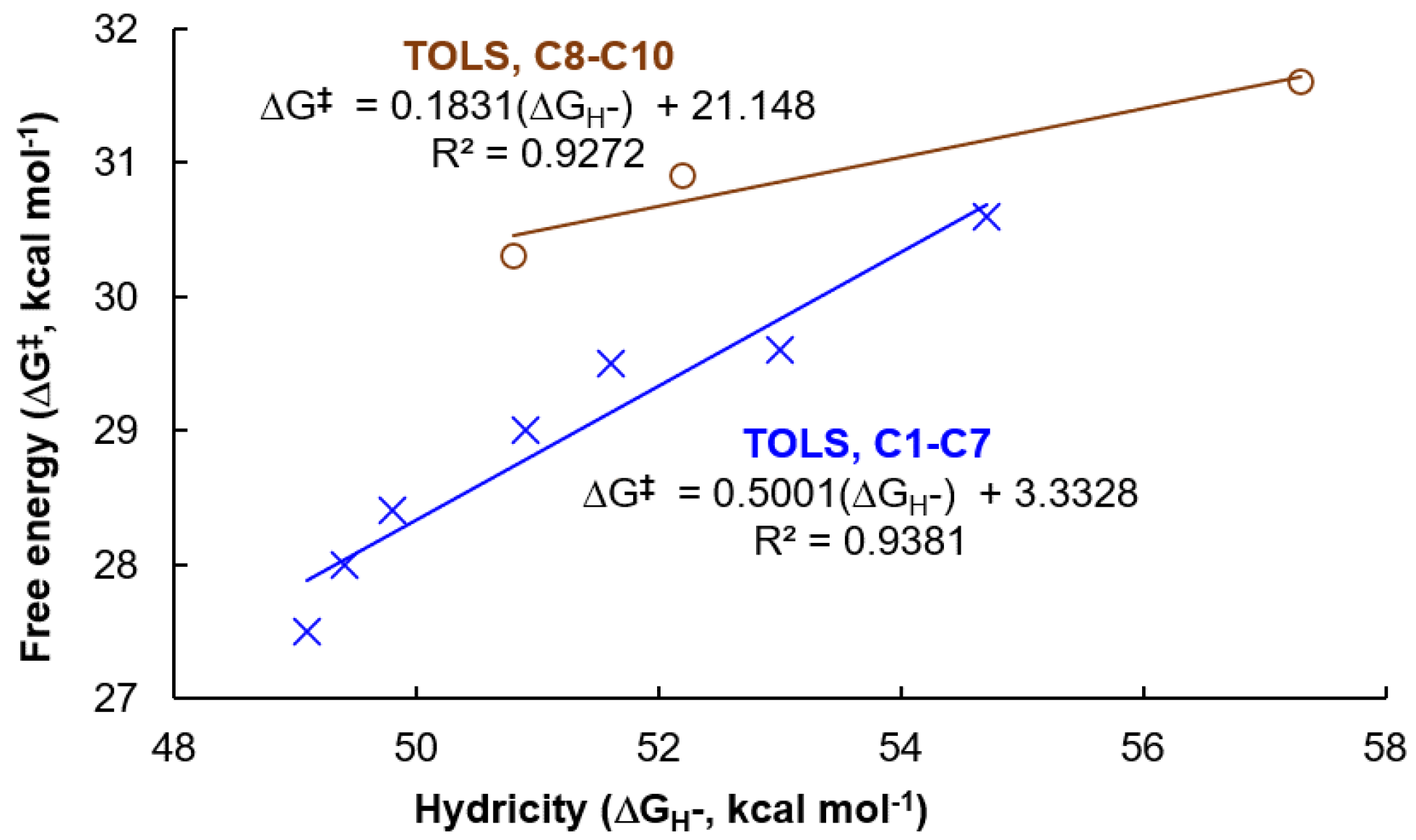
3.2. Hydricity as A Parameter to Predict the Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References and Note
- Ravishankara, A.R.; Daniel, J.S.; Portmann, R.W. Nitrous Oxide (N2O): The Dominant Ozone-Depleting Substance Emitted in the 21st Century. Science 2009, 326, 123–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montzka, S.A.; Dlugokencky, E.J.; Butler, J.H. Non-CO2 greenhouse gases and climate change. Nature 2011, 476, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Chipperfield, M. Nitrous oxide delays ozone recovery. Nat. Geosci. 2009, 2, 742–743. [Google Scholar] [CrossRef]
- Griffis, T.J.; Chen, Z.; Baker, J.M.; Wood, J.D.; Millet, D.B.; Lee, X.; Venterea, R.T.; Turner, P.A. Nitrous oxide emissions are enhanced in a warmer and wetter world. Proc. Natl. Acad. Sci. USA 2017, 114, 12081–12085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Lee, J.W.; Chandran, K.; Kim, S.; Khanal, S.K. Nitrous Oxide (N2O) Emission from Aquaculture: A Review. Environ. Sci. Technol. 2012, 46, 6470–6480. [Google Scholar] [CrossRef] [PubMed]
- IPCC. Climate Change 2013: The Physical Science Basis: Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 2013. [Google Scholar]
- Yang, G.; Peng, Y.; Marushchak, M.E.; Chen, Y.; Wang, G.; Li, F.; Zhang, D.; Wang, J.; Yu, J.; Liu, L.; et al. Magnitude and Pathways of Increased Nitrous Oxide Emissions from Uplands Following Permafrost Thaw. Environ. Sci. Technol. 2018, 52, 9162–9169. [Google Scholar] [CrossRef]
- Richard, S.S.; Anne, R.D.; Luke, D.O.; Darryn, W.W. Impact of future nitrous oxide and carbon dioxide emissions on the stratospheric ozone layer. Environ. Res. Lett. 2015, 10, 034011. [Google Scholar] [CrossRef]
- Kanter, D.; Mauzerall, D.L.; Ravishankara, A.R.; Daniel, J.S.; Portmann, R.W.; Grabiel, P.M.; Moomaw, W.R.; Galloway, J.N. A post-Kyoto partner: Considering the stratospheric ozone regime as a tool to manage nitrous oxide. Proc. Natl. Acad. Sci. USA 2013, 110, 4451–4457. [Google Scholar] [CrossRef] [Green Version]
- Tolman, W.B. Binding and Activation of N2O at Transition-Metal Centers: Recent Mechanistic Insights. Angew. Chem. Int. Ed. 2010, 49, 1018–1024. [Google Scholar] [CrossRef] [Green Version]
- Severin, K. Synthetic chemistry with nitrous oxide. Chem. Soc. Rev. 2015, 44, 6375–6386. [Google Scholar] [CrossRef] [Green Version]
- You, Y.; Chang, H.; Ma, L.; Guo, L.; Qin, X.; Li, J.; Li, J. Enhancement of N2O decomposition performance by N2O pretreatment over Ce-Co-O catalyst. Chem. Eng. J. 2018, 347, 184–192. [Google Scholar] [CrossRef]
- Konsolakis, M. Recent Advances on Nitrous Oxide (N2O) Decomposition over Non-Noble-Metal Oxide Catalysts: Catalytic Performance, Mechanistic Considerations, and Surface Chemistry Aspects. ACS Catal. 2015, 5, 6397–6421. [Google Scholar] [CrossRef]
- Kaczmarczyk, J.; Zasada, F.; Janas, J.; Indyka, P.; Piskorz, W.; Kotarba, A.; Sojka, Z. Thermodynamic Stability, Redox Properties, and Reactivity of Mn3O4, Fe3O4, and Co3O4 Model Catalysts for N2O Decomposition: Resolving the Origins of Steady Turnover. ACS Catal. 2016, 6, 1235–1246. [Google Scholar] [CrossRef]
- Yan, L.; Zhang, X.; Ren, T.; Zhang, H.; Wang, X.; Suo, J. Superior performance of nano-Au supported over Co3O4 catalyst in direct N2O decomposition. Chem. Commun. 2002, 860–861. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Feller, M.; Ben-David, Y.; Milstein, D. Hydrogenation and Hydrosilylation of Nitrous Oxide Homogeneously Catalyzed by a Metal Complex. J. Am. Chem. Soc. 2017, 139, 5720–5723. [Google Scholar] [CrossRef]
- Vaughan, G.A.; Rupert, P.B.; Hillhouse, G.L. Selective O-atom transfer from nitrous oxide to hydride and aryl ligands of bis(pentamethylcyclopentadienyl)hafnium derivatives. J. Am. Chem. Soc. 1987, 109, 5538–5539. [Google Scholar] [CrossRef]
- Matsunaga, P.T.; Hillhouse, G.L.; Rheingold, A.L. Oxygen-atom transfer from nitrous oxide to a nickel metallacycle. Synthesis, structure, and reactions of (2,2’-bipyridine)Ni(OCH2CH2CH2CH2). J. Am. Chem. Soc. 1993, 115, 2075–2077. [Google Scholar] [CrossRef]
- Kaplan, A.W.; Bergman, R.G. Nitrous Oxide Mediated Oxygen Atom Insertion into a Ruthenium−Hydride Bond. Synthesis and Reactivity of the Monomeric Hydroxoruthenium Complex (DMPE)2Ru(H)(OH). Organometallics 1997, 16, 1106–1108. [Google Scholar] [CrossRef]
- Kaplan, A.W.; Bergman, R.G. Nitrous Oxide Mediated Synthesis of Monomeric Hydroxoruthenium Complexes. Reactivity of (DMPE)2Ru(H)(OH) and the Synthesis of a Silica-Bound Ruthenium Complex. Organometallics 1998, 17, 5072–5085. [Google Scholar] [CrossRef]
- Koo, K.; Hillhouse, G.L. Formation of a Substituted Tetrahydrofuran by Formal [2 + 2 + 1] Coupling of an Oxygen Atom with Two Olefins at a Nickel Center. Organometallics 1998, 17, 2924–2925. [Google Scholar] [CrossRef]
- Tskhovrebov, A.G.; Solari, E.; Scopelliti, R.; Severin, K. Reactions of Grignard Reagents with Nitrous Oxide. Organometallics 2014, 33, 2405–2408. [Google Scholar] [CrossRef] [Green Version]
- Laplaza, C.E.; Odom, A.L.; Davis, W.M.; Cummins, C.C.; Protasiewicz, J.D. Cleavage of the Nitrous Oxide NN Bond by a Tris(amido)molybdenum(III) Complex. J. Am. Chem. Soc. 1995, 117, 4999–5000. [Google Scholar] [CrossRef]
- Ni, C.; Ellis, B.D.; Long, G.J.; Power, P.P. Reactions of Ar’CrCrAr’ with N2O or N3(1-Ad): Complete cleavage of the Cr-Cr quintuple interaction. Chem. Commun. 2009, 2332–2334. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Michel, R.; Koley, D.; Roesky, H.W.; Stalke, D. Reactivity Studies of a Disilene with N2O and Elemental Sulfur. Inorg. Chem. 2011, 50, 10878–10883. [Google Scholar] [CrossRef]
- Poh, S.; Hernandez, R.; Inagaki, M.; Jessop, P.G. Oxidation of Phosphines by Supercritical Nitrous Oxide. Org. Lett. 1999, 1, 583–586. [Google Scholar] [CrossRef]
- Ben-Daniel, R.; Neumann, R. Activation of Nitrous Oxide and Selective Oxidation of Alcohols and Alkylarenes Catalyzed by the [PV2Mo10O40]5− Polyoxometalate Ion. Angew. Chem. Int. Ed. 2003, 42, 92–95. [Google Scholar] [CrossRef]
- Tskhovrebov, A.G.; Naested, L.C.E.; Solari, E.; Scopelliti, R.; Severin, K. Synthesis of Azoimidazolium Dyes with Nitrous Oxide. Angew. Chem. Int. Ed. 2015, 54, 1289–1292. [Google Scholar] [CrossRef]
- Lide, D.R. (Ed.) CRC Handbook of Chemistry and Physics, 83rd ed.; CRC Press LLC: Boca Raton, FL, USA, 2002. [Google Scholar]
- Lee, J.-D.; Fang, W.-P.; Li, C.-S.; Cheng, C.-H. Catalytic reduction of nitrous oxide by carbon monoxide in the presence of rhodium carbonyl and hydroxide. Evidence for an electron-transfer and an oxygen-transfer mechanism. J. Chem. Soc. Dalton Trans. 1991, 1923–1927. [Google Scholar] [CrossRef] [Green Version]
- Zeng, R.; Feller, M.; Diskin-Posner, Y.; Shimon, L.J.W.; Ben-David, Y.; Milstein, D. CO Oxidation by N2O Homogeneously Catalyzed by Ruthenium Hydride Pincer Complexes Indicating a New Mechanism. J. Am. Chem. Soc. 2018, 140, 7061–7064. [Google Scholar] [CrossRef]
- Pacultová, K.; Obalová, L.; Kovanda, F.; Jirátová, K. Catalytic reduction of nitrous oxide with carbon monoxide over calcined Co–Mn–Al hydrotalcite. Catal. Today 2008, 137, 385–389. [Google Scholar] [CrossRef]
- Ketrat, S.; Maihom, T.; Wannakao, S.; Probst, M.; Nokbin, S.; Limtrakul, J. Coordinatively Unsaturated Metal–Organic Frameworks M3(btc)2 (M = Cr, Fe, Co, Ni, Cu, and Zn) Catalyzing the Oxidation of CO by N2O: Insight from DFT Calculations. Inorg. Chem. 2017, 56, 14005–14012. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Zhang, Y.; Xiang, C.; Li, Y.; Fan, T.; Lei, Q.; Fang, W. Non-innocent PNN ligand is important for CO oxidation by N2O catalyzed by a (PNN)Ru–H pincer complex: Insights from DFT calculations. Dalton Trans. 2018, 47, 15324–15330. [Google Scholar] [CrossRef] [PubMed]
- Luque-Urrutia, J.A.; Poater, A. The Fundamental Noninnocent Role of Water for the Hydrogenation of Nitrous Oxide by PNP Pincer Ru-based Catalysts. Inorg. Chem. 2017, 56, 14383–14387. [Google Scholar] [CrossRef] [PubMed]
- Escayola, S.; Solà, M.; Poater, A. Mechanism of the Facile Nitrous Oxide Fixation by Homogeneous Ruthenium Hydride Pincer Catalysts. Inorg. Chem. 2020, 59, 9374–9383. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter. 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results Obtained With The Correlation Energy Density Functionals Of Becke And Lee, Yang And Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Couty, M.; Hall, M.B. Basis sets for transition metals: Optimized outer p functions. J. Comput. Chem. 1996, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Check, C.E.; Faust, T.O.; Bailey, J.M.; Wright, B.J.; Gilbert, T.M.; Sunderlin, L.S. Addition of polarization and diffuse functions to the LANL2DZ basis set for p-block elements. J. Phys. Chem. A 2001, 105, 8111–8116. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular-Orbital Studies of Organic-Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. Influence of Polarization Functions on Molecular-Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- In Gaussian software, the 6-31G(d’) basis set has the exponent of d polarization functions for C, N, O taken from the 6-311G(d) basis sets, instead of the original arbitrarily assigned exponent of 0.8 used in the 6-31G(d) basis sets. For H, the 6-31G(d’) keyword in Gaussian software utilizes the 6-31G(d) basis sets.
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Liang, G.; Hollis, T.K.; Webster, C.E. Computational Analysis of the Intramolecular Oxidative Amination of an Alkene Catalyzed by the Extreme π-loading N-Heterocyclic Carbene Pincer Tantalum(V) Bis(imido) Complex. Organometallics 2018, 37, 1671–1681. [Google Scholar] [CrossRef]
- Zhang, M.; Liang, G. Understanding the Sigmatropic Shifts of Cyclopenta-2,4-dien-1-yltrimethylsilane in its Diels-Alder Addition. Org. Biomol. Chem. 2021, 19, 1732–1737. [Google Scholar] [CrossRef] [PubMed]
- Witte, J.; Mardirossian, N.; Neaton, J.B.; Head-Gordon, M. Assessing DFT-D3 Damping Functions Across Widely Used Density Functionals: Can We Do Better? J. Chem. Theory Comput. 2017, 13, 2043–2052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; An, K.; Wang, Y.; Wu, Y.-D.; Zhang, X.; Yu, Z.-X.; He, W. A Combined Computational and Experimental Study of Rh-Catalyzed C–H Silylation with Silacyclobutanes: Insights Leading to a More Efficient Catalyst System. J. Am. Chem. Soc. 2021, 143, 3571–3582. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Wang, F. Performing Molecular Dynamics Simulations and Computing Hydration Free Energies on the B3LYP-D3(BJ) Potential Energy Surface with Adaptive Force Matching: A Benchmark Study with Seven Alcohols and One Amine. ACS Phys. Chem. Au 2021, 1, 14–24. [Google Scholar] [CrossRef]
- Xie, H.; Li, Y.; Huang, L.; Nong, F.; Ren, G.; Fan, T.; Lei, Q.; Fang, W. Dehydrogenation of benzyl alcohol with N2O as the hydrogen acceptor catalyzed by the rhodium(I) carbene complex: Insights from quantum chemistry calculations. Dalton Trans. 2016, 45, 16485–16491. [Google Scholar] [CrossRef]
- Yu, H.; Jia, G.; Lin, Z. Theoretical Studies on O-Insertion Reactions of Nitrous Oxide with Ruthenium Hydride Complexes. Organometallics 2008, 27, 3825–3833. [Google Scholar] [CrossRef]
- Yao, L.; Li, Y.; Huang, L.; Guo, K.; Ren, G.; Wu, Z.; Lei, Q.; Fang, W.; Xie, H. A DFT study on the mechanisms of hydrogenation and hydrosilylation of nitrous oxide catalyzed by a ruthenium PNP pincer complex. Comput. Theor. Chem. 2018, 1128, 48–55. [Google Scholar] [CrossRef]
- Murdoch, J.R. What is the rate-limiting step of a multistep reaction? J. Chem. Educ. 1981, 58, 32. [Google Scholar] [CrossRef]
- Kozuch, S.; Shaik, S. How to Conceptualize Catalytic Cycles? The Energetic Span Model. Acc. Chem. Res. 2011, 44, 101–110. [Google Scholar] [CrossRef]
- Wiedner, E.S.; Chambers, M.B.; Pitman, C.L.; Bullock, R.M.; Miller, A.J.M.; Appel, A.M. Thermodynamic hydricity of transition metal hydrides. Chem. Rev. 2016, 116, 8655–8692. [Google Scholar] [CrossRef]
- Ilic, S.; Alherz, A.; Musgrave, C.B.; Glusac, K.D. Thermodynamic and kinetic hydricities of metal-free hydrides. Chem. Soc. Rev. 2018, 47, 2809–2836. [Google Scholar] [CrossRef] [PubMed]
- Jeletic, M.S.; Hulley, E.B.; Helm, M.L.; Mock, M.T.; Appel, A.M.; Wiedner, E.S.; Linehan, J.C. Understanding the Relationship Between Kinetics and Thermodynamics in CO2 Hydrogenation Catalysis. ACS Catal. 2017, 7, 6008–6017. [Google Scholar] [CrossRef]
- Ostericher, A.L.; Waldie, K.M.; Kubiak, C.P. Utilization of Thermodynamic Scaling Relationships in Hydricity To Develop Nickel Hydrogen Evolution Reaction Electrocatalysts with Weak Acids and Low Overpotentials. ACS Catal. 2018, 8, 9596–9603. [Google Scholar] [CrossRef]
- Waldie, K.M.; Ostericher, A.L.; Reineke, M.H.; Sasayama, A.F.; Kubiak, C.P. Hydricity of transition-metal hydrides: Thermodynamic considerations for CO2 reduction. ACS Catal. 2018, 8, 1313–1324. [Google Scholar] [CrossRef] [Green Version]
- Goodfellow, A.S.; Bühl, M. Hydricity of 3D Transition Metal Complexes from Density Functional Theory: A Benchmarking Study. Molecules 2021, 26, 4072. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Liu, Z.; Tang, L.; Li, J.; Yang, Z. Trans Influence of Boryl Ligands in CO2 Hydrogenation on Ruthenium Complexes: Theoretical Prediction of Highly Active Catalysts for CO2 Reduction. Catalysts 2021, 11, 1356. [Google Scholar] [CrossRef]
- Mondal, B.; Neese, F.; Ye, S. Toward Rational Design of 3D Transition Metal Catalysts for CO2 Hydrogenation Based on Insights into Hydricity-Controlled Rate-Determining Steps. Inorg. Chem. 2016, 55, 5438–5444. [Google Scholar] [CrossRef]
- Muckerman, J.T.; Achord, P.; Creutz, C.; Polyansky, D.E.; Fujita, E. Calculation of thermodynamic hydricities and the design of hydride donors for CO2 reduction. Proc. Natl. Acad. Sci. USA 2012, 109, 15657–15662. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
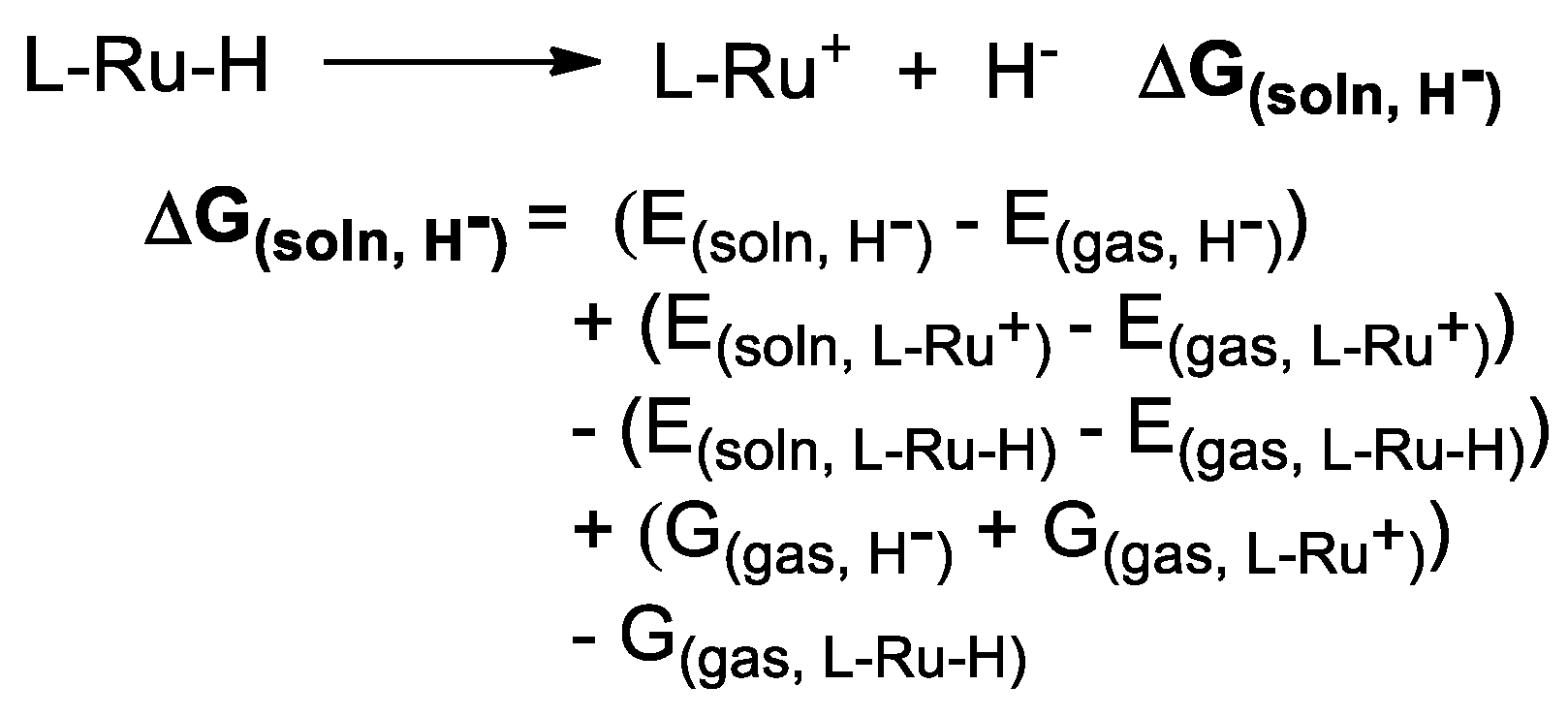{kind=link}
| Catalyst | Hydricity, ΔGH– (kcal mol−1) | Ru-H (cm−1) | TS-1-2 (kcal mol−1) | TS-3-4 (kcal mol−1) | TOLS (kcal mol−1) |
|---|---|---|---|---|---|
| C1, (PCF3NCF3N)-Ru-H | 54.7 | 1950.9 | 24.9 | 27.3 | 30.6 |
| C2, (PCF3NN)-Ru-H | 53.0 | 1947.1 | 24.4 | 26.7 | 29.5 |
| C3, (PNCF3N)-Ru-H | 51.6 | 1946.1 | 24.2 | 26.9 | 29.6 |
| C4, (PNN)-Ru-H | 50.9 | 1943.3 | 24.1 | 26.6 | 29.0 |
| C5, (PCH3NN)-Ru-H | 49.4 | 1941.3 | 24.1 | 26.4 | 28.0 |
| C6, (PNCH3N)-Ru-H | 49.8 | 1942.4 | 23.7 | 26.1 | 28.4 |
| C7, (PCH3NCH3N)-Ru-H | 49.1 | 1941.9 | 23.6 | 26.0 | 27.5 |
| C8, (PNC)-Ru-H | 52.2 | 1910.7 | 24.0 | 28.4 | 30.9 |
| C9, (PCH3NCH3C)-Ru-H | 50.8 | 1906.5 | 23.9 | 27.9 | 30.3 |
| C10, (PCF3NCF3C)-Ru-H | 57.3 | 1914.0 | 24.6 | 29.3 | 31.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, G.; Zhang, M.; Webster, C.E. Mechanistic Studies of Oxygen-Atom Transfer (OAT) in the Homogeneous Conversion of N2O by Ru Pincer Complexes. Inorganics 2022, 10, 69. https://doi.org/10.3390/inorganics10060069
Liang G, Zhang M, Webster CE. Mechanistic Studies of Oxygen-Atom Transfer (OAT) in the Homogeneous Conversion of N2O by Ru Pincer Complexes. Inorganics. 2022; 10(6):69. https://doi.org/10.3390/inorganics10060069
Chicago/Turabian StyleLiang, Guangchao, Min Zhang, and Charles Edwin Webster. 2022. "Mechanistic Studies of Oxygen-Atom Transfer (OAT) in the Homogeneous Conversion of N2O by Ru Pincer Complexes" Inorganics 10, no. 6: 69. https://doi.org/10.3390/inorganics10060069
APA StyleLiang, G., Zhang, M., & Webster, C. E. (2022). Mechanistic Studies of Oxygen-Atom Transfer (OAT) in the Homogeneous Conversion of N2O by Ru Pincer Complexes. Inorganics, 10(6), 69. https://doi.org/10.3390/inorganics10060069