1. Introduction
Carbonic acid, H
2CO
3, is the simple hydrate of carbon dioxide and a ubiquitous molecule whose name has been mistakenly used for carbon dioxide itself at the end of the 19th century [
1]; the H
2CO
3 molecule must have been touched by the alchemists already. As given in any standard chemistry textbook [
2,
3], carbonic acid forms directly from water and carbon dioxide according to the equilibrium equation:
The two acid constants are
KA1 = 1.3 · 10
−4 and
KA2 = 4.84 · 10
−11. Since H
2CO
3 not only behaves as a weak acid but seemingly decays quite easily into the two starting molecules and, hence, makes bubbles in everyday soda water, preparing “free” carbonic acid has been falsely considered plainly impossible for many decades. Already in 1965, however, a pioneering low-temperature direct synthesis in dimethyl ether according to
was successfully carried out [
4] on the gram scale, including an estimate of its exothermic formation enthalpy (−649 ± 42 kJ mol
−1). The astonishing kinetic stability of carbonic acid devoid of any coordinating molecules has also been demonstrated from both experiment and quantum chemistry [
5], including the crucial role of excess water molecules in catalyzing the H
2CO
3 decay, essentially an autocatalytic process.
It does not come as a surprise that characterizing carbonic acid has therefore been mainly performed by spectroscopic methods, in particular as regards infrared data [
6,
7], and even two different polymorphs (α and β) of carbonic acid were claimed based on matrix IR techniques [
7,
8,
9] given prior sublimation to prepare the pure compound. Only recently, however, “α-H
2CO
3” was characterized as being a monomethyl ester of H
2CO
3 while β-H
2CO
3 appears to be the only form of carbonic acid known up to the present day [
10]. Admittedly, parts of the (molecular) literature look slightly controversial, possibly due to spectroscopic ambiguities (frequency and phase assignments, see above), including hidden priority claims.
Despite the fundamental character of H
2CO
3 and its (biochemical) paramount importance for the regulation of the blood’s pH, the acidification of the world oceans, its predicted chemistry in the outer planets, and a true plethora of other issues (see references above and further references within), its crystal structure is still unknown, seemingly mirroring tremendous experimental challenges. Hence, the shape of the molecule in the crystal, the intermolecular connectivity, the details of the hydrogen bonds, the entire solid-state energetics and other related chemical and also physical questions are still unsolved, at least experimentally. With regard to theory, however, there have been density-functional/force-field attempts [
11] and a groundbreaking DFT prediction for H
2CO
3 having orthorhombic
Pnma symmetry at about 1 GPa pressure (and beyond) based on evolutionary algorithms [
12], in harmony with high-pressure spectroscopic data [
13]. This has been followed by the proposal of a unit cell for high-pressure water-CO
2 phases [
14] being of triclinic, not orthorhombic symmetry, however. The present study aims to experimentally determine the crystal structure of carbonic acid at high pressure.
2. Results and Discussion
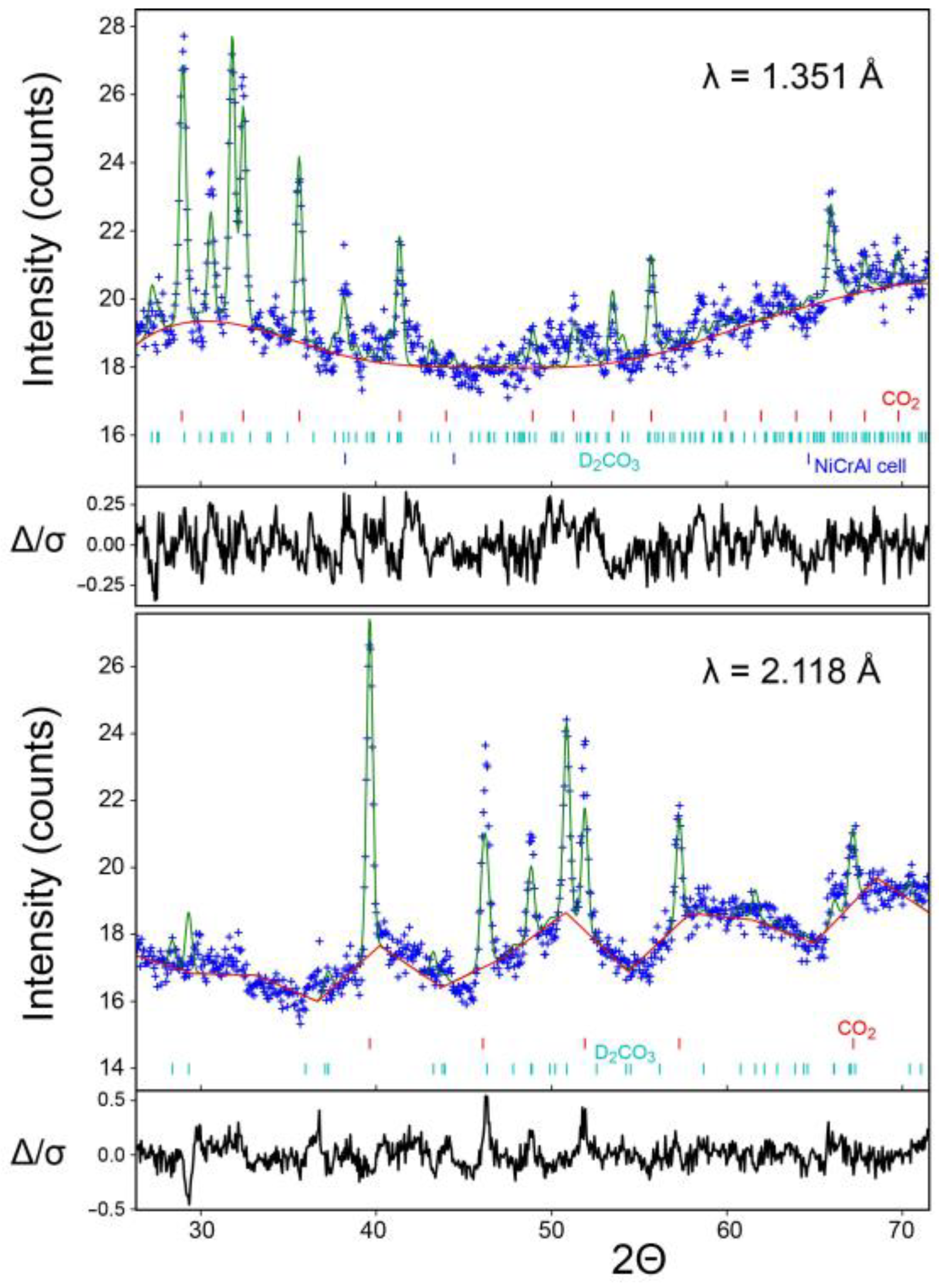
To solve the puzzle, in particular the necessity of correctly localizing the hydrogen atoms and prevent the notorious problem of incoherent neutron-protium scattering, a neutron-diffraction study on
deuterated carbonic acid, D
2CO
3, seemed similar to an optimum choice but this would require a comparatively large sample volume of about 0.4 mL for GPa conditions not being commercially available; earlier unpublished calculations of ours assuming approximate isotypism of H
2CO
3 with urea firmly indicated stability slightly below 2 GPa [
15]. Hence, we started with almost equimolar solid mixtures of crystalline H
2O and CO
2 manipulated at liquid-nitrogen temperature [
16] for orienting tests using tiny amounts with a standard diamond anvil cell and laboratory X-ray diffraction. For the decisive neutron-diffraction experiment, a much larger D
2O/CO
2 mixture with an intentional CO
2 excess was ground and inserted into a unique self-built pre-cooled hybrid pressure cell [
17] which was closed and pressurized with a load of 4 tons. After returning to room temperature, it was further pressurized to a final pressure of 1.85 GPa as measured in situ by ruby fluorescence [
17]. Neutron powder diffraction patterns were collected at the STRESS-SPEC instrument [
18]. Three phases (D
2CO
3, CO
2, and the Ni-Cr-Al fcc alloy of the container) were simultaneously taken into account for the neutron-data analysis, with no experimental evidence whatsoever for additional phases. Manual structure solution inspired by the previous structure proposal [
12] and further Rietveld refinements were carried out [
19] but a reduction to the
translationengleich subgroup
P112
1/
a (=
P2
1/
c) was needed and yielded unit cell parameters as given in
Table A1. Excess CO
2-I served as an internal standard for the isotropic atomic displacement parameters while the
Uiso parameters of the deuterium atoms were based on the D
2O-VIII neutron study [
20]. All refined atomic parameters are found in
Table A2. An overview of the final Rietveld result is provided in
Figure 1. The ruby-determined pressure of 1.85 GPa was also independently verified [
21,
22,
23].
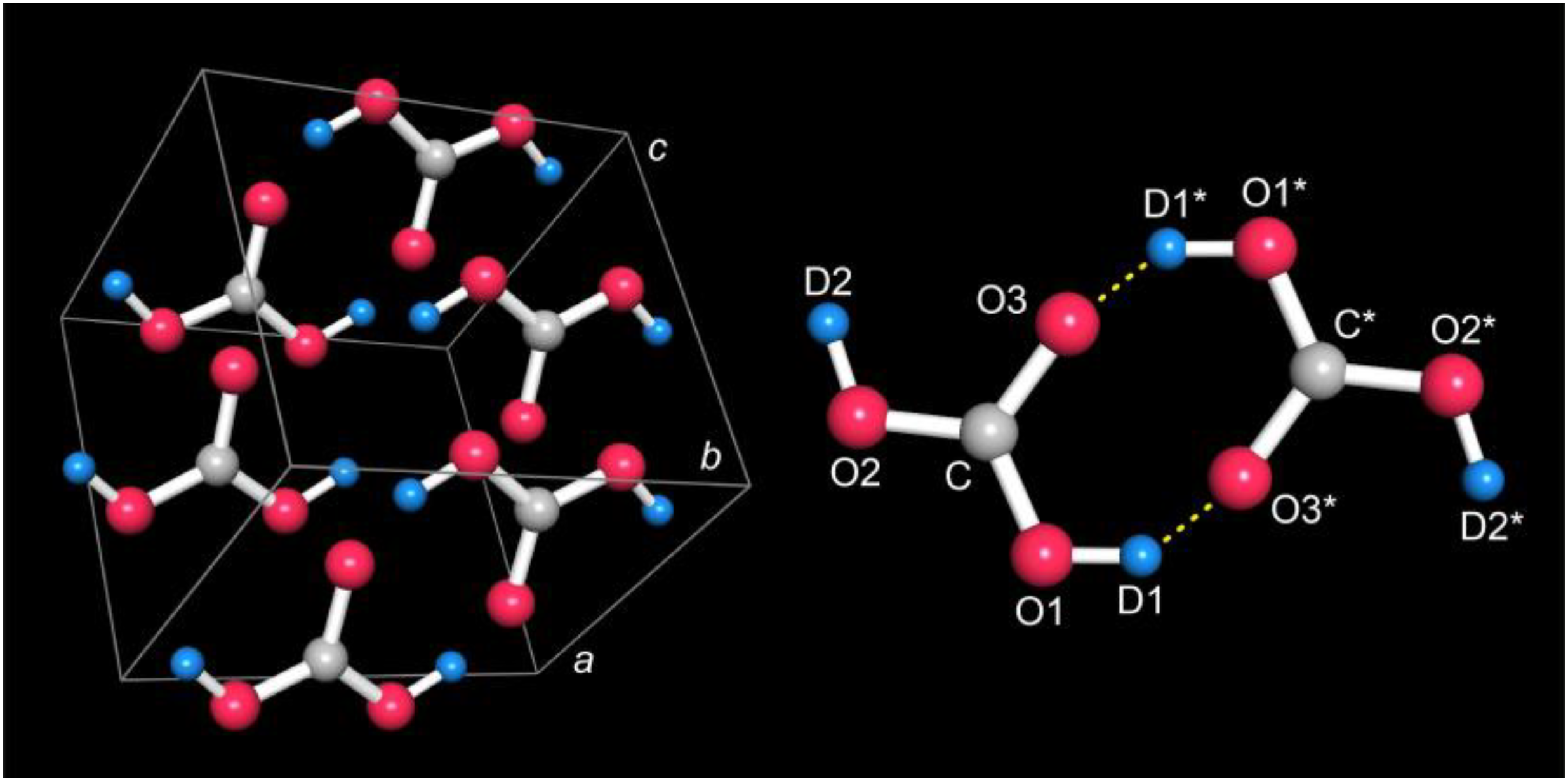
As regards the crystal structure itself, the unit cell of carbonic acid contains just one symmetry-independent D
2CO
3 molecule adopting the
anti-anti conformation and four molecular units in total, see
Figure 2 (left). In the D
2CO
3 molecule, the central carbon atom is bonded to the O1 and O2 hydroxyl atoms with
d = 1.33(2) and 1.34(2) Å, clearly too short if compared to a standard C–O single bond (1.43 Å); for atomic labels, see
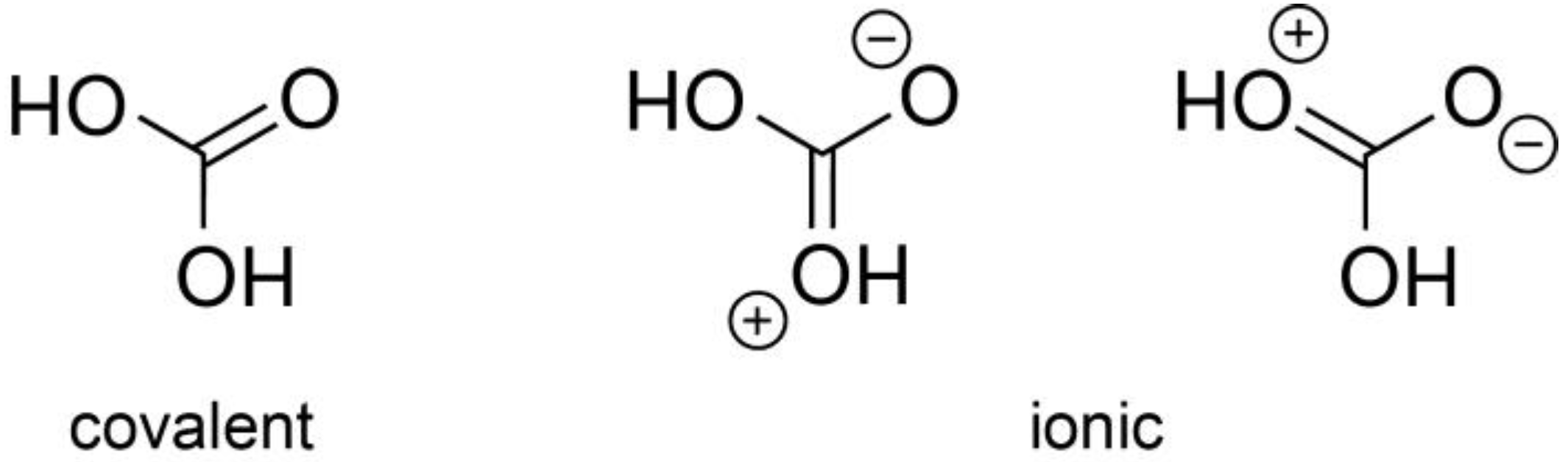
Figure 2 (right). The two hydroxyl O atoms further bond to the D1 and D2 atoms at 0.99(2) Å both. The formally double-bonded O3 atom is found at about the same C–O distance of 1.34(2) Å, this time too long compared to a standard C=O = 1.23 Å double bond. Even given the restricted resolution of the data due to the low neutron counts, both bond-length irregularities must be taken seriously and directly point towards other mesomeric structures in the spirit of valence-bond theory, see
Figure 3, involving delocalized π bonding in the central CO
3 triangle through well-overlapping C 2
pz and O 2
pz atomic orbitals. Hence, it is not surprising that even with a dramatically reduced restraining weight factor, the atoms within D
2CO
3 are in an almost perfect planar arrangement, the π bonding being in control of the molecular shape. We note that this phenomenon has been found before in related molecular units, for example, in guanidinate salts involving a central CN
3 core such as in SrC(NH)
3 [
24].
Two D
2CO
3 molecules arrange in dimers, see
Figure 2 (right), connected via two C–
O1–D1∙∙∙O3*=C* hydrogen bonds with a short O1–O3* distance of 2.13(3) Å and a rather acute O1–D1···O3* angle of 136(5)°. The molecular planes are slightly shifted within the dimer to a distance of about 0.45 Å. We note that the symmetric double hydrogen-bridging bonds would be classified as strong in terms of distance and medium in terms of angle [
26], the angle being less reliable in this case, however (see below). The dimeric structure seemingly resembles the most stable
molecular arrangement of carbonic acid already theoretically predicted in 1997 [
27] and further theoretically explored recently [
28,
29]. At the same time, this very dimer shape makes alternative D split positions with half occupancy less likely, as also corroborated experimentally from significantly worse neutron-data refinements.
Due to the restricted resolution, it is instructive and also advantageous to compare the experimental structural data with those
theoretical ones derived from DFT optimization using plane waves, PAW pseudopotentials, and a DFT-D3(BJ) or HSE exchange-correlation functional, also using
P2
1/
c space-group symmetry [
30,
31,
32,
33,
34,
35,
36,
37,
38,
39,
40]. All geometrical data are found in
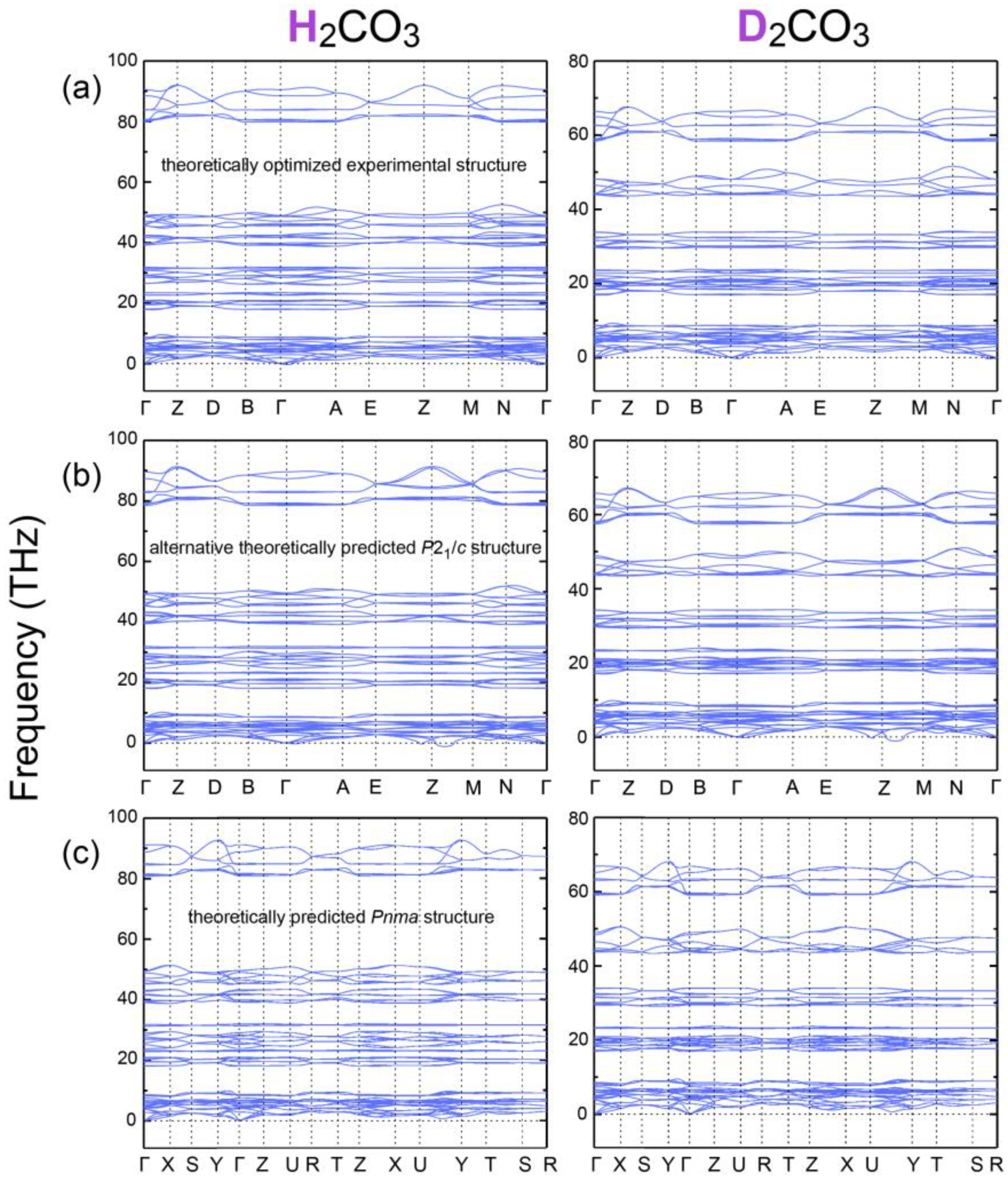
Table A3. While this DFT-optimized experimental structure is mechanically stable in terms of phonons [
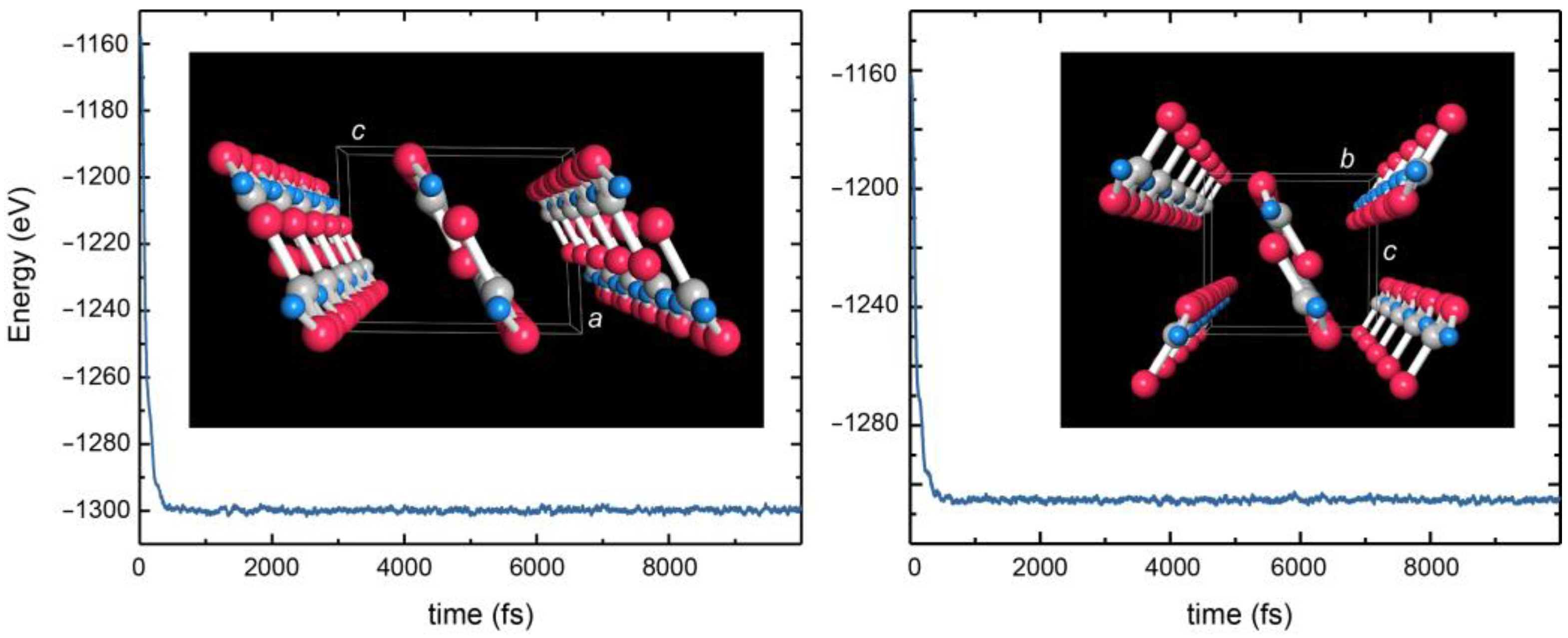
41] showing no imaginary frequencies (
Figure A2), also in an
NPT molecular-dynamics (MD) simulation [
42], see
Figure A3, the C–O1/O2 bonds arrive at an even shorter 1.30 Å instead of the expected single-bond 1.43 Å whereas O–D is confirmed as 1.02 Å. The theoretical O1–O3* distance and O1–D1···O3* angle (as well as O2–D2···O3* because the H-bonding mode becomes slightly more symmetric) widen to 2.57 Å and 176°. Most significant, however, is the C=O3 bond which then comes out shorter (1.26 Å) but not as short as in the prototype double bond. As such, π bonding over the entire CO
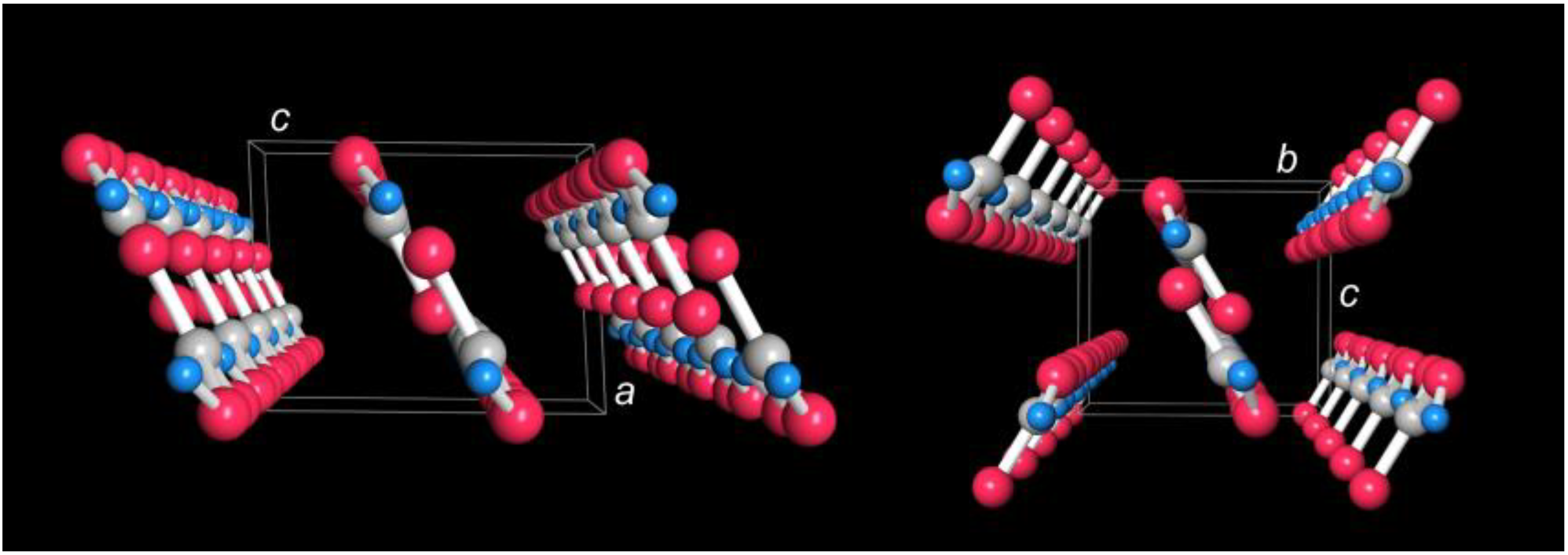
3 triangle is nicely corroborated from DFT, as analyzed in more detail below. In what follows, we will refer to these theoretical bond distances because they look superior to the experimental ones (due to the limited neutron count) while still capturing all the experimentally determined structural motifs. Before doing so, a look at
Figure 4 (left) shows the packing of the dimers which essentially form infinite stripes along the
b direction.
The theoretical free energy of formation of D
2CO
3 at 2 GPa including zero-point energies arrives at −684 kJ mol
−1 at 300 K and slightly lowers to −687 kJ mol
−1 as the temperature cools down to 0 K, assuming a direct reaction from solid D
2 in
P6
3/
mmc [
43], diamond-like C [
44], and solid O
2 in
Cmcm [
44]. This theoretical energy exceeds the 1965 experimental estimate by only 6% and is within a single standard deviation of that value, thus highlighting the applaudable precision of the early contribution.
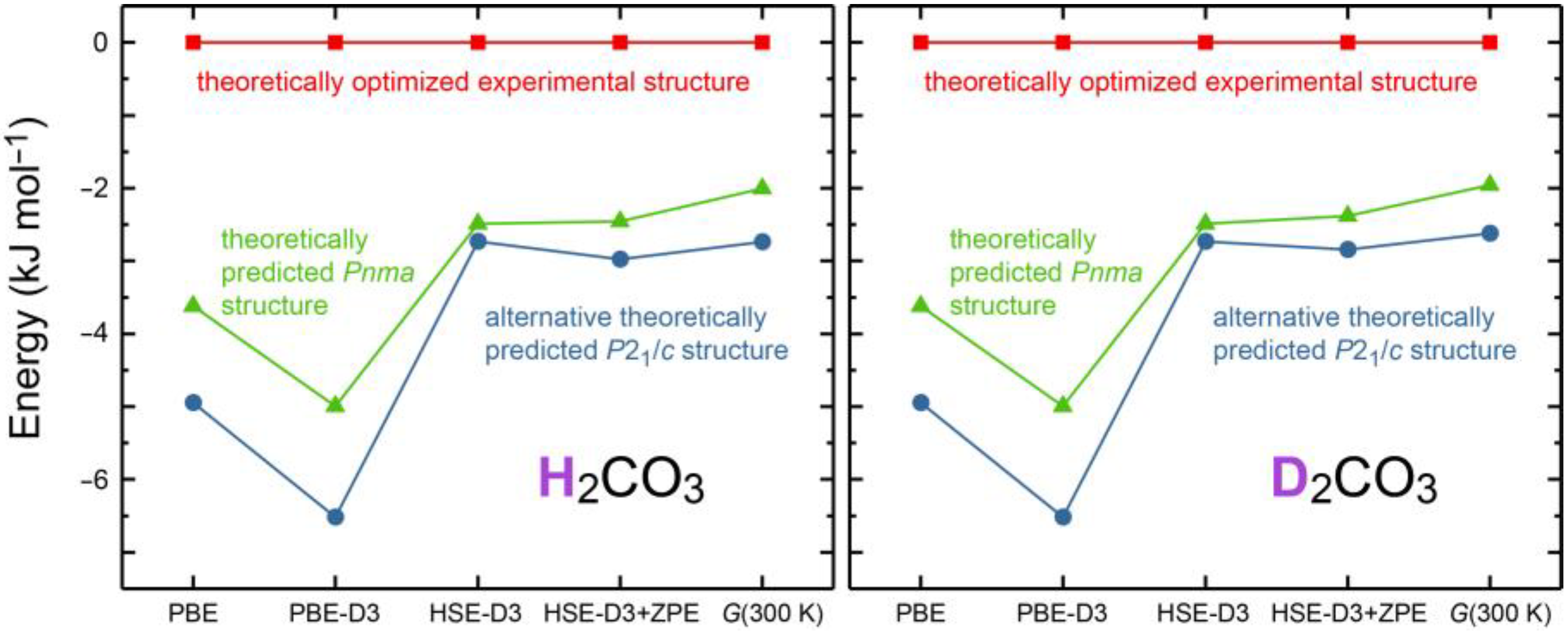
There are two more puzzling theoretical findings which relate to the existence of hypothetic molecular polymorphs. First, the theoretically predicted crystal structure of H
2CO
3 in
Pnma space-group symmetry [
12], also found here by means of structural searches via particle swarm optimization [
45,
46], can be firmly ruled out given the experimental data. It nonetheless turns out as being electronically lower in energy, stabilized by about 5 kJ mol
−1 as calculated by PBE-D3, surprisingly enough. While this small energy difference clearly challenges density-functional theory by its minute size (note that the atomic connectivity is identical), it is at least thinkable that the
Pnma polymorph may exist and can be made using other growing conditions, yet to be found, but only if sufficient activation barriers exist between the polymorphs. Likewise, there is yet another monoclinic
P2
1/
c polymorph including the same D
2CO
3 molecule with identical interatomic distances and O–D···O* angle; its data have been compiled in
Table A4. This hypothetical polymorph, also stable in terms of phonons and
NPT MD simulations (see
Figure A2 and
Figure A3), turns out as being even lower by 1.5 kJ mol
−1 (PBE-D3) and by almost 1 kJ mol
−1 in terms of Gibbs free energies; it exhibits identical dimers and the same connectivity leading to molecular stripes but this time allowing for another stacking, running along the
a direction as shown in
Figure 4 (right); only the difference in stacking is causing the minute energetic change. Further experimental and theoretical studies may evidence whether those two hypothetical polymorphs go back to theoretical deficiencies (say, inaccurate dispersion correction and/or neglect of nuclear anharmonic vibrational contributions) or must be considered real; for an overview of relative energies including various functional approaches, zero-point energy and other corrections as well as (negligible) H/D mass impact by phonon contributions, see
Figure A1. The latter figure makes different polymorphs for H
2CO
3 and D
2CO
3 very unlikely. Likewise, more accurate non-local vdW-DF functionals such as optB88 do not lead to quantitatively different energy courses. If the here found experimental structure were only metastable, Ostwald’s step rule would be at play and render it as the polymorph with the smallest energy barrier for crystallization [
47].
In terms of chemical bonding, an analytical unitary transformation from the delocalized plane-wave picture into a localized representation was carried out [
48,
49,
50,
51,
52]. The various bonds are easily evaluated by their integrated crystal-orbital bond indices (ICOBI) given in
Table A5 [
53]. As anticipated already from the mesomeric structures in
Figure 3 and the bond lengths themselves, the “single” bonds (ICOBI ≈ 1.21, not 1) come out stronger, and the “double” bond (ICOBI ≈ 1.39, not 2) is weaker. Indeed, the π bonding originating from the
pz–
pz interaction alone amounts to a bond order of about ⅓, in harmony with MO theory [
24], see also below. The O–D bonds (ICOBI ≈ 0.66) appear as slightly weaker than expected, a clear consequence of the astonishingly strong hydrogen-bridging bonds (ICOBI ≈ 0.18) into the opposite direction. Upon looking at the isolated monomer (also
Table A5), the “single” bonds become closer to single (ICOBI ≈ 1.08) and the “double” bond becomes closer to double bond character (ICOBI ≈ 1.72) while the isolated O–D hydroxyl bonds (devoid of H bonding) also become more typical (ICOBI ≈ 0.79). Clearly, the bonding of carbonic acid is heavily affected by the periodic structure since the crystal field equilibrates the C–O and O–D bond orders.
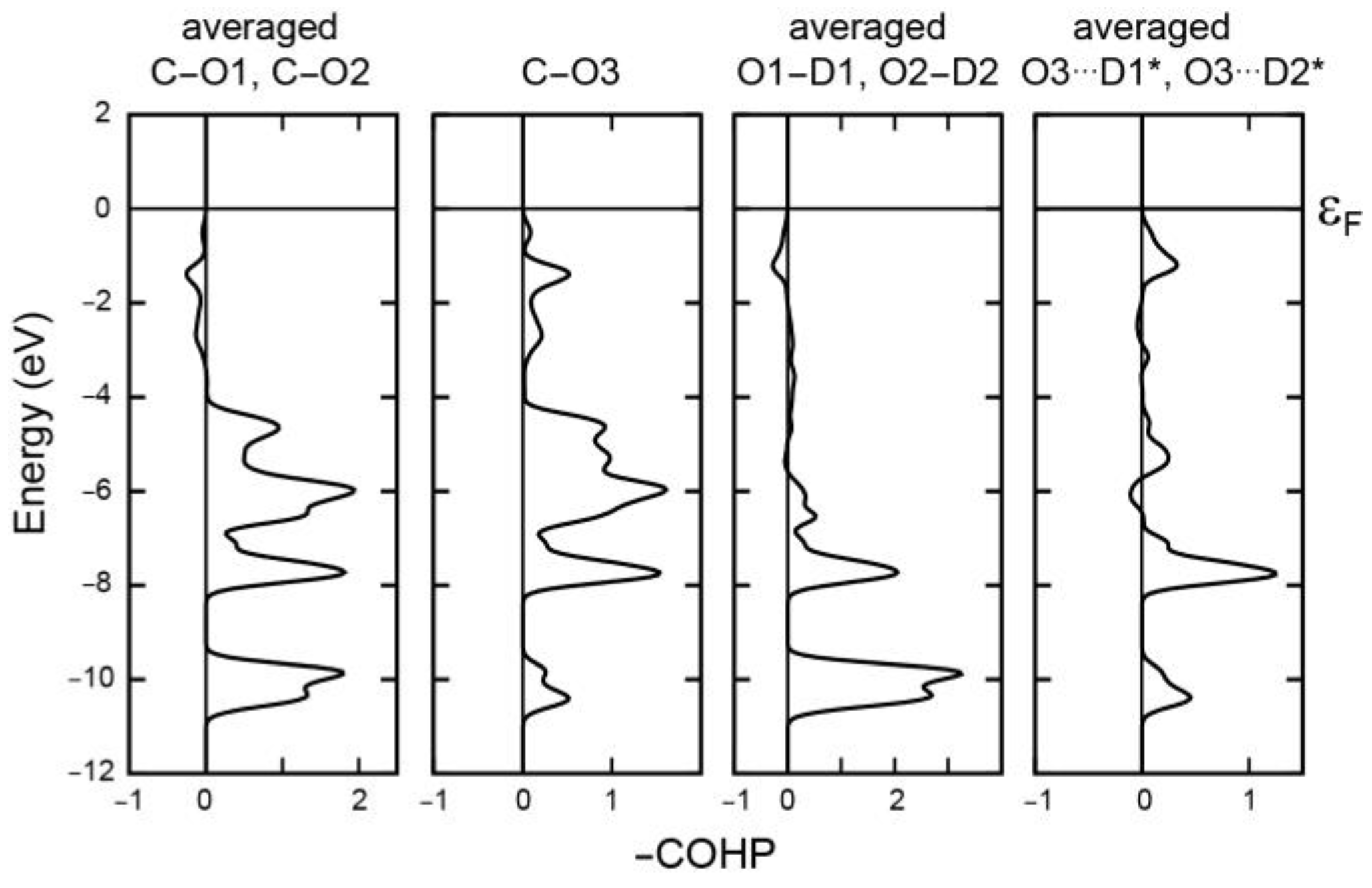
Energywise, similar conclusions can be drawn from Crystal Orbital Hamilton Population (COHP) plots depicted in
Figure 5 whose numerical data are found in
Table A6. The strongest C–O3 “double” bond exclusively exhibits bonding levels over the entire energy region, and the highest π part, between −3 eV and the Fermi level, shows up antibondingly for the “single” C–O1 and C–O2 bonds. A small antibonding level of the hydroxyl O1–D1 and O2–D2 single bonds in the proximity of the Fermi level reappears bondingly in the hydrogen-bridging bonds O3···D1* and O3···D2* which come out exceedingly strong, with ICOHP values of about −1.84 eV (
Table A6) corresponding to ca. 178 kJ mol
−1. A while ago we have shown how covalency contributes to hydrogen bonding, measured by ICOHP [
54], and the current value is even larger than for the H bond in the DFT-optimized benzoylacetone molecule [
54]. While ICOHP does not exactly equal the covalent bond energy but measures the covalent part of the band-structure energy, it nonetheless serves as a reliable numerical criterion, even when density-based schemes (say, bond-critical points etc.) may fail. Clearly, this hydrogen bond is extraordinarily strong.
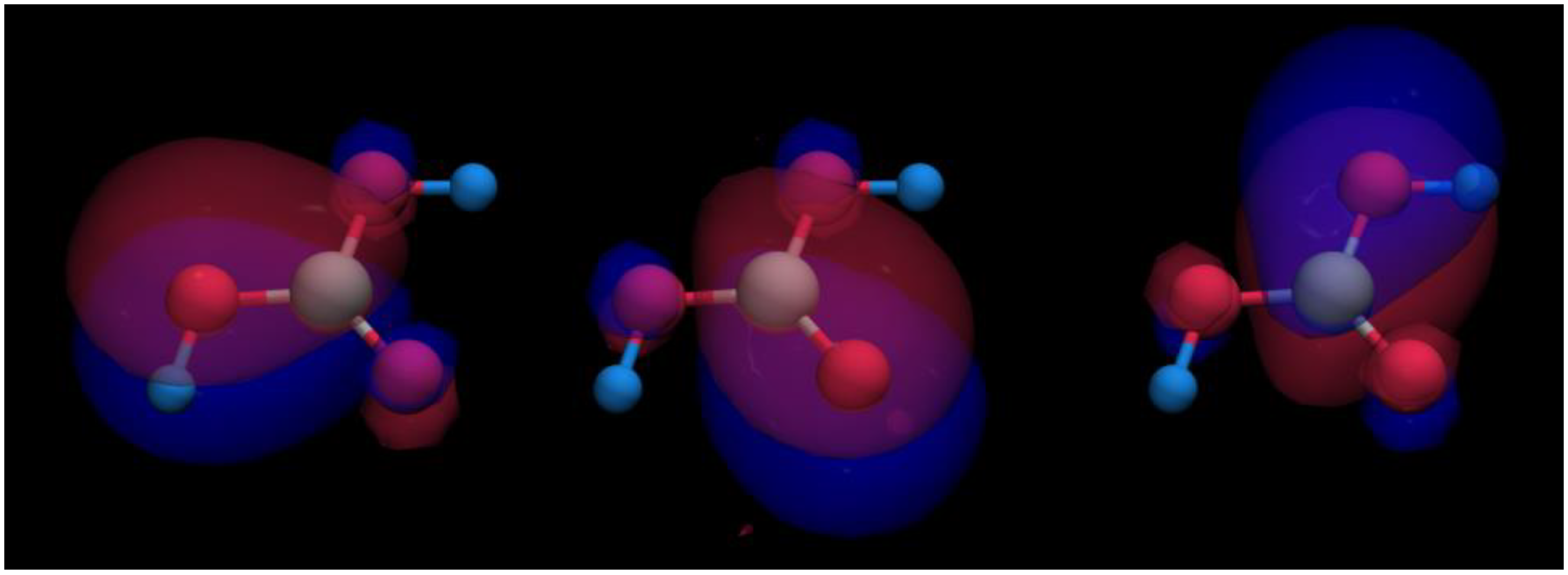
The π bonding already alluded to in
Figure 5 is easily visualized by transforming those bonding interactions between −3 eV and the Fermi level from reciprocal into real space using a molecular-orbital based representation depicted in
Figure A4 [
55,
56]. In the crystal, one finds three visually identical π orbitals of mostly bonding nature, as expected, localized on the three carbon–oxygen bonds, in harmony with the three mesomeric structures depicted in
Figure 3.
In terms of a general comparison of the covalent bonding energetics in carbonic acid in the crystal and in the isolated monomer (without any H bonding), the numerical entries (
Table A6) provide an unexpected finding. When the monomer condenses into the crystal, all intramolecular bonds strengthen quite a lot energywise, in particular the C–O “single” bonds (ca. + 30%) but also the C=O “double” bond (ca. + 2%) and also the O–D hydroxyl bonds (ca. + 4%). As anticipated from the aforementioned bond-order data, the crystal field also affects the intramolecular bond energies, in this case by stabilizing everything collectively.
As regards ionic contributions, the wavefunction-based Löwdin charges as projected from plane waves are given in
Table A7 and arrive at +0.64 for C, −0.42 for the hydroxyl O atoms, −0.50 for the carbonyl O atom, and +0.35 for hydrogen; for the isolated molecule not profiting from a periodic Madelung field, the charges must be somewhat higher (and they are) to establish about the same Coulomb forces. In any case, either solid-state or molecular, ionicity is also at play, besides the dominating covalency. For the crystalline solid, ionicity straightforwardly leads to the aforementioned Madelung field and a (electrostatically defined) lattice energy for crystalline carbonic acid. This electrostatic part of the lattice energy is easily calculated based on Löwdin charges [
53], and it arrives at about −980 kJ mol
−1, a truly substantial amount for a molecular crystal, on the order of rock salt, NaCl. Admittedly, such point-charge model for a molecular solid must be considered with some caution. Interestingly enough, the alternative lowest-energy polymorph proposed by DFT has a 1.9% larger lattice energy, so its stabilization does not only go back to better packing but reflects a collective ionicity-based electrostatic effect.
If we focus on an isolated dimer of carbonic acid in vacuo, the transition state for the concerted deuterium transfer was localized from ab initio molecular-orbital calculations [
34,
57,
58]. The associated barrier arrives at Δ
G = +0.5 kJ mol
−1, which would result in rapid conversion at room temperature. In the extended solid, however, there is a stabilization of the ground state or a destabilization of the transition state by the surrounding periodic units, that is, by the aforementioned Madelung field. Attempts localizing the product resulting from such concerted proton transfer on the periodic potential-energy surface failed, indicating that it is indeed destabilized to the point of not appearing as a stationary point on the latter, in perfect harmony with the neutron data.
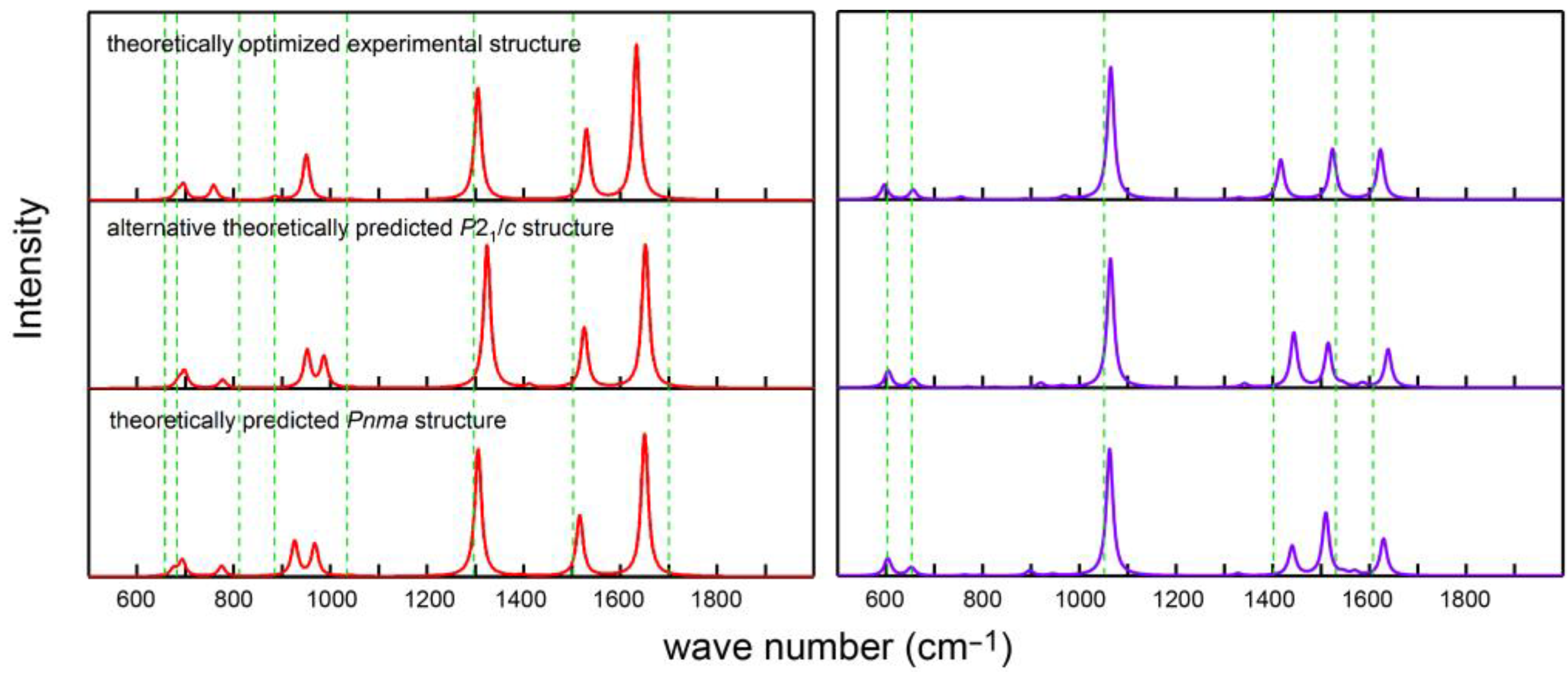
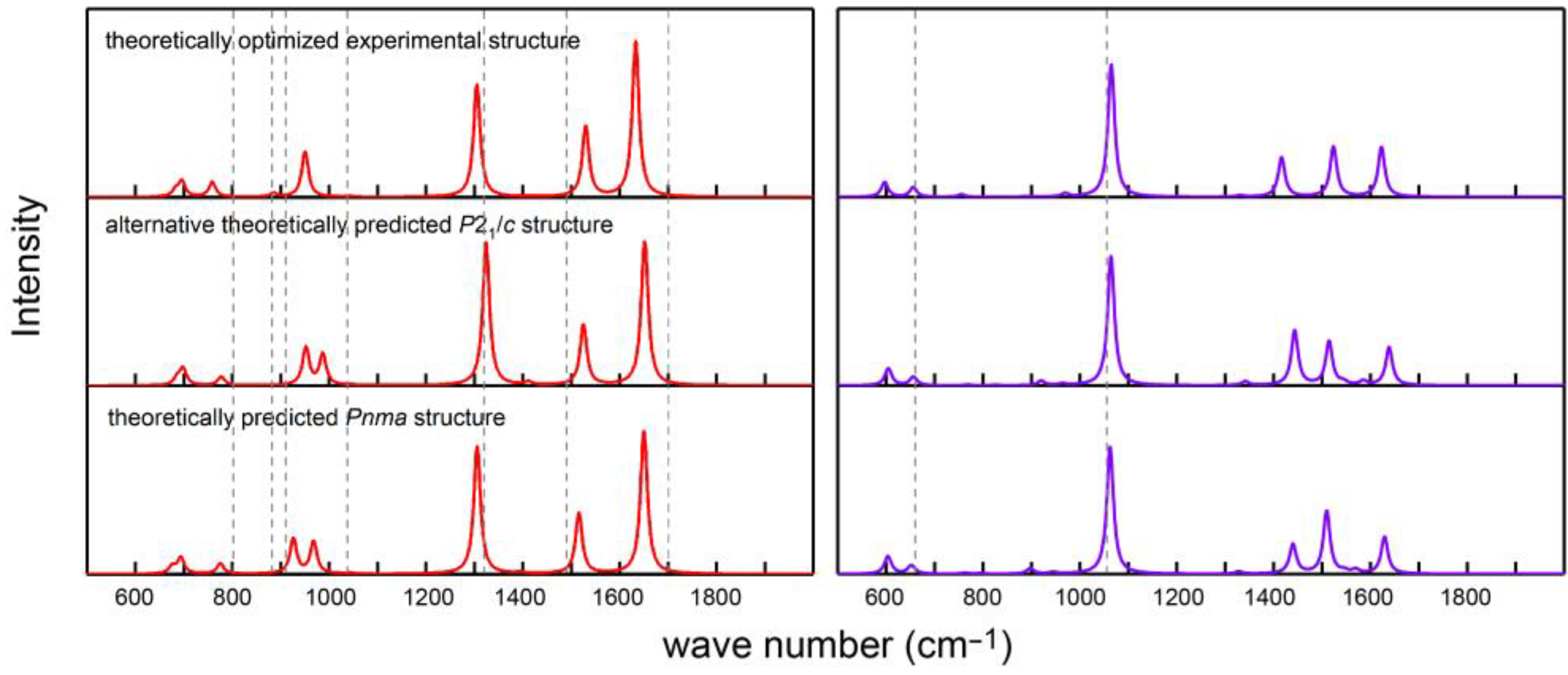
It is instructive to compare the calculated IR and Raman spectra of carbonic acid with those that have been published before.
Figure 6 shows such a comparison with the experimental results [
59] at ambient pressure indicating moderate (IR) to good (Raman) agreement, in particular for the experimental DFT-optimized structure; residual deviations may be related to the neglect of anharmonicity, temperature, in addition to other factors, as previously reported [
12,
60]. On the other hand, the calculations clearly evidence that the positions of the IR and Raman signals are almost unaffected by the crystal symmetry, thereby unsurprisingly highlighting the local nature of the molecular vibrations. It is somewhat surprising, however, that the match between theory and experiment does not become better if one analyzes the high-pressure experimental data [
13] depicted in
Figure A5, in particular because these lack some of the important Raman signals, probably due to experimental difficulties. Nonetheless, the influence of the pressure on the peak positions must be considered weak, simply by visually comparing
Figure 6 with
Figure A5.
The crystal structure of carbonic acid is fundamental in character and also unique, as other simple species for potential comparison are rather rare. One exception would be given by formic acid, H
2CO
2 = HCOOH, also exhibiting symmetric double hydrogen bonding in all known physical phases [
61], namely gas, liquid, solid, matrix, and molecular beam, and its enthalpy of dimerization by two H bonds is 150.7 ± 0.8 kJ mol
−1. A probably even more fitting species to compare to is given by trithio carbonic acid, H
2CS
3, thus including the higher homologue of oxygen. That molecule likewise crystallizes at lower temperature, but this time without external pressure, hence it was structurally clarified decades ago already [
62,
63]. In that pioneering study one finds planar SC(SH)
2 molecules in the same
P2
1/
c space group with mean C=S = 1.65 and C–S = 1.73 Å distances, also linked through hydrogen-bridging bonds but leading to a helical arrangement of chains along the
a direction and three-dimensionally connected by weaker contacts. We also note that this single-crystal X-ray structure analysis of H
2CS
3 made the authors recognize the C–S “single” bonds being shorter than the normal value (1.78 Å) and the C=S “double” bonds longer than anticipated (1.61 Å) such that significant π bonding in the CS
3 triangle originating from C 2
pz and S 3
pz orbitals is indicated, a simple consequence of the molecular shape.
4. Materials and Methods
Neutron powder diffraction patterns were collected at the STRESS-SPEC instrument at the FRM II neutron source in Garching [
18] using two wavelengths, 1.351 and 2.118 Å, and two 2θ ranges of 22.441–75.543° and 22.387–75.599°, respectively. To avoid reflections caused by the pressure apparatus, a radial collimator (field view = 2 mm) was used. Despite the (111), (200), and (220) reflections of the inner-cylinder alloy being visible at 38.25°, 44.45°, and 64.67° in the 1.351 Å diffraction pattern, no other pressure-cell signals were recorded. Three phases (D
2CO
3, CO
2, and the Ni-Cr-Al fcc alloy of the container) were simultaneously taken into account for the neutron-data analysis. There was no evidence whatsoever for additional phases.
Manual structure solution inspired by the previous structure proposal [
12] in space group
Pnma (reproduced by our structure prediction) and further Rietveld refinement were carried out using the GSAS-II program system [
19]. Despite some qualitative similarity of the data with the predicted
Pnma structure for the strongest reflections around
d = 2.35–2.6 Å, orthorhombic symmetry was clearly missing, so a reduction to the
translationengleich subgroup
P112
1/
a (=
P2
1/
c) was needed which eventually yielded unit cell parameters of
a = 5.392(2) Å,
b = 6.660(3) Å,
c = 5.690(1) Å, and
β = 92.66(3)°. Hence, the
Z = 4 unit-cell volume is 204.1 Å
3 and corresponds to a 30.1 cm
3 molar D
2CO
3 volume, thereby questioning the previous triclinic unit cell [
14]. To save parameters in the Rietveld refinement, the atomic distances and angles were softly restrained to C–O = 1.3 Å, O–D = 0.98 Å, O–C–O = 120°, and C–O–D = 109.54°, with all atoms also restrained to be in-plane. An alternative C–O restraining model using C–O = 1.43 Å and C=O = 1.23 Å proved inferior for quantum-chemical reasons, see text. Likewise, due to the intended presence of excess carbon dioxide in the form of CO
2-I, its pattern served as an internal standard for the isotropic atomic displacement parameters under identical conditions in terms of
p and
T [
17], namely
Uiso = 0.040 Å
2 for all carbon and 0.056 Å
2 for all oxygen atoms. The
Uiso parameters of the deuterium atoms were set to the 1.47-fold value of those of the neighboring oxygen atoms, as derived from the neutron study on D
2O-VIII at about the same conditions [
20]. Full details concerning the structure determination including two sets of raw diffraction data are available in CIF format and have been deposited as CCDC 2176140–2176141. These data can be obtained free of charge via
www.ccdc.cam.ac.uk/data_request/cif accessed on 14 August 2022, or by emailing
[email protected], or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44-1223-336033.
To verify the pressure measured by ruby fluorescence, it was additionally determined from the reflections of CO
2-I using a three parameter Vinet equation-of-state model [
21,
22] namely,
where
X = (
V/
V0)
⅓,
η =
3/
2(
B0′ − 1),
V0 = 50.63 Å
3,
B0 = 2.54 GPa and
B0′ = 9.23. To obtain reasonable values of
B0′,
B0 and
V0, the molecular volume of CO
2-I was extrapolated to ambient pressure using experimental data [
23]. As such, the molecular volume of 39.633 Å
3 is equivalent to a calculated pressure of 1.85 GPa, in perfect numerical agreement with the pressure determined from ruby fluorescence.
Periodic DFT calculations were performed utilizing VASP 5.4.4.18 [
30,
31,
32,
33] using either the PBE exchange–correlation [
34] or the HSE functional [
35] and hard PAW pseudopotentials [
36,
37] together with dispersion corrections by the DFT-D3(BJ) approach [
38,
39]. Plane-wave basis functions up to an energy cutoff of 850 eV were included in the calculation and an external pressure of 2 GPa was considered throughout. For geometry optimizations, accurate convergence precision and a
k-point spacing of 0.2 Å
−1 were used; single-point calculations were performed using a 6 × 6 × 6 Monkhorst–Pack mesh [
40].
Phonon calculations were conducted using the supercell approach in the PHONOPY code [
41], with calculational parameters as in the static calculation. The phonon calculations also served to calculate the Born charges and dielectric constants for simulating the IR and Raman spectra [
42]. Molecular dynamics (MD) simulations were carried out using
NPT ensembles with a time step of 1 fs as implemented in MedeA VASP code [
32,
33,
37]. A plane-wave energy cutoff of 500 eV and a single Γ point were used. The simulations were performed in 2 × 2 × 2 supercells containing 192 atoms for all structures at 2 GPa and 298 K.
Structural searches were conducted through a particle swarm optimization algorithm implemented in the CALYPSO code [
45,
46] resting on density-functional theory (see above). To ensure the diversity and reliability of the predicted structures, structure simulations were performed with models containing up to four formula units at 1, 2, 10, 20, 30, and 40 GPa, respectively.
The periodic single-point delocalized wavefunctions were unitarily transformed into a complete set of atomic-orbital-like wavefunctions of contracted Slater type by means of LOBSTER 4.1.0 [
48,
49,
50,
51,
52]. The results were further analyzed by calculating Crystal Orbital Bond Indices (COBI), projected Crystal Orbital Hamilton Populations (COHP), wavefunction-based Löwdin charges, and automatic Madelung energies based on the latter.
Molecular bonding orbitals were generated from the plane-wave data using an accurate top-down frozen-density embedding approach [
55] and a subsequent localization following the Pipek–Mezey algorithm [
56], implemented into a development version of LOBSTER.
Molecular optimizations of ground-state and transition-state structures were carried out using Turbomole 7.5.1 [
57] employing a PBE0-D3 hybrid density-functional approach [
34] based on the def2-QZVP basis set [
58]. Stationary points were verified by frequency analysis.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






