
Copper Complexes of Silicon Pyridine-2-olates RSi(pyO)3 (R = Me, Ph, Bn, Allyl) and Ph2Si(pyO)2


Abstract
:
1. Introduction
2. Results and Discussion
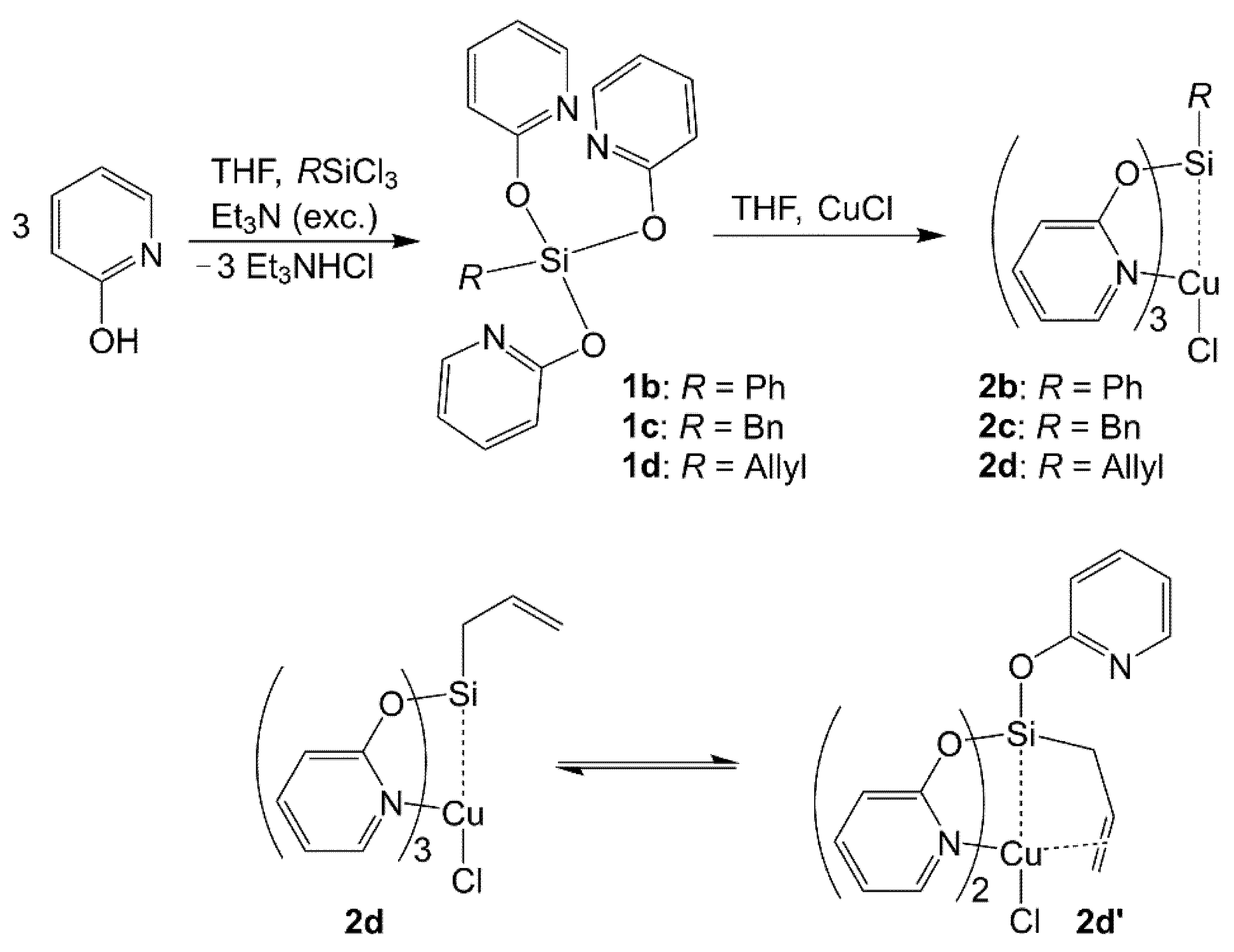
2.1. Syntheses and Characterization of Cu(I) Compounds RSi(μ2-pyO)3CuCl
2.1.1. Syntheses of RSi(μ2-pyO)3CuCl
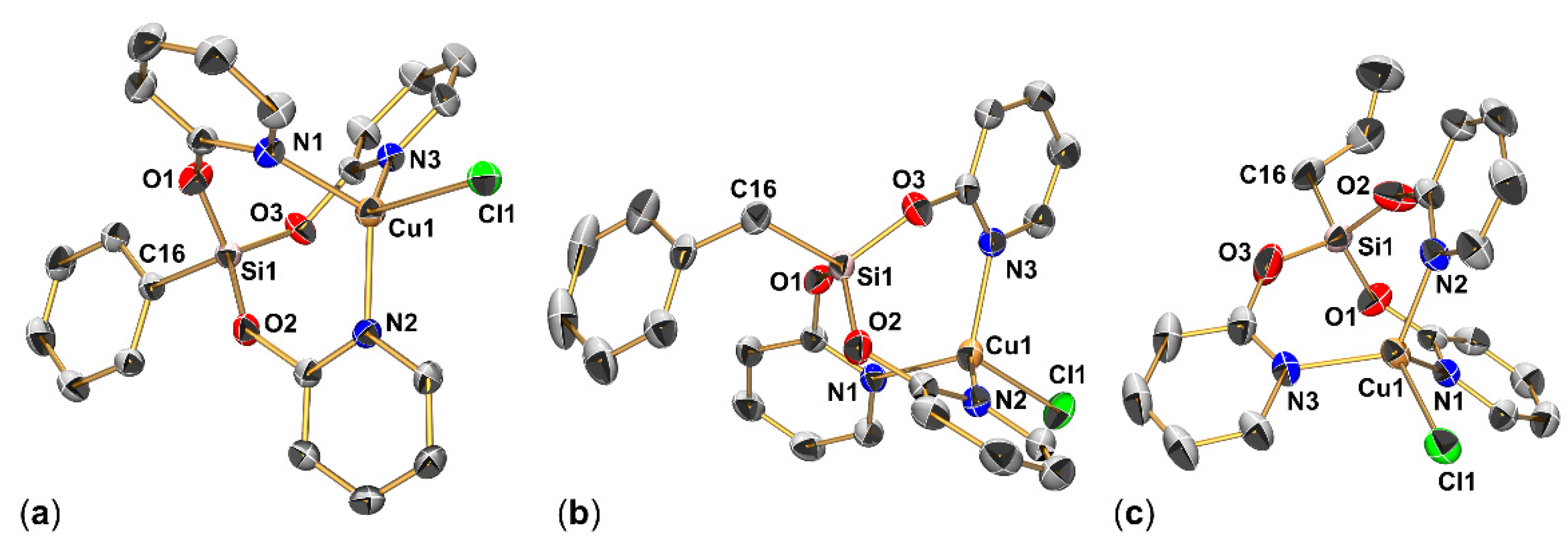
2.1.2. Molecular Structures of RSi(μ2-pyO)3CuCl (R = Me (2a), Ph (2b), Bn (2c), Allyl (2d))
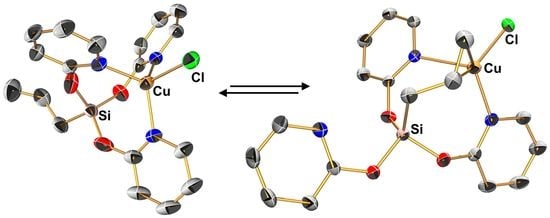
2.1.3. Molecular Structure of (κO-pyO)Si(μ2-pyO)2(Allyl)CuCl (2d′)
2.1.4. Computational Analysis of the Isomerization of 2d and 2d’
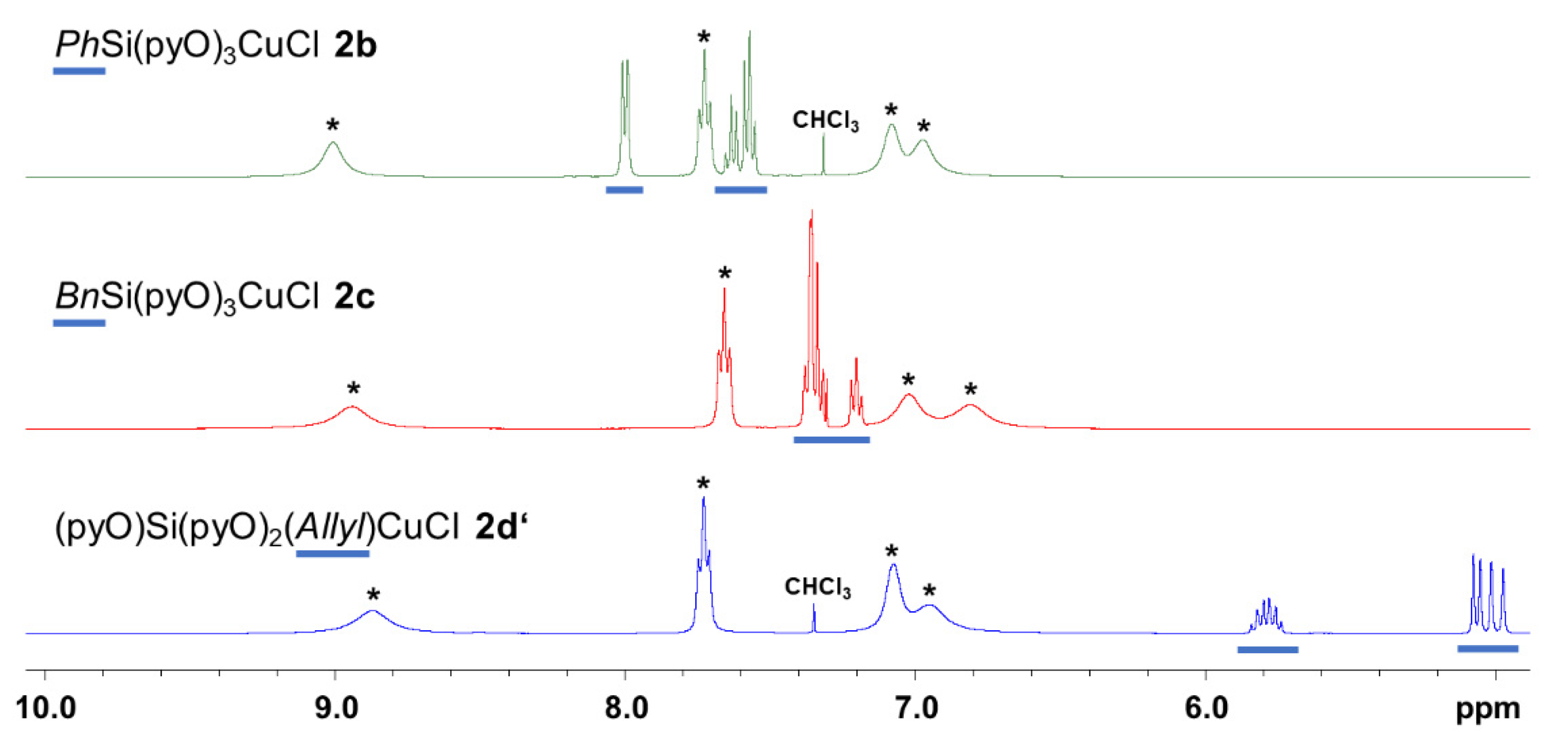
2.1.5. NMR Spectroscopic Analyses of 2b, 2c and 2d′
2.2. Syntheses and Crystallographic Characterization of Cu(II) Compounds RSi(μ2-pyO)4CuCl (R = Me (3a), Ph (3b), Bn (3c), Allyl (3d))
2.3. Reaction of Ph2Si(pyO)2 and CuCl
3. Materials and Methods
3.1. General Considerations
3.2. Syntheses and Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 1c | 1d 1 | (2b)2 (THF) |
|---|---|---|---|
| Formula | C22H19N3O3Si | C18H17N3S3Si | C46H42Cl2Cu2N6O7Si2 |
| Mr | 401.49 | 351.43 | 1045.04 |
| T(K) | 180(2) | 180(2) | 200(2) |
| λ(Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | monoclinic | triclinic | monoclinic |
| Space group | P21/c | P | P21/n |
| a(Å) | 10.6121(6) | 9.7675(8) | 9.1764(4) |
| b(Å) | 17.1874(9) | 10.6315(8) | 18.4453(10) |
| c(Å) | 11.2970(6) | 17.7868(16) | 13.3241(5) |
| α(°) | 90 | 78.951(6) | 90 |
| β(°) | 92.385(4) | 88.972(7) | 91.991(3) |
| γ(°) | 90 | 77.779(6) | 90 |
| V(Å3) | 2058.72(19) | 1771.2(3) | 2253.89(18) |
| Z | 4 | 4 | 2 |
| ρcalc(g·cm−1) | 1.30 | 1.32 | 1.54 |
| μMoKα (mm−1) | 0.1 | 0.2 | 1.2 |
| F(000) | 840 | 736 | 1072 |
| θmax(°), Rint | 28.0, 0.0309 | 25.0, 0.0560 | 28.0, 0.0395 |
| Completeness | 99.9% | 99.8% | 99.9% |
| Reflns collected | 24,896 | 16,879 | 23,599 |
| Reflns unique | 4961 | 4242 | 5437 |
| Restraints | 0 | 20 | 4 |
| Parameters | 263 | 477 | 316 |
| GoF | 1.118 | 1.025 | 1.068 |
| R1, wR2 [I > 2σ(I)] | 0.0423, 0.1032 | 0.0435, 0.0905 | 0.0341, 0.0770 |
| R1, wR2 (all data) | 0.0613, 0.1176 | 0.0794, 0.1011 | 0.0507, 0.0848 |
| Largest peak/hole (e·Å−3) | 0.28, −0.36 | 0.19, −0.26 | 0.39, −0.37 |
| Parameter | 2c 1 | 2d 2 | 2d′ |
|---|---|---|---|
| Formula | C22H19ClCuN3O3Si | C18H17ClCuN3O3Si | C18H17ClCuN3O3Si |
| Mr | 500.48 | 450.42 | 450.42 |
| T(K) | 200(2) | 200(2) | 180(2) |
| λ(Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | orthorhombic | monoclinic | triclinic |
| Space group | Pca21 | P21/c | P |
| a(Å) | 15.3617(7) | 9.0369(2) | 8.7413(3) |
| b(Å) | 8.6072(5) | 23.5711(6) | 10.4028(3) |
| c(Å) | 16.3936(7) | 9.6583(2) | 10.7168(3) |
| α(°) | 90 | 90 | 80.052(3) |
| β(°) | 90 | 108.839(2) | 80.903(2) |
| γ(°) | 90 | 90 | 89.273(3) |
| V(Å3) | 2167.58(18) | 1947.10(8) | 947.69(5) |
| Z | 4 | 4 | 2 |
| ρcalc(g·cm−1) | 1.53 | 1.54 | 1.58 |
| μMoKα (mm−1) | 1.2 | 1.3 | 1.4 |
| F(000) | 1024 | 920 | 460 |
| θmax(°), Rint | 28.0, 0.0273 | 28.0, 0.0375 | 28.0, 0.0299 |
| Completeness | 99.9% | 100% | 100% |
| Reflns collected | 20,601 | 32,981 | 19,166 |
| Reflns unique | 5100 | 4701 | 4570 |
| Restraints | 1 | 27 | 0 |
| Parameters | 280 | 285 | 244 |
| GoF | 1.087 | 1.081 | 1.101 |
| R1, wR2 [I > 2σ(I)] | 0.0235, 0.0557 | 0.0336, 0.0790 | 0.0303, 0.0732 |
| R1, wR2 (all data) | 0.0276, 0.0583 | 0.0414, 0.0826 | 0.0353, 0.0753 |
| Largest peak/hole (e·Å−3) | 0.22, −0.24 | 0.51, −0.32 | 0.31, −0.43 |
| Parameter | 3b | 3c | 3d 1 |
|---|---|---|---|
| Formula | C26H21ClCuN4O4Si | C27H23ClCuN4O4Si | C23H21ClCuN4O4Si |
| Mr | 580.55 | 594.57 | 544.52 |
| T(K) | 200(2) | 200(2) | 200(2) |
| λ(Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | orthorhombic | monoclinic | monoclinic |
| Space group | Pbca | P21/c | P21/c |
| a(Å) | 15.7556(8) | 15.8215(9) | 15.2140(3) |
| b(Å) | 18.2565(7) | 9.8221(4) | 9.6459(2) |
| c(Å) | 17.2013(6) | 17.4885(10) | 17.0865(3) |
| α(°) | 90 | 90 | 90 |
| β(°) | 90 | 109.961(4) | 108.677(1) |
| γ(°) | 90 | 90 | 90 |
| V(Å3) | 4947.8(4) | 2554.5(2) | 2375.44(8) |
| Z | 8 | 4 | 4 |
| ρcalc(g·cm−1) | 1.56 | 1.55 | 1.52 |
| μMoKα (mm−1) | 1.1 | 1.0 | 1.1 |
| F(000) | 2376 | 1220 | 1116 |
| θmax(°), Rint | 28.0, 0.0550 | 26.0, 0.0312 | 26.0, 0.0677 |
| Completeness | 99.6% | 99.8% | 100% |
| Reflns collected | 26,190 | 26,505 | 33,151 |
| Reflns unique | 5951 | 5012 | 4670 |
| Restraints | 0 | 0 | 2 |
| Parameters | 334 | 343 | 326 |
| GoF | 1.011 | 1.081 | 1.075 |
| R1, wR2 [I > 2σ(I)] | 0.0356, 0.0787 | 0.0291, 0.0704 | 0.0367, 0.0769 |
| R1, wR2 (all data) | 0.0656, 0.0877 | 0.0412, 0.0747 | 0.0517, 0.0817 |
| Largest peak/hole (e·Å−3) | 0.48, −0.61 | 0.38, −0.41 | 0.31, −0.44 |
| Parameter | 52 | (54) (THF)2 1 | (53) (CHCl3) 2 |
|---|---|---|---|
| Formula | C44H36Cl2Cu2N4O4Si2 | C52H52Cl4Cu4N4O6Si2 | C45H37Cl6Cu3N4O4Si2 |
| Mr | 938.93 | 1281.11 | 1157.28 |
| T(K) | 180(2) | 180(2) | 180(2) |
| λ(Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | triclinic | triclinic | monoclinic |
| Space group | P | P | Pn |
| a(Å) | 9.6434(6) | 9.6236(3) | 8.9766(2) |
| b(Å) | 10.4351(6) | 12.6110(4) | 19.8415(3) |
| c(Å) | 12.0659(7) | 12.8592(4) | 13.5256(3) |
| α(°) | 100.595(5) | 110.726(2) | 90 |
| β(°) | 110.075(5) | 107.692(2) | 92.846(2) |
| γ(°) | 106.546(5) | 98.836(2) | 90 |
| V(Å3) | 1038.34(12) | 1329.53(8) | 2406.06(8) |
| Z | 1 | 1 | 2 |
| ρcalc(g·cm−1) | 1.50 | 1.60 | 1.60 |
| μMoKα (mm−1) | 1.3 | 1.9 | 1.7 |
| F(000) | 480 | 652 | 1168 |
| θmax(°), Rint | 28.0, 0.0293 | 28.0, 0.0387 | 28.0, 0.0547 |
| Completeness | 100% | 100% | 99.9% |
| Reflns collected | 16,743 | 25,647 | 52,253 |
| Reflns unique | 5010 | 6424 | 11,597 |
| Restraints | 0 | 0 | 32 |
| Parameters | 262 | 316 | 603 |
| GoF | 1.072 | 1.038 | 1.086 |
| R1, wR2 [I > 2σ(I)] | 0.0334, 0.0838 | 0.0346, 0.0833 | 0.0330, 0.0697 |
| R1, wR2 (all data) | 0.0391, 0.0865 | 0.0469, 0.0876 | 0.0418, 0.0737 |
| Largest peak/hole (e·Å−3) | 0.90, −0.45 | 0.40, −0.48 | 0.25, −0.46 |
References
- Avent, A.G.; Gehrhus, B.; Hitchcock, P.B.; Lappert, M.F.; Maciejewski, H. Synthesis and characterisation of bis(amino)silylene–nickel(0), –palladium(II), –platinum(0), –platinum(II) and copper(I) complexes. J. Organomet. Chem. 2003, 686, 321–331. [Google Scholar] [CrossRef]
- Paesch, A.N.; Kreyenschmidt, A.-K.; Herbst-Irmer, R.; Stalke, D. Side-Arm Functionalized Silylene Copper(I) Complexes in Catalysis. Inorg. Chem. 2019, 58, 7000–7009. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, Y.; Xie, Y.; Wei, P.; Gilliard, R.J., Jr.; Schwartz, N.A.; Schaefer, H.F., III; von Schleyer, R.P.; Robinson, G.H. Dynamic Complexation of Copper(I) Chloride by Carbene-Stabilized Disilicon. Chem. Eur. J. 2014, 20, 9208–9211. [Google Scholar] [CrossRef]
- Gualco, P.; Amgoune, A.; Miqueu, K.; Ladeira, S.; Bourissou, D. A Crystalline σ Complex of Copper. J. Am. Chem. Soc. 2011, 133, 4257–4259. [Google Scholar] [CrossRef] [PubMed]
- Plotzitzka, J.; Kleeberg, C. [(NHC)CuI−ER3 ] Complexes (ER3 = SiMe2Ph, SiPh3, SnMe3): From Linear, Mononuclear Complexes to Polynuclear Complexes with Ultrashort CuI···CuI Distances. Inorg. Chem. 2016, 55, 4813–4823. [Google Scholar] [CrossRef] [PubMed]
- Plotzitzka, J.; Kleeberg, C. [(18-C-6)K][(N≡C)CuI−SiMe2Ph], a Potassium Silylcyanocuprate as a Catalyst Model for Silylation Reactions with Silylboranes: Syntheses, Structures, and Catalytic Properties. Inorg. Chem. 2017, 56, 6671–6680. [Google Scholar] [CrossRef]
- Klett, J.; Klinkhammer, K.W.; Niemeyer, M. Ligand Exchange between Arylcopper Compounds and Bis(hypersilyl)tin or Bis(hypersilyl)lead: Synthesis and Characterization of Hypersilylcopper and a Stannanediyl Complex with a Cu−Sn Bond. Chem. Eur. J. 1999, 5, 2531–2536. [Google Scholar] [CrossRef]
- Frogley, B.J.; Hill, A.F.; Sharma, M.; Sinha, A.; Ward, J.S. Semi-bridging σ-silyls as Z-type ligands. Chem. Commun. 2020, 56, 3532–3535. [Google Scholar] [CrossRef]
- Nova, A.; Suh, H.-W.; Schmeier, T.J.; Guard, L.M.; Eisenstein, O.; Hazari, N.; Maseras, F. An Unusual Example of Hypervalent Silicon: A Five-Coordinate Silyl Group Bridging Two Palladium or Nickel Centers through a Nonsymmetrical Four-Center Two-Electron Bond. Angew. Chem. Int. Ed. 2014, 53, 1103–1108. [Google Scholar] [CrossRef]
- Green, M.L.H. A new approach to the formal classification of covalent compounds of the elements. J. Organomet. Chem. 1995, 500, 127–148. [Google Scholar] [CrossRef]
- Chuit, C.; Corriu, R.J.P.; Reye, C.; Young, J.C. Reactivity of Penta- and Hexacoordinate Silicon Compounds and Their Role as Reaction Intermediates. Chem. Rev. 1993, 93, 1371–1448. [Google Scholar] [CrossRef]
- Wagler, J.; Böhme, U.; Kroke, E. Higher Coordinated Molecular Silicon Compounds. Struct. Bond. 2014, 155, 29–105. [Google Scholar] [CrossRef]
- Lemière, G.; Millanvois, A.; Ollivier, C.; Fensterbank, L. A Parisian Vision of the Chemistry of Hypercoordinated Silicon Derivatives. Chem. Rec. 2021, 21, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Grobe, J.; Wehmschulte, R.; Krebs, B.; Läge, M. Alternativ-Liganden. XXXII Neue Tetraphosphan-Nickelkomplexe mit Tripod-Liganden des Typs XM’(OCH2PMe2)n(CH2CH2PR2)3-n (M‘ = Si, Ge; n = 0–3). Z. Anorg. Allg. Chem. 1995, 621, 583–596. [Google Scholar] [CrossRef]
- Grobe, J.; Krummen, N.; Wehmschulte, R.; Krebs, B.; Läge, M. Alternativ-Liganden. XXXI Nickelcarbonylkomplexe mit Tripod-Liganden des Typs XM’(OCH2PMe2)n(CH2CH2PR2)3-n (M‘ = Si, Ge; n = 0–3). Z. Anorg. Allg. Chem. 1994, 620, 1645–1658. [Google Scholar] [CrossRef]
- Grobe, J.; Lütke-Brochtrup, K.; Krebs, B.; Läge, M.; Niemeyer, H.-H.; Würthwein, E.-U. Alternativ-Liganden XXXVIII. Neue Versuche zur Synthese von Pd(0)- und Pt(0)-Komplexen des Tripod-Phosphanliganden FSi(CH2CH2PMe2)3. Z. Naturforsch. 2007, 62, 55–65. [Google Scholar] [CrossRef]
- Gualco, P.; Lin, T.-P.; Sircoglou, M.; Mercy, M.; Ladeira, S.; Bouhadir, G.; Pérez, L.M.; Amgoune, A.; Maron, L.; Gabbaï, F.P.; et al. Gold–Silane and Gold–Stannane Complexes: Saturated Molecules as σ-Acceptor Ligands. Angew. Chem. Int. Ed. 2009, 48, 9892–9895. [Google Scholar] [CrossRef]
- Wagler, J.; Brendler, E. Metallasilatranes: Palladium(II) and Platinum(II) as Lone-Pair Donors to Silicon(IV). Angew. Chem. Int. Ed. 2010, 49, 624–627. [Google Scholar] [CrossRef]
- Wahlicht, S.; Brendler, E.; Heine, T.; Zhechkov, L.; Wagler, J. 7-Azaindol-1-yl(organo)silanes and Their PdCl2 Complexes: Pd-Capped Tetrahedral Silicon Coordination Spheres and Paddlewheels with a Pd-Si Axis. Organometallics 2014, 33, 2479–2488. [Google Scholar] [CrossRef]
- Ehrlich, L.; Gericke, R.; Brendler, E.; Wagler, J. (2-Pyridyloxy)silanes as Ligands in Transition Metal Coordination Chemistry. Inorganics 2018, 6, 119. [Google Scholar] [CrossRef]
- Lyssenko, K.A.; Korlyukov, A.A.; Antipin, M.Y.; Knyazev, S.P.; Kirin, V.N.; Alexeev, N.V.; Chernyshev, E.A. The nature of the intramolecular transannular Si···N interaction in crystalline 1-methylsilatrane, as found from X-ray diffraction data. Mendeleev Commun. 2000, 10, 88–90. [Google Scholar] [CrossRef]
- Párkányi, L.; Simon, K.; Nagy, J. Crystal and molecular structure of [beta]-1-phenylsilatrane, C12H17NSi. Acta Crystallogr. B 1974, 30, 2328–2332. [Google Scholar] [CrossRef]
- Párkányi, L.; Nagy, J.; Simon, K. Crystal and molecular structures of γ-1-phenylsilatrane: Some structural features of silatranes. J. Organomet. Chem. 1975, 101, 11–18. [Google Scholar] [CrossRef]
- White, J.M.; Jones, S. Low-temperature structure of allyl silatrane. Acta Crystallogr. C 1999, 55, 962–963. [Google Scholar] [CrossRef]
- Kuß, S.; Brendler, E.; Wagler, J. Molecular Structures of the Pyridine-2-olates PhE(pyO)3 (E = Si, Ge, Sn) − [4+3]-Coordination at Si, Ge vs. Heptacoordination at Sn. Crystals 2022, 12, 1802. [Google Scholar] [CrossRef]
- Seidel, A.; Weigel, M.; Ehrlich, L.; Gericke, R.; Brendler, E.; Wagler, J. Molecular Structures of the Silicon Pyridine-2-(thi)olates Me3Si(pyX), Me2Si(pyX)2 and Ph2Si(pyX)2 (py = 2-Pyridyl, X = O, S), and Their Intra- and Intermolecular Ligand Exchange in Solution. Crystals 2022, 12, 1054. [Google Scholar] [CrossRef]
- Okuniewski, A.; Rosiak, D.; Chojnaki, J.; Becker, B. Coordination polymers and molecular structures among complexes of mercury(II) halides with selected 1-benzoylthioureas. Polyhedron 2015, 90, 47–57. [Google Scholar] [CrossRef]
- Dużak, T.; Kinzhybalo, V.; Ślepokura, K.; Olijnyk, V. Tetraallylsilane π−Complexation: Synthesis and Structure of [Cu5Cl5(CH2−CH=CH2)4Si]. Z. Anorg. Allg. Chem. 2009, 635, 2324–2327. [Google Scholar] [CrossRef]
- Kamei, T.; Fujita, K.; Itami, K.; Yoshida, J. Copper-Catalyzed Allylation of Carbonyl Derivatives Using Allyl(2-pyridyl)silanes. Org. Lett. 2005, 7, 4725–4728. [Google Scholar] [CrossRef]
- Shi, C.; Xi, X.; Hou, Z.; Liu, E.; Wang, W.; Jin, S.; Wu, Y.; Wu, G. Atomic-Level Characterization of Dynamics of Copper Ions in CuAgSe. J. Phys. Chem. C 2016, 120, 3229–3234. [Google Scholar] [CrossRef]
- Endo, K.; Yamamoto, K.; Deguchi, K.; Matsushita, K. 63Cu High Resolution NMR of Cu(I) Halides in Aqueous Solution and Suspension. Bull. Chem. Soc. Jpn. 1987, 60, 2803–2807. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.K.; Becker, E.D.; Cabral de Menzenes, S.M.; Goodfellow, R.; Granger, P. Nuclear spin properties and conventions for the chemical shift. Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar] [CrossRef]
- Sakida, S.; Kato, N.; Kawamoto, Y. 63Cu magic angle spinning (MAS) nuclear magnetic resonance (NMR) study of CuX crystals (X = Cl, Br, and I) and CuX-based glasses (X = Cl, Br, and I). Mat. Res. Bull. 2002, 37, 2263–2274. [Google Scholar] [CrossRef]
- Tang, J.A.; Ellis, B.D.; Warren, T.H.; Hanna, J.V.; Macdonald, C.L.B.; Schurko, R.W. Solid-State 63Cu and 65Cu NMR Spectroscopy of Inorganic and Organometallic Copper(I) Complexes. J. Am. Chem. Soc. 2007, 129, 13049–13065. [Google Scholar] [CrossRef]
- Massiot, D.; Fayon, F.; Capron, M.; King, I.; Le Calvlé, S.; Alonso, B.; Durand, J.-O.; Bujoli, B.; Gan, Z.; Hoatson, G. Modeling one- and two-dimensional Solid State NMR spectra. Magn. Reson. Chem. 2002, 40, 70–76. [Google Scholar] [CrossRef]
- Gericke, R.; Wagler, J. Coordination and Electrochemical Switching on Paddle-Wheel Complexes Containing an As−Ru or a Sb−Ru Axis. Inorg. Chem. 2021, 60, 18122–18132. [Google Scholar] [CrossRef]
- Dattelbaum, A.M.; Martin, J.D. Benzene−Copper(I) Coordination in a Bimetallic Chain Complex. Inorg. Chem. 1999, 38, 6200–6205. [Google Scholar] [CrossRef] [PubMed]
- Dattelbaum, A.M.; Martin, J.D. Benzene and ethylene binding to copper(I)–ziconium(IV) chloride materials: The crystal structure and solid-state reactivity of ((bz)2Cu)2Zr2Cl10 · bz. Polyhedron 2006, 25, 349–359. [Google Scholar] [CrossRef]
- Daly, S.; Haddow, M.F.; Orpen, A.G.; Rolls, G.T.A.; Wass, D.F.; Wingad, R.L. Copper(I) Diphosphine Catalysts for C-N Bond Formation: Synthesis, Structure, and Ligand Effects. Organometallics 2008, 27, 3196–3202. [Google Scholar] [CrossRef]
- Chen, B.-L.; Mok, K.-F.; Ng, S.-C. Synthesis, crystal structures and dynamic NMR studies of novel trinuclear copper(I) halide complexes with 2,5-bis[(diphenylphosphino)-methyl]thiophene. J. Chem. Soc., Dalton Trans. 1998, 2861–2866. [Google Scholar] [CrossRef]
- Collins, L.R.; Lowe, J.P.; Mahon, M.F.; Poulten, R.C.; Whittlesey, M.K. Copper Diamidocarbene Complexes: Characterization of Monomeric to Tetrameric Species. Inorg. Chem. 2014, 53, 2699–2707. [Google Scholar] [CrossRef] [PubMed]
- Crestani, M.G.; Manbeck, G.F.; Brennessel, W.W.; McCormick, T.M.; Eisenberg, R. Synthesis and Characterization of Neutral Luminescent Diphosphine Pyrrole- and Indole-Aldimine Copper(I) Complexes. Inorg. Chem. 2011, 50, 7172–7188. [Google Scholar] [CrossRef] [PubMed]
- Köhn, R.D.; Laudo, L.D.; Pan, Z.; Speiser, F.; Kociok-Köhn, G. Triangular tricopper(I) clusters supported by donor-substituted triazacyclohexanes. Dalton Trans. 2009, 4556–4568. [Google Scholar] [CrossRef] [PubMed]
- Dyason, J.C.; Healy, P.C.; Engelhardt, L.M.; Pakawatchai, C.; Patrick, V.A.; Raston, C.L.; White, A.H. Lewis-base adducts of Group 1B metal(I) compounds. Part 16. Synthesis, structure, and solid-state phosphorus-31 nuclear magnetic resonance spectra of some novel [Cu4X4L4](X = halogen, L = N, P base) ‘cubane’ clusters. J. Chem. Soc. Dalton Trans. 1985, 14, 831–838. [Google Scholar] [CrossRef] [Green Version]
- Noor, A.; Shahzad, A.; Khan, E.; Tahir, M.N.; Khan, G.S.; ur Rashid, A.; Said, M. Polynuclear Cu(I) and Ag(I) Complexes of 1,3-Diisobutyl Thiourea, Synthesis, Crystal Structure and Antioxidant Potentials. Inorganics 2022, 10, 185. [Google Scholar] [CrossRef]
- Kämpfe, A.; Brendler, E.; Kroke, E.; Wagler, J. Tp*Cu(I)–CN–SiL2–NC–Cu(I)Tp*—A hexacoordinate Si-complex as connector for redox active metals via π-conjugated ligands. Dalton Trans. 2015, 44, 4744–4750. [Google Scholar] [CrossRef] [Green Version]
- Gericke, R.; Wagler, J. Ruthenium complexes of phosphino derivatives of carboxylic amides: Synthesis and characterization of tridentate P,E2 and tetradentate P,E3 (E = N,O) ligands and their reactivity towards [RuCl2(PPh3)3]. Polyhedron 2017, 125, 57–67. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Program for the Solution of Crystal Structures; SHELXS-97; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Program for the Refinement of Crystal Structures; SHELXL-2014/7; University of Göttingen: Göttingen, Germany, 2014. [Google Scholar]
- Sheldrick, G.M. Program for the Refinement of Crystal Structures; SHELXL-2018/3; University of Göttingen: Göttingen, Germany, 2018. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, L.J. ORTEP-3 for windows—A version of ORTEP-III with a graphical user interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- POV-RAY, (Version 3.7). Trademark of Persistence of Vision Raytracer Pty. Hallam Oaks Pty. Ltd.: Williamstown, Australia, 1991–2013. Available online: http://www.povray.org/download/(accessed on 28 June 2021).
- Neese, F. Software update: The ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 2022, 8, e1606. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, D.A.; Neese, F. All-electron basis sets for heavy elements. WIREs Comput. Mol. Sci. 2014, 4, 363–374. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597. [Google Scholar] [CrossRef]
- Van Wüllen, C. Molecular density functional calculations in the regular relativistic approximation: Method, application to coinage metal diatomics, hydrides, fluorides and chlorides, and comparison with first-order relativistic calculations. J. Chem. Phys. 1998, 109, 392. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Stoychev, G.L.; Auer, A.A.; Neese, F. Automatic Generation of Auxiliary Basis Sets. J. Chem. Theory Comput. 2017, 13, 554–562. [Google Scholar] [CrossRef]
- Chemcraft, Version 1.8 (Build 164); 2016. Available online: http://www.chemcraftprog.com/ (accessed on 19 September 2015).
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. C 2015, 71, 9–18. [Google Scholar] [CrossRef] [PubMed]
















| 2a | 2b | 2c | 2d | |
|---|---|---|---|---|
| Si1–O1 | 1.626(2) | 1.633(2) | 1.628(2) | 1.621(2) |
| Si1–O2 | 1.629(2) | 1.625(2) | 1.626(2) | 1.613(2) |
| Si1–O3 | 1.618(2) | 1.638(2) | 1.635(2) | 1.612(2) |
| Si1–C16 | 1.834(3) | 1.841(2) | 1.855(3) | 1.845(2) |
| Cu1–N1 | 2.038(2) | 2.060(2) | 2.037(2) | 2.061(2) |
| Cu1–N2 | 2.039(2) | 2.016(2) | 2.049(2) | 2.033(2) |
| Cu1–N3 | 2.023(2) | 2.055(2) | 2.073(2) | 2.021(2) |
| Cu1–Cl1 | 2.361(1) | 2.359(1) | 2.334(1) | 2.349(1) |
| Cu1···Si1 | 3.204(1) | 3.211(1) | 3.227(1) | 3.196(1) |
| O1–Si1–O2 | 111.41(13) | 113.18(9) | 112.21(10) | 110.99(12) |
| O1–Si1–O3 | 113.91(12) | 108.96(9) | 109.94(10) | 111.99(13) |
| O2–Si1–O3 | 111.89(12) | 113.92(9) | 110.54(11) | 114.26(14) |
| N1–Cu1–N2 | 112.39(8) | 115.97(7) | 111.40(9) | 108.02(11) |
| N1–Cu1–N3 | 113.67(8) | 103.62(7) | 111.64(8) | 113.16(11) |
| N2–Cu1–N3 | 114.46(8) | 119.47(7) | 113.13(9) | 118.75(12) |
| Cl1–Cu1···Si1 | 177.35(2) | 179.13(2) | 177.70(3) | 179.60(2) |
| Cu1···Si1–C16 | 177.55(13) | 174.58(7) | 174.01(9) | 178.09(10) |
| R–Si = | δ 29Si (RSi(pyO)3) [CDCl3] 1 | δ 29Si (RSi(pyO)3CuCl) [CDCl3] 2 | Δδ 29Si (1vs2) | δiso 29Si (RSi(pyO)3CuCl) [CP/MAS] 2 | Δδ 29Si (2) |
|---|---|---|---|---|---|
| Me–Si a | −46.5 [20] | −64.1 [20] | −17.6 | −70.0 [20] −71.4 [20] | −5.9 −7.3 |
| Ph–Si b | −64.7 [25] | −80.9 | −16.2 | −82.3 | −1.4 |
| Bn–Si c | −54.7 | −72.1 | −17.4 | −67.6 | +4.5 |
| Allyl–Si d/d′ | −53.4 | −65.4 | −12.0 | −52.6 | +12.8 |
| 3a | 3b | 3c | 3d | |
|---|---|---|---|---|
| Si1–O1 | 1.753(2) | 1.765(2) | 1.744(2) | 1.742(2) |
| Si1–O2 | 1.757(2) | 1.733(2) | 1.750(2) | 1.757(2) |
| Si1–O3 | 1.753(2) | 1.764(2) | 1.756(2) | 1.752(2) |
| Si1–O4 | 1.755(2) | 1.746(2) | 1.746(2) | 1.746(2) |
| Si1–C21 | 1.847(2) | 1.861(2) | 1.870(2) | 1.857(3) |
| Cu1–N1 | 2.023(2) | 2.047(2) | 2.042(2) | 2.028(2) |
| Cu1–N2 | 2.055(2) | 2.029(2) | 2.057(2) | 2.035(2) |
| Cu1–N3 | 2.025(2) | 2.038(2) | 2.055(2) | 2.052(2) |
| Cu1–N4 | 2.049(2) | 2.034(2) | 2.044(2) | 2.019(2) |
| Cu1–Cl1 | 2.403(1) | 2.389(1) | 2.403(1) | 2.395(1) |
| Cu1···Si1 | 2.919(1) | 2.888(1) | 2.915(1) | 2.897(1) |
| O1–Si1–O3 | 154.22(6) | 157.60(8) | 154.98(8) | 155.82(9) |
| O2–Si1–O4 | 155.83(6) | 154.21(8) | 154.14(8) | 153.49(9) |
| N1–Cu1–N3 | 159.32(5) | 159.23(8) | 158.15(7) | 157.76(9) |
| N2–Cu1–N4 | 158.66(5) | 160.00(8) | 159.05(7) | 159.26(9) |
| Cl1–Cu1···Si1 | 178.24(2) | 177.27(2) | 179.43(2) | 178.15(3) |
| Cu1···Si1–C21 | 177.51(6) | 177.99(8) | 176.47(8) | 177.16(11) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seidel, A.; Gericke, R.; Brendler, E.; Wagler, J. Copper Complexes of Silicon Pyridine-2-olates RSi(pyO)3 (R = Me, Ph, Bn, Allyl) and Ph2Si(pyO)2. Inorganics 2023, 11, 2. https://doi.org/10.3390/inorganics11010002
Seidel A, Gericke R, Brendler E, Wagler J. Copper Complexes of Silicon Pyridine-2-olates RSi(pyO)3 (R = Me, Ph, Bn, Allyl) and Ph2Si(pyO)2. Inorganics. 2023; 11(1):2. https://doi.org/10.3390/inorganics11010002
Chicago/Turabian StyleSeidel, Anne, Robert Gericke, Erica Brendler, and Jörg Wagler. 2023. "Copper Complexes of Silicon Pyridine-2-olates RSi(pyO)3 (R = Me, Ph, Bn, Allyl) and Ph2Si(pyO)2" Inorganics 11, no. 1: 2. https://doi.org/10.3390/inorganics11010002
APA StyleSeidel, A., Gericke, R., Brendler, E., & Wagler, J. (2023). Copper Complexes of Silicon Pyridine-2-olates RSi(pyO)3 (R = Me, Ph, Bn, Allyl) and Ph2Si(pyO)2. Inorganics, 11(1), 2. https://doi.org/10.3390/inorganics11010002