





Integration of CO2 Capture and Conversion by Employing Metal Oxides as Dual Function Materials: Recent Development and Future Outlook


Abstract
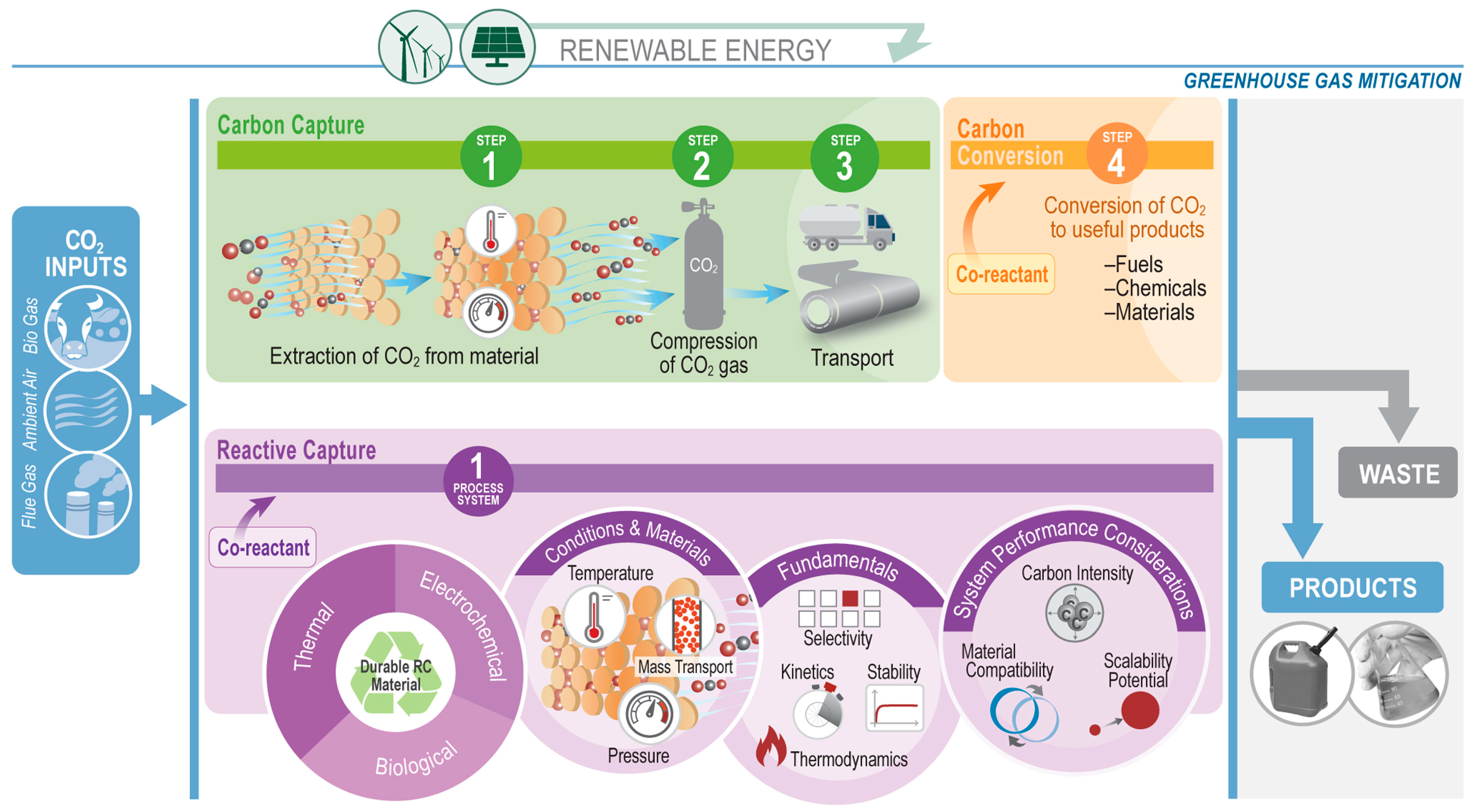
:1. Introduction
- “dual function material” AND “CO2 capture” AND “CO2 conversion”
- “dual function material” AND “CO2 capture” AND “CO2 utilization”
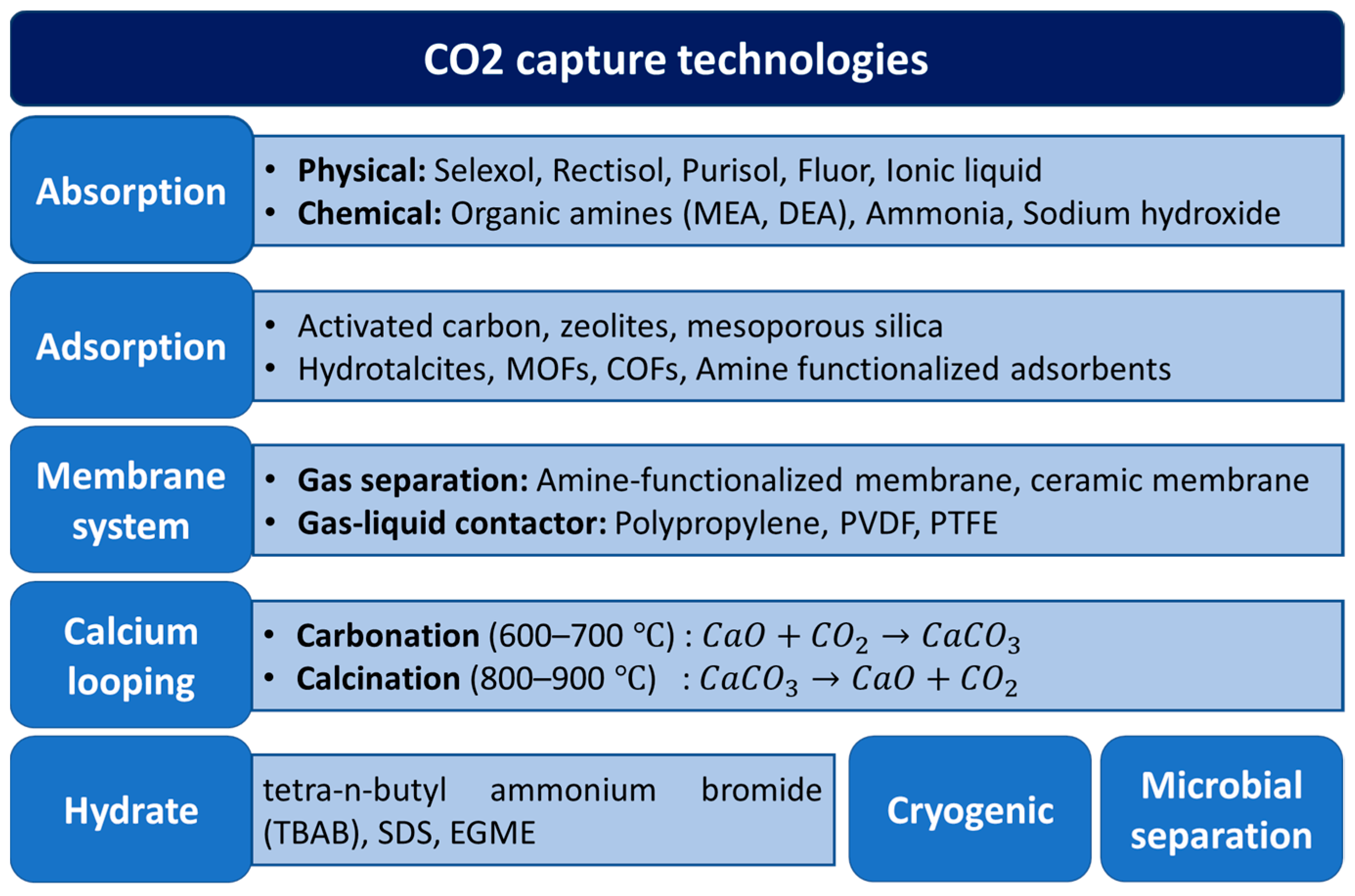
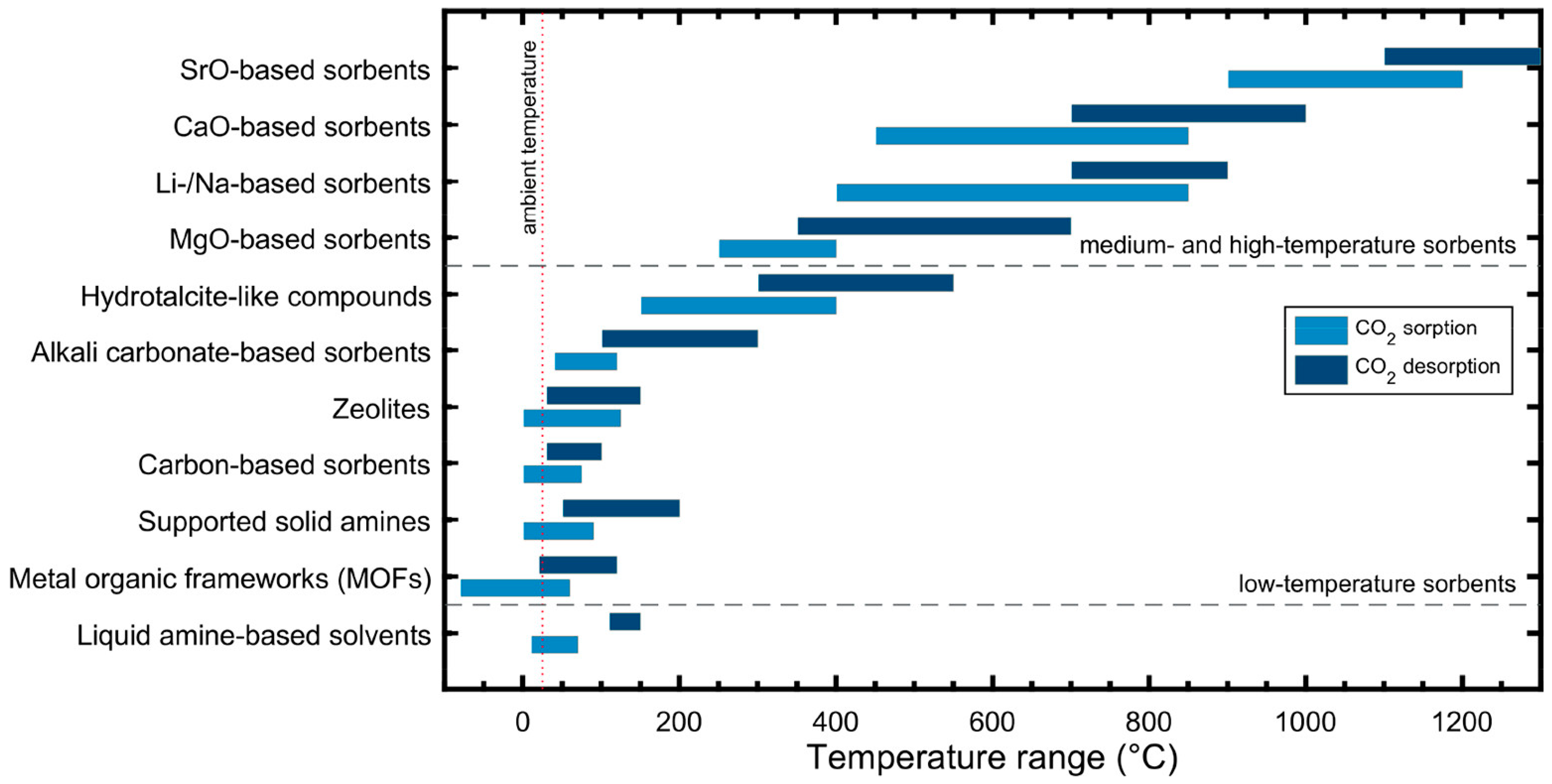
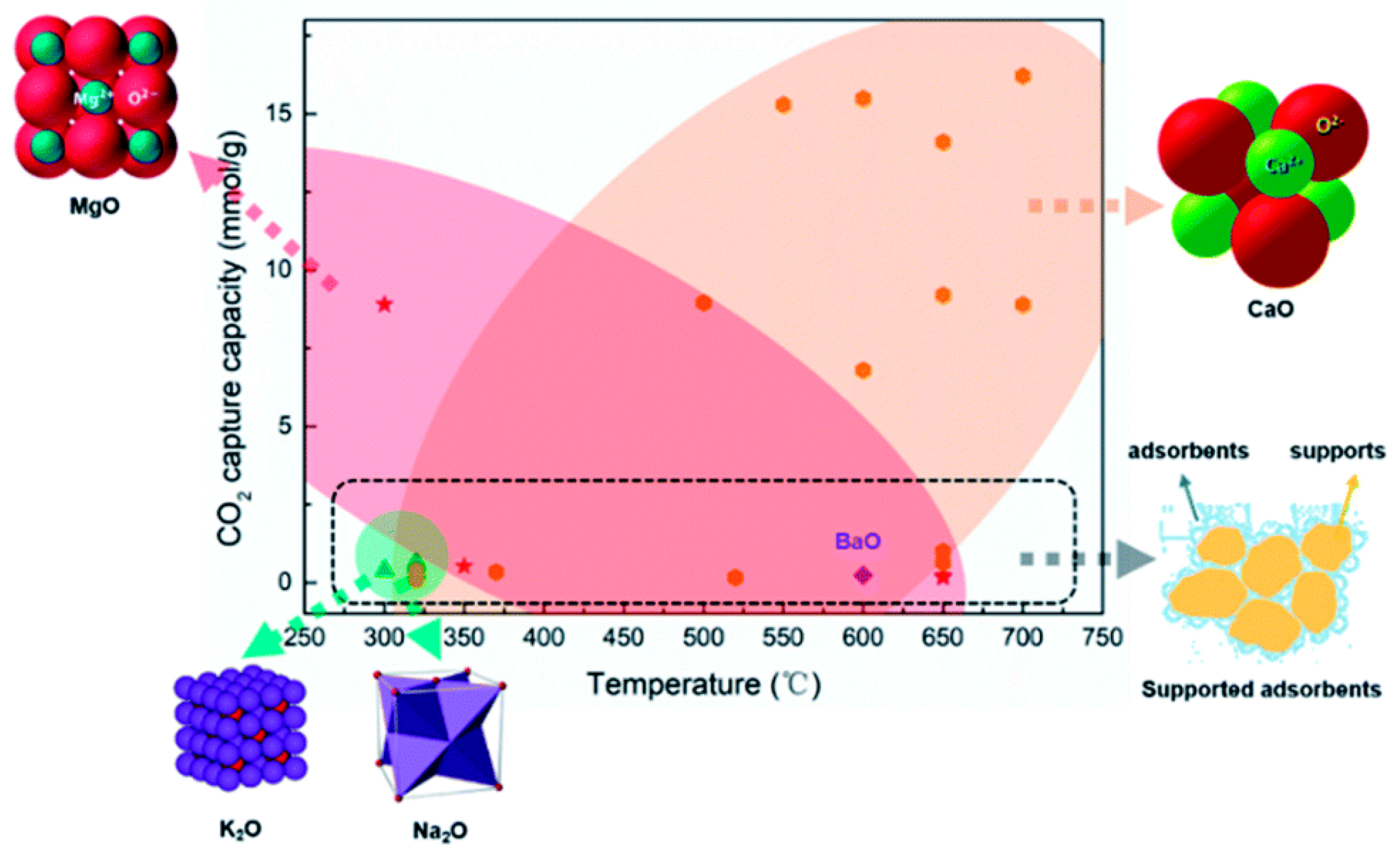
2. Metal Oxides as Sorbents for CO2 Capture
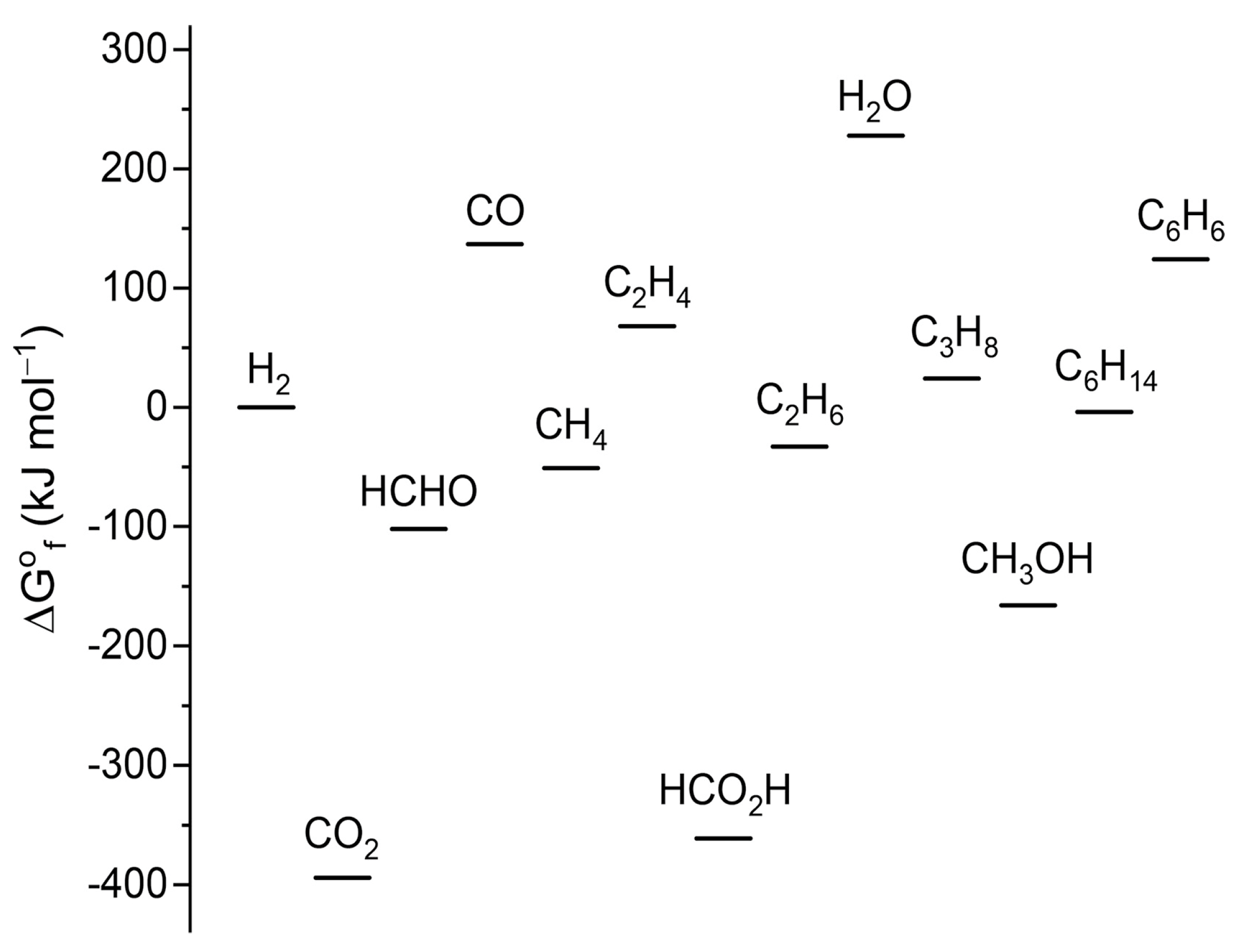
3. Overview of CO2 Catalytic Conversion
4. Dual Function Materials: Synthetic Methods
4.1. Incipient Wetness Impregnation
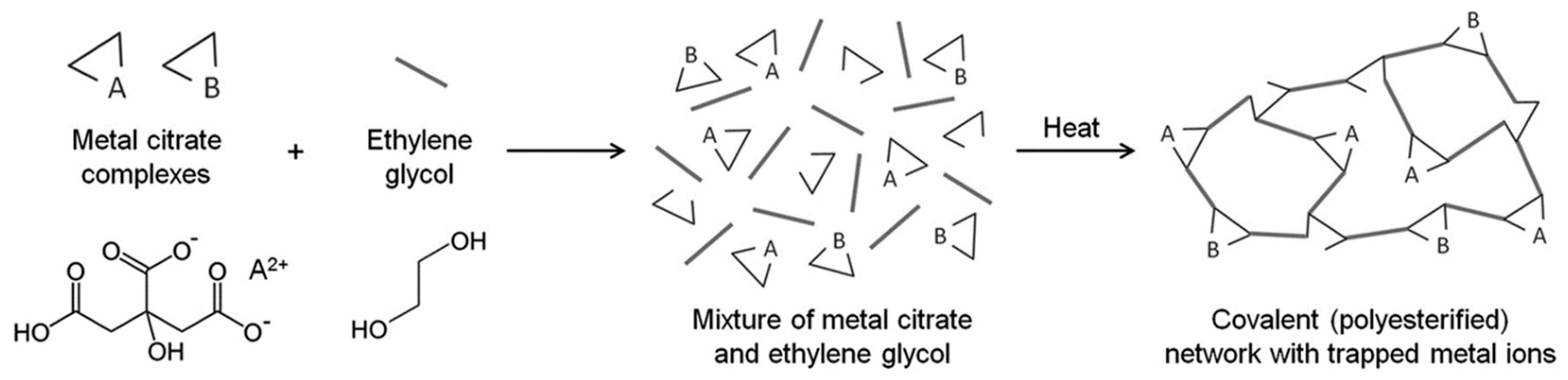
4.2. Sol–Gel Method
4.3. Co-Precipitation Method
4.4. Mixing Method
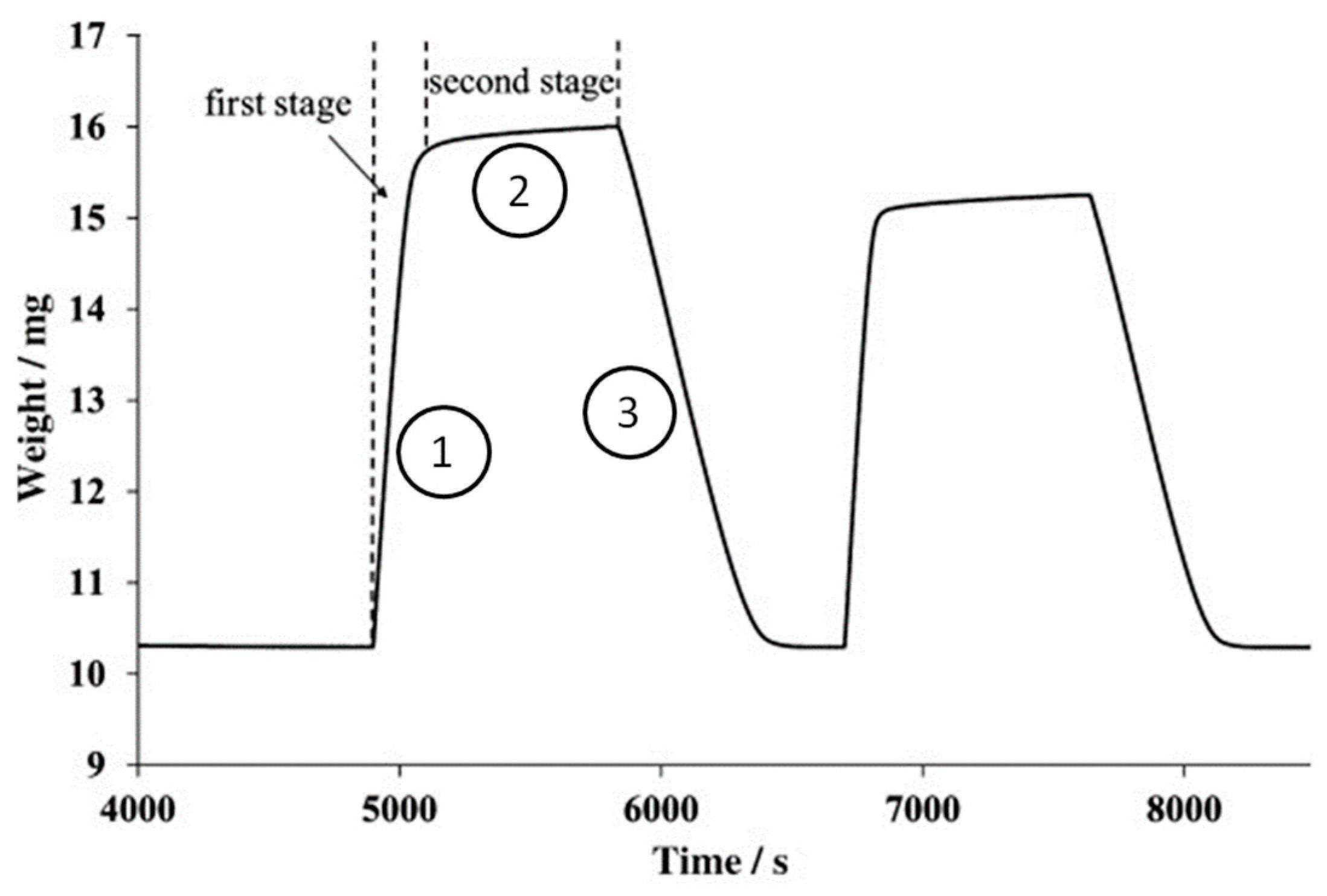
5. Performance Evaluation of Dual Function Materials
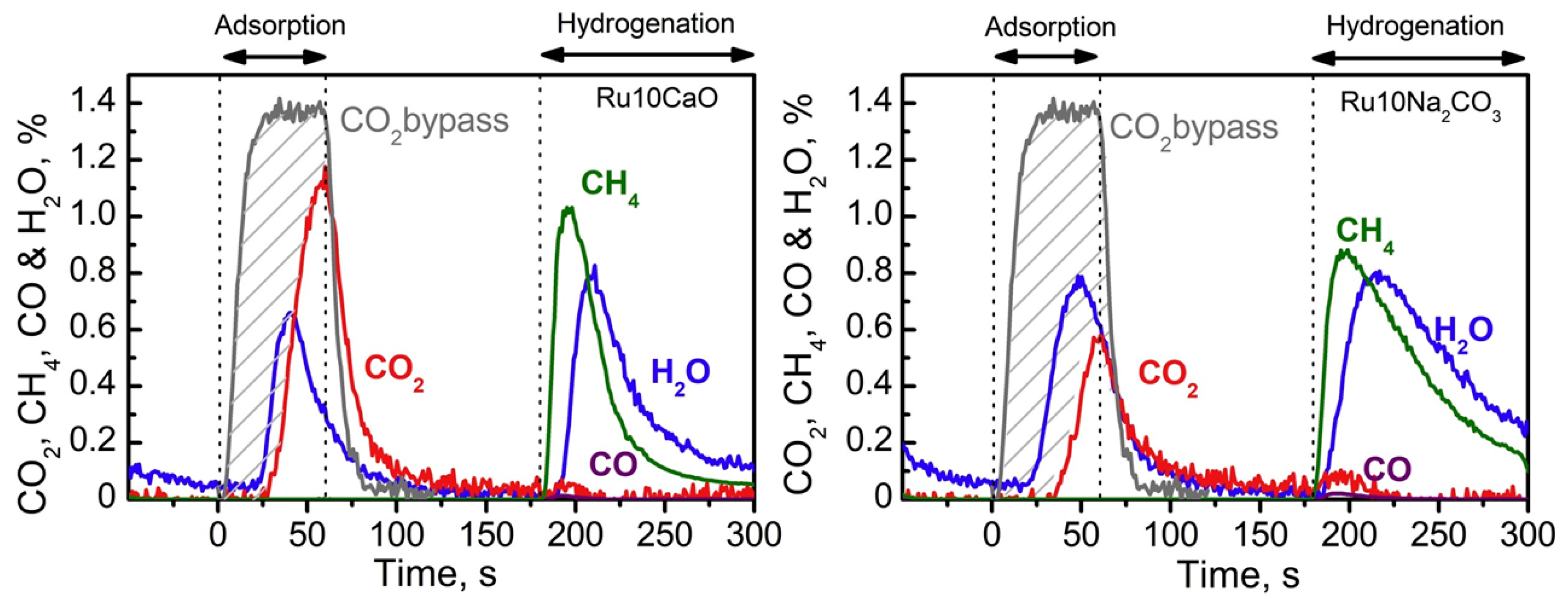
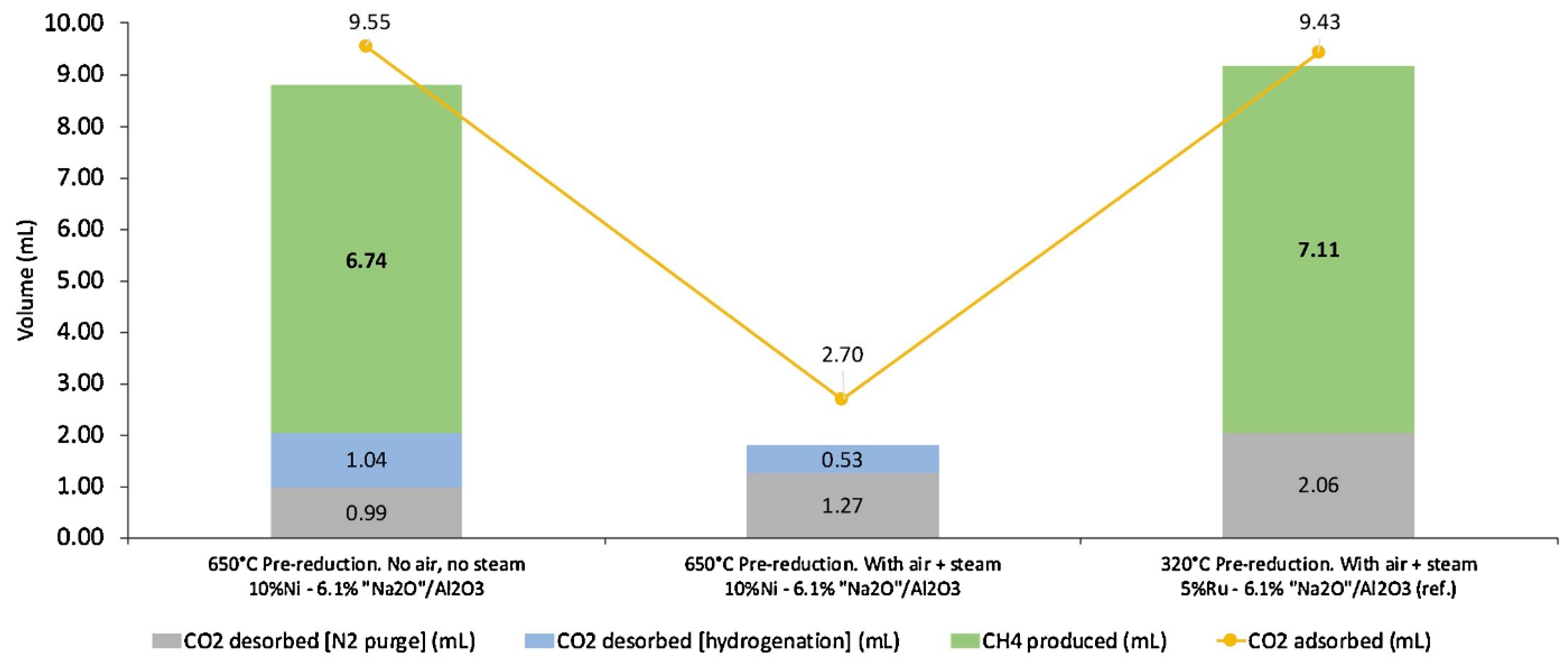
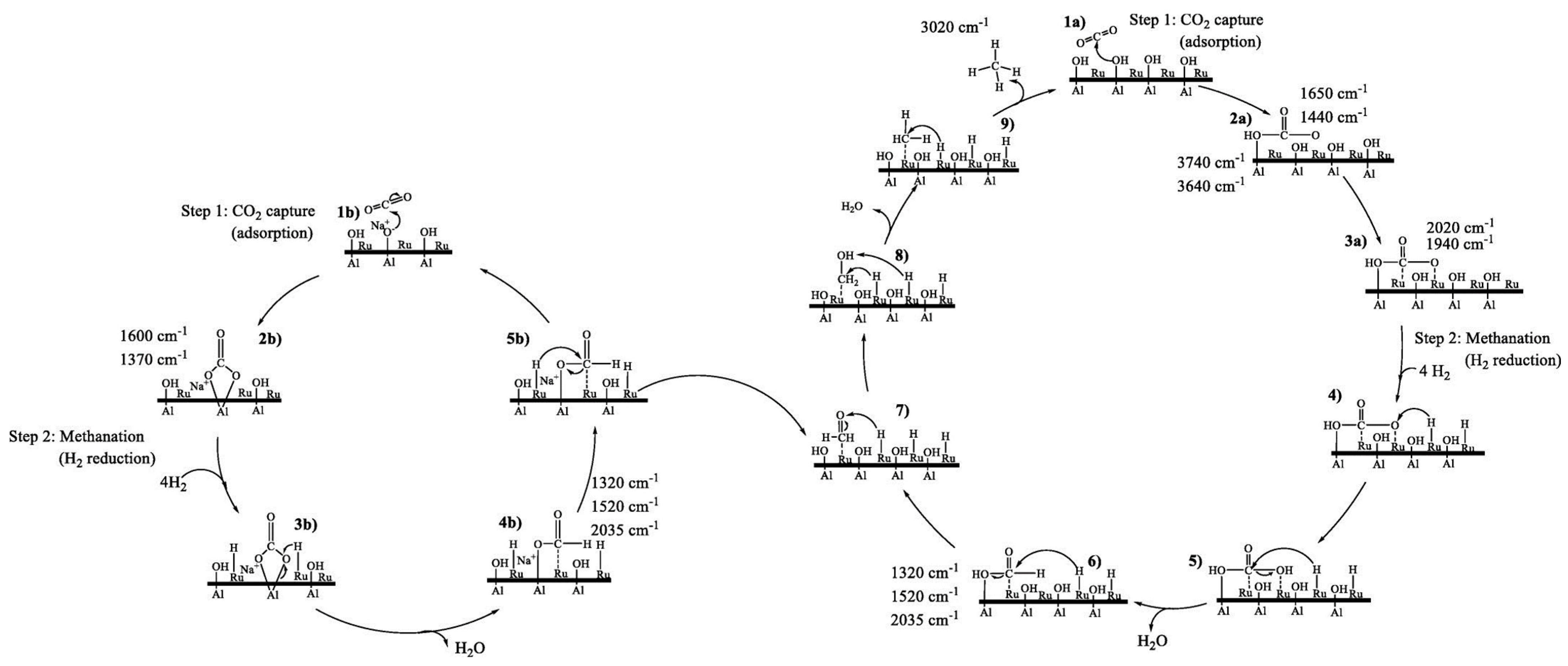
5.1. ICCC with Methanation Reaction
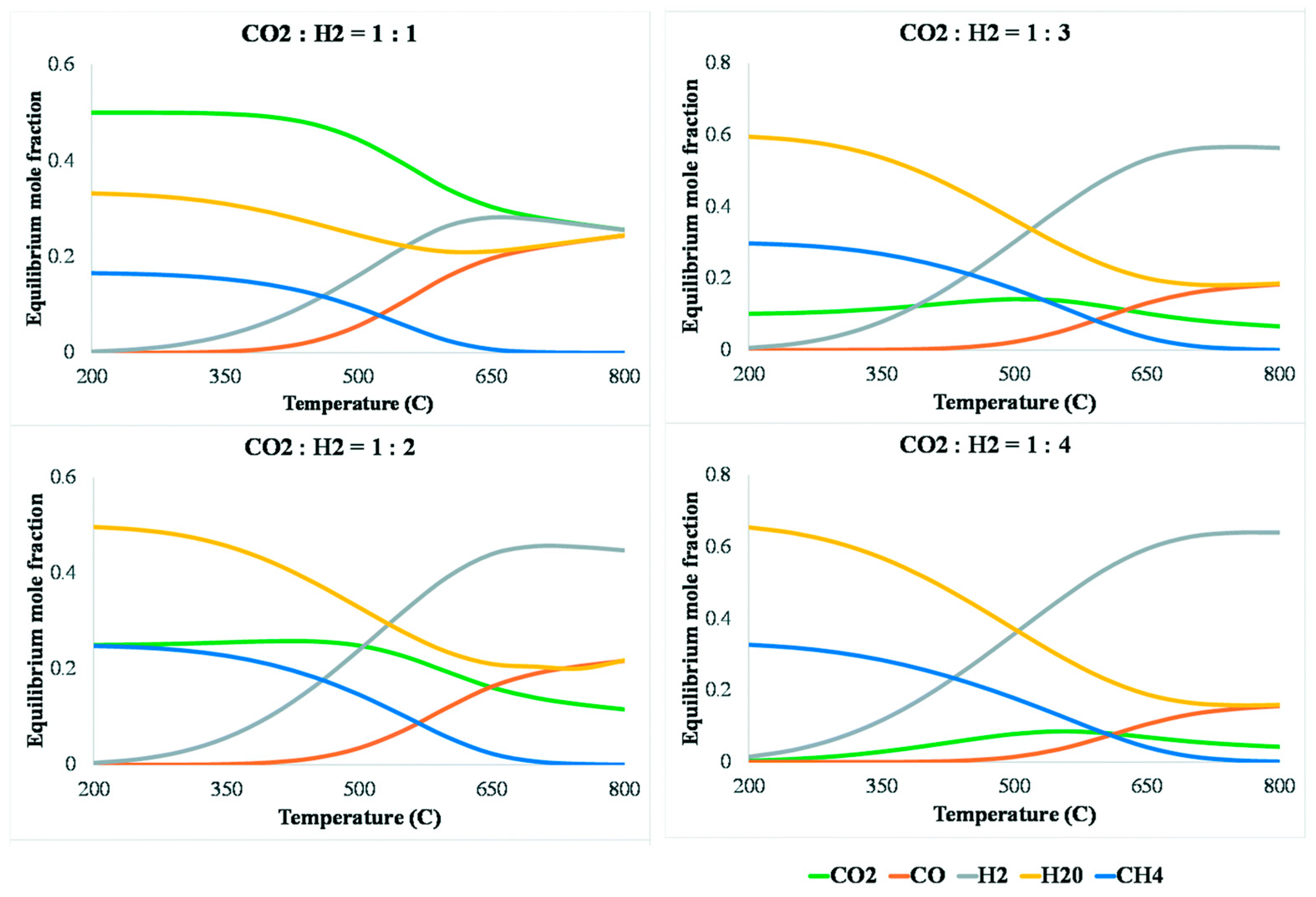
5.2. ICCC with Reverse Water–Gas Shift (RWGS) Reaction
5.3. ICCC with Dry Reforming of Methane (DRM)
6. Perspectives and Future Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- An IPCC Special Report on the Impacts of Global Warming of 1.5 °C above Pre-Industrial Levels and Related Global Greenhouse Gas Emission Pathways, in the Context of Strengthening the Global Response to the Threat of Climate Change, Sustainable Development, and Efforts to Eradicate Poverty; Intergovernmental Panel on Climate Change: Geneva, Switzerland, 2018.
- Koytsoumpa, E.I.; Bergins, C.; Kakaras, E. The CO2 economy: Review of CO2 capture and reuse technologies. J. Supercrit. Fluids 2018, 132, 3–16. [Google Scholar] [CrossRef]
- Leeson, D.; Mac Dowell, N.; Shah, N.; Petit, C.; Fennell, P.S. A Techno-economic analysis and systematic review of carbon capture and storage (CCS) applied to the iron and steel, cement, oil refining and pulp and paper industries, as well as other high purity sources. Int. J. Greenh. Gas Control 2017, 61, 71–84. [Google Scholar] [CrossRef]
- Naims, H. Economics of carbon dioxide capture and utilization—A supply and demand perspective. Environ. Sci. Pollut. Res. 2016, 23, 22226–22241. [Google Scholar] [CrossRef] [PubMed]
- Wilberforce, T.; Baroutaji, A.; Soudan, B.; Al-Alami, A.H.; Olabi, A.G. Outlook of carbon capture technology and challenges. Sci. Total Environ. 2019, 657, 56–72. [Google Scholar] [CrossRef]
- Omodolor, I.S.; Otor, H.O.; Andonegui, J.A.; Allen, B.J.; Alba-Rubio, A.C. Dual-Function Materials for CO2 Capture and Conversion: A Review. Ind. Eng. Chem. Res. 2020, 59, 17612–17631. [Google Scholar] [CrossRef]
- von der Assen, N.; Bardow, A. Life cycle assessment of polyols for polyurethane production using CO2 as feedstock: Insights from an industrial case study. Green Chem. 2014, 16, 3272–3280. [Google Scholar] [CrossRef]
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable Conversion of Carbon Dioxide: An Integrated Review of Catalysis and Life Cycle Assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef]
- Huang, C.-H.; Tan, C.-S. A Review: CO2 Utilization. Aerosol Air Qual. Res. 2014, 14, 480–499. [Google Scholar] [CrossRef]
- Freyman, M.C.; Huang, Z.; Ravikumar, D.; Duoss, E.B.; Li, Y.; Baker, S.E.; Pang, S.H.; Schaidle, J.A. Reactive CO2 capture: A path forward for process integration in carbon management. Joule 2023, 7, 631–651. [Google Scholar] [CrossRef]
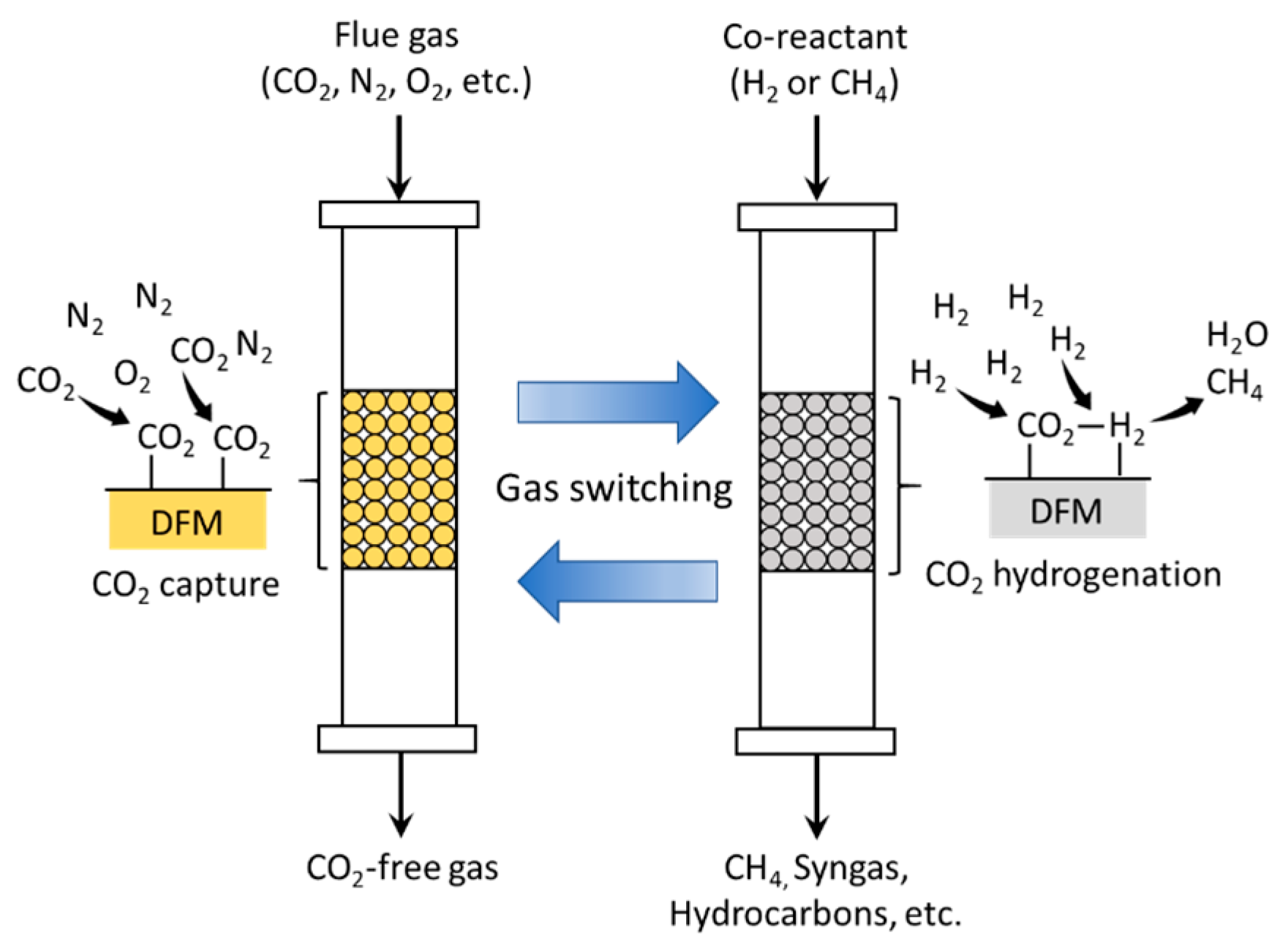
- Duyar, M.S.; Treviño, M.A.A.; Farrauto, R.J. Dual function materials for CO2 capture and conversion using renewable H2. Appl. Catal. B Environ. 2015, 168–169, 370–376. [Google Scholar] [CrossRef]
- Melo Bravo, P.; Debecker, D.P. Combining CO2 capture and catalytic conversion to methane. Waste Dispos. Sustain. Energy 2019, 1, 53–65. [Google Scholar] [CrossRef]
- Merkouri, L.-P.; Reina, T.R.; Duyar, M.S. Closing the Carbon Cycle with Dual Function Materials. Energy Fuels 2021, 35, 19859–19880. [Google Scholar] [CrossRef]
- Sun, S.; Sun, H.; Williams, P.T.; Wu, C. Recent advances in integrated CO2 capture and utilization: A review. Sustain. Energy Fuels 2021, 5, 4546–4559. [Google Scholar] [CrossRef]
- Sabri, M.A.; Al Jitan, S.; Bahamon, D.; Vega, L.F.; Palmisano, G. Current and future perspectives on catalytic-based integrated carbon capture and utilization. Sci. Total Environ. 2021, 790, 148081. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Dang, C.; Yang, G.; Cao, Y.; Wang, H.; Peng, F.; Yu, H. Bi-functional particles for integrated thermo-chemical processes: Catalysis and beyond. Particuology 2021, 56, 10–32. [Google Scholar] [CrossRef]
- Bhatta, L.K.G.; Subramanyam, S.; Chengala, M.D.; Olivera, S.; Venkatesh, K. Progress in hydrotalcite like compounds and metal-based oxides for CO2 capture: A review. J. Clean. Prod. 2015, 103, 171–196. [Google Scholar] [CrossRef]
- Khalilpour, R.; Mumford, K.; Zhai, H.; Abbas, A.; Stevens, G.; Rubin, E.S. Membrane-based carbon capture from flue gas: A review. J. Clean. Prod. 2015, 103, 286–300. [Google Scholar] [CrossRef]
- Al-Mamoori, A.; Krishnamurthy, A.; Rownaghi, A.A.; Rezaei, F. Carbon Capture and Utilization Update. Energy Technol. 2017, 5, 834–849. [Google Scholar] [CrossRef]
- Yang, Z.-Z.; Wei, J.-J.; Zeng, G.-M.; Zhang, H.-Q.; Tan, X.-F.; Ma, C.; Li, X.-C.; Li, Z.-H.; Zhang, C. A review on strategies to LDH-based materials to improve adsorption capacity and photoreduction efficiency for CO2. Coord. Chem. Rev. 2019, 386, 154–182. [Google Scholar] [CrossRef]
- Xie, K.; Fu, Q.; Qiao, G.G.; Webley, P.A. Recent progress on fabrication methods of polymeric thin film gas separation membranes for CO2 capture. J. Membr. Sci. 2019, 572, 38–60. [Google Scholar] [CrossRef]
- Gao, W.; Liang, S.; Wang, R.; Jiang, Q.; Zhang, Y.; Zheng, Q.; Xie, B.; Toe, C.Y.; Zhu, X.; Wang, J.; et al. Industrial carbon dioxide capture and utilization: State of the art and future challenges. Chem. Soc. Rev. 2020, 49, 8584–8686. [Google Scholar] [CrossRef] [PubMed]
- Younas, M.; Rezakazemi, M.; Daud, M.; Wazir, M.B.; Ahmad, S.; Ullah, N.; Inamuddin; Ramakrishna, S. Recent progress and remaining challenges in post-combustion CO2 capture using metal-organic frameworks (MOFs). Prog. Energy Combust. Sci. 2020, 80, 100849. [Google Scholar] [CrossRef]
- Zheng, J.; Chong, Z.R.; Qureshi, M.F.; Linga, P. Carbon Dioxide Sequestration via Gas Hydrates: A Potential Pathway toward Decarbonization. Energy Fuels 2020, 34, 10529–10546. [Google Scholar] [CrossRef]
- Dunstan, M.T.; Donat, F.; Bork, A.H.; Grey, C.P.; Müller, C.R. CO2 Capture at Medium to High Temperature Using Solid Oxide-Based Sorbents: Fundamental Aspects, Mechanistic Insights, and Recent Advances. Chem. Rev. 2021, 121, 12681–12745. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Ren, Z.; Si, W.; Ma, Q.; Huang, W.; Liao, K.; Huang, Z.; Wang, Y.; Li, J.; Xu, P. Research progress on CO2 capture and utilization technology. J. CO2 Util. 2022, 66, 102260. [Google Scholar] [CrossRef]
- Dubey, A.; Arora, A. Advancements in carbon capture technologies: A review. J. Clean. Prod. 2022, 373, 133932. [Google Scholar] [CrossRef]
- Peres, C.B.; Resende, P.M.R.; Nunes, L.J.R.; Morais, L.C.d. Advances in Carbon Capture and Use (CCU) Technologies: A Comprehensive Review and CO2 Mitigation Potential Analysis. Clean Technol. 2022, 4, 1193–1207. [Google Scholar] [CrossRef]
- Idem, R.; Supap, T.; Shi, H.; Gelowitz, D.; Ball, M.; Campbell, C.; Tontiwachwuthikul, P. Practical experience in post-combustion CO2 capture using reactive solvents in large pilot and demonstration plants. Int. J. Greenh. Gas Control 2015, 40, 6–25. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Yu, J. Applications of Zeolites in Sustainable Chemistry. Chem 2017, 3, 928–949. [Google Scholar] [CrossRef]
- Suescum-Morales, D.; Jiménez, J.R.; Fernández-Rodríguez, J.M. Review of the Application of Hydrotalcite as CO2 Sinks for Climate Change Mitigation. ChemEngineering 2022, 6, 50. [Google Scholar] [CrossRef]
- Veerabhadrappa, M.G.; Maroto-Valer, M.M.; Chen, Y.; Garcia, S. Layered Double Hydroxides-Based Mixed Metal Oxides: Development of Novel Structured Sorbents for CO2 Capture Applications. ACS Appl. Mater. Interfaces 2021, 13, 11805–11813. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Zhou, T.; Gao, Y.; Louis, B.; O’Hare, D.; Wang, Q. Molten salts-modified MgO-based adsorbents for intermediate-temperature CO2 capture: A review. J. Energy Chem. 2017, 26, 830–838. [Google Scholar] [CrossRef]
- Kwak, J.-S.; Kim, K.-Y.; Oh, K.-R.; Kwon, Y.-U. Performance enhancement of all-solid CO2 absorbent based on Na2CO3-promoted MgO by using ZrO2 dispersant. Int. J. Greenh. Gas Control 2019, 81, 38–43. [Google Scholar] [CrossRef]
- Joo, H.; Cho, S.J.; Na, K. Control of CO2 absorption capacity and kinetics by MgO-based dry sorbents promoted with carbonate and nitrate salts. J. CO2 Util. 2017, 19, 194–201. [Google Scholar] [CrossRef]
- Jeon, H.; Triviño, M.L.T.; Hwang, S.; Moon, J.H.; Yoo, J.; Seo, J.G. Unveiling the carbonation mechanism in molten salt-promoted MgO-Al2O3 sorbents. J. CO2 Util. 2020, 39, 101153. [Google Scholar] [CrossRef]
- Hu, Z.; Wang, Y.; Shah, B.B.; Zhao, D. CO2 Capture in Metal–Organic Framework Adsorbents: An Engineering Perspective. Adv. Sustain. Syst. 2019, 3, 1800080. [Google Scholar] [CrossRef]
- Song, K.S.; Fritz, P.W.; Coskun, A. Porous organic polymers for CO2 capture, separation and conversion. Chem. Soc. Rev. 2022, 51, 9831–9852. [Google Scholar] [CrossRef]
- Wang, J.; Wang, L.; Wang, Y.; Zhang, D.; Xiao, Q.; Huang, J.; Liu, Y.-N. Recent progress in porous organic polymers and their application for CO2 capture. Chin. J. Chem. Eng. 2022, 42, 91–103. [Google Scholar] [CrossRef]
- Luo, R.; Chen, M.; Liu, X.; Xu, W.; Li, J.; Liu, B.; Fang, Y. Recent advances in CO2 capture and simultaneous conversion into cyclic carbonates over porous organic polymers having accessible metal sites. J. Mater. Chem. A 2020, 8, 18408–18424. [Google Scholar] [CrossRef]
- Silva, J.M.; Trujillano, R.; Rives, V.; Soria, M.A.; Madeira, L.M. High temperature CO2 sorption over modified hydrotalcites. Chem. Eng. J. 2017, 325, 25–34. [Google Scholar] [CrossRef]
- Miguel, C.V.; Trujillano, R.; Rives, V.; Vicente, M.A.; Ferreira, A.F.P.; Rodrigues, A.E.; Mendes, A.; Madeira, L.M. High temperature CO2 sorption with gallium-substituted and promoted hydrotalcites. Sep. Purif. Technol. 2014, 127, 202–211. [Google Scholar] [CrossRef]
- Faria, A.C.; Trujillano, R.; Rives, V.; Miguel, C.V.; Rodrigues, A.E.; Madeira, L.M. Alkali metal (Na, Cs and K) promoted hydrotalcites for high temperature CO2 capture from flue gas in cyclic adsorption processes. Chem. Eng. J. 2022, 427, 131502. [Google Scholar] [CrossRef]
- Lee, J.M.; Min, Y.J.; Lee, K.B.; Jeon, S.G.; Na, J.G.; Ryu, H.J. Enhancement of CO2 Sorption Uptake on Hydrotalcite by Impregnation with K2CO3. Langmuir 2010, 26, 18788–18797. [Google Scholar] [CrossRef]
- Halabi, M.H.; de Croon, M.H.J.M.; van der Schaaf, J.; Cobden, P.D.; Schouten, J.C. High capacity potassium-promoted hydrotalcite for CO2 capture in H2 production. Int. J. Hydrogen Energy 2012, 37, 4516–4525. [Google Scholar] [CrossRef]
- Kim, S.; Lee, K.B. Impregnation of hydrotalcite with NaNO3 for enhanced high-temperature CO2 sorption uptake. Chem. Eng. J. 2019, 356, 964–972. [Google Scholar] [CrossRef]
- Oliveira, E.L.G.; Grande, C.A.; Rodrigues, A.E. CO2 sorption on hydrotalcite and alkali-modified (K and Cs) hydrotalcites at high temperatures. Sep. Purif. Technol. 2008, 62, 137–147. [Google Scholar] [CrossRef]
- Perejón, A.; Romeo, L.M.; Lara, Y.; Lisbona, P.; Martínez, A.; Valverde, J.M. The Calcium-Looping technology for CO2 capture: On the important roles of energy integration and sorbent behavior. Appl. Energy 2016, 162, 787–807. [Google Scholar] [CrossRef]
- Benhelal, E.; Shamsaei, E.; Rashid, M.I. Challenges against CO2 abatement strategies in cement industry: A review. J. Environ. Sci. 2021, 104, 84–101. [Google Scholar] [CrossRef]
- Plaza, M.G.; Martínez, S.; Rubiera, F. CO2 Capture, Use, and Storage in the Cement Industry: State of the Art and Expectations. Energies 2020, 13, 5692. [Google Scholar] [CrossRef]
- MacKenzie, A.; Granatstein, D.L.; Anthony, E.J.; Abanades, J.C. Economics of CO2 Capture Using the Calcium Cycle with a Pressurized Fluidized Bed Combustor. Energy Fuels 2007, 21, 920–926. [Google Scholar] [CrossRef]
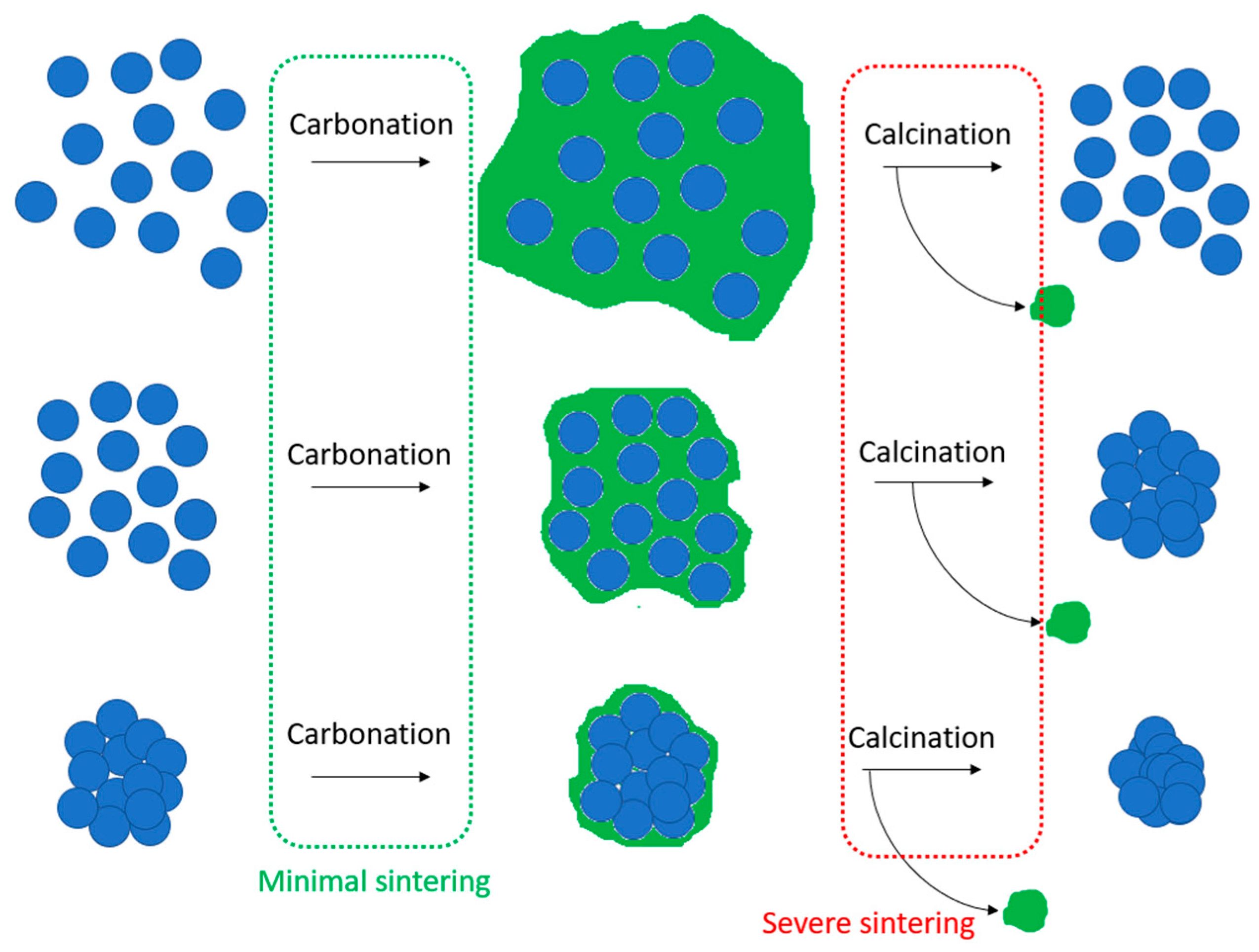
- Geng, Y.-q.; Guo, Y.-x.; Fan, B.; Cheng, F.-q.; Cheng, H.-g. Research progress of calcium-based adsorbents for CO2 capture and anti-sintering modification. J. Fuel Chem. Technol. 2021, 49, 998–1013. [Google Scholar] [CrossRef]
- Hu, Y.; Lu, H.; Liu, W.; Yang, Y.; Li, H. Incorporation of CaO into inert supports for enhanced CO2 capture: A review. Chem. Eng. J. 2020, 396, 125253. [Google Scholar] [CrossRef]
- Krödel, M.; Landuyt, A.; Abdala, P.M.; Müller, C.R. Mechanistic Understanding of CaO-Based Sorbents for High-Temperature CO2 Capture: Advanced Characterization and Prospects. ChemSusChem 2020, 13, 6259–6272. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wu, C.; Shen, B.; Zhang, X.; Zhang, Y.; Huang, J. Progress in the development and application of CaO-based adsorbents for CO2 capture—A review. Mater. Today Sustain. 2018; 1–2, 1–27. [Google Scholar] [CrossRef]
- Lu, H.; Khan, A.; Pratsinis, S.E.; Smirniotis, P.G. Flame-Made Durable Doped-CaO Nanosorbents for CO2 Capture. Energy Fuels 2009, 23, 1093–1100. [Google Scholar] [CrossRef]
- Li, L.; King, D.L.; Nie, Z.; Howard, C. Magnesia-Stabilized Calcium Oxide Absorbents with Improved Durability for High Temperature CO2 Capture. Ind. Eng. Chem. Res. 2009, 48, 10604–10613. [Google Scholar] [CrossRef]
- Lysikov, A.I.; Salanov, A.N.; Okunev, A.G. Change of CO2 Carrying Capacity of CaO in Isothermal Recarbonation—Decomposition Cycles. Ind. Eng. Chem. Res. 2007, 46, 4633–4638. [Google Scholar] [CrossRef]
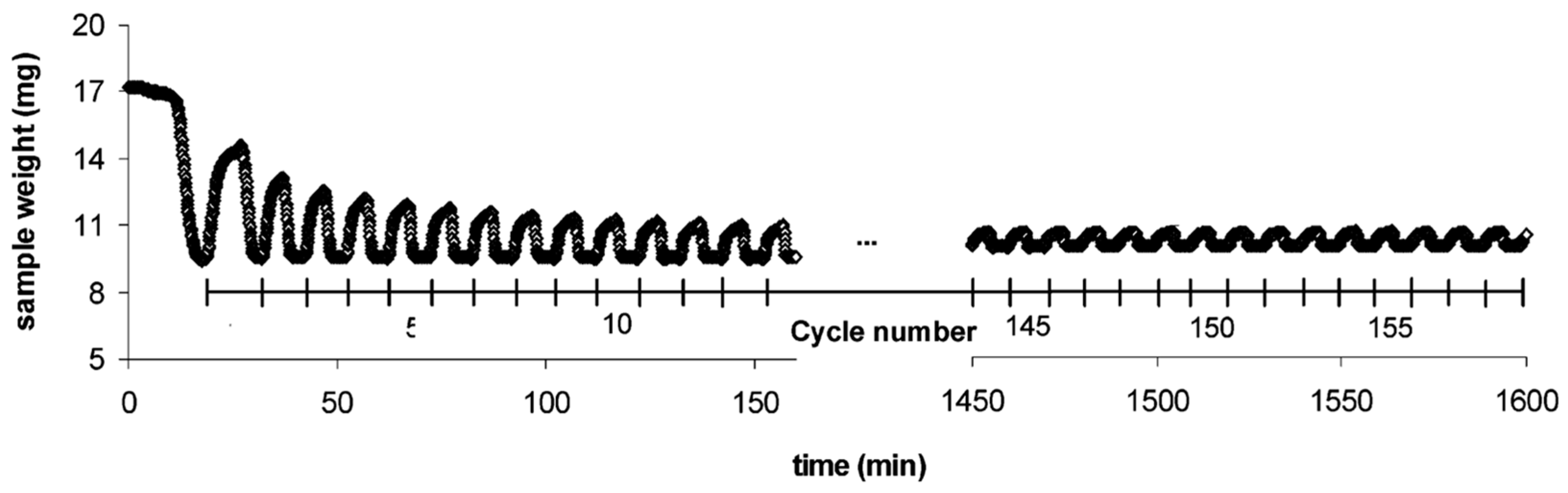
- Grasa, G.S.; Abanades, J.C. CO2 Capture Capacity of CaO in Long Series of Carbonation/Calcination Cycles. Ind. Eng. Chem. Res. 2006, 45, 8846–8851. [Google Scholar] [CrossRef]
- Kierzkowska, A.M.; Pacciani, R.; Müller, C.R. CaO-Based CO2 Sorbents: From Fundamentals to the Development of New, Highly Effective Materials. ChemSusChem 2013, 6, 1130–1148. [Google Scholar] [CrossRef]
- Jing, J.-Y.; Li, T.-Y.; Zhang, X.-W.; Wang, S.-D.; Feng, J.; Turmel, W.A.; Li, W.-Y. Enhanced CO2 sorption performance of CaO/Ca3Al2O6 sorbents and its sintering-resistance mechanism. Appl. Energy 2017, 199, 225–233. [Google Scholar] [CrossRef]
- Zhou, Z.; Qi, Y.; Xie, M.; Cheng, Z.; Yuan, W. Synthesis of CaO-based sorbents through incorporation of alumina/aluminate and their CO2 capture performance. Chem. Eng. Sci. 2012, 74, 172–180. [Google Scholar] [CrossRef]
- Han, R.; Gao, J.; Wei, S.; Su, Y.; Qin, Y. Development of highly effective CaO@Al2O3 with hierarchical architecture CO2 sorbents via a scalable limited-space chemical vapor deposition technique. J. Mater. Chem. A 2018, 6, 3462–3470. [Google Scholar] [CrossRef]
- Wang, N.; Feng, Y.; Liu, L.; Guo, X. Effects of preparation methods on the structure and property of Al-stabilized CaO-based sorbents for CO2 capture. Fuel Process. Technol. 2018, 173, 276–284. [Google Scholar] [CrossRef]
- López, J.M.; Grasa, G.; Murillo, R. Evaluation of the effect of inert support on the carbonation reaction of synthetic CaO-based CO2 sorbents. Chem. Eng. J. 2018, 350, 559–572. [Google Scholar] [CrossRef]
- Yu, Y.S.; Liu, W.Q.; An, H.; Yang, F.S.; Wang, G.X.; Feng, B.; Zhang, Z.X.; Rudolph, V. Modeling of the carbonation behavior of a calcium based sorbent for CO2 capture. Int. J. Greenh. Gas Control 2012, 10, 510–519. [Google Scholar] [CrossRef]
- Kurlov, A.; Broda, M.; Hosseini, D.; Mitchell, S.J.; Pérez-Ramírez, J.; Müller, C.R. Mechanochemically Activated, Calcium Oxide-Based, Magnesium Oxide-Stabilized Carbon Dioxide Sorbents. ChemSusChem 2016, 9, 2380–2390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Z.; Peng, Y.; Su, W.; Sun, X.; Li, J. Investigation on a novel CaO–Y2O3 sorbent for efficient CO2 mitigation. Chem. Eng. J. 2014, 243, 297–304. [Google Scholar] [CrossRef]
- Naeem, M.A.; Armutlulu, A.; Imtiaz, Q.; Müller, C.R. CaO-Based CO2 Sorbents Effectively Stabilized by Metal Oxides. ChemPhysChem 2017, 18, 3280–3285. [Google Scholar] [CrossRef]
- Kim, S.M.; Kierzkowska, A.M.; Broda, M.; Müller, C.R. Sol-gel synthesis of MgAl2O4-stabilized CaO for CO2 capture. Energy Procedia 2017, 114, 220–229. [Google Scholar] [CrossRef]
- Koirala, R.; Reddy, G.K.; Smirniotis, P.G. Single nozzle flame-made highly durable metal doped Ca-based sorbents for CO2 capture at high temperature. Energy Fuels 2012, 26, 3103–3109. [Google Scholar] [CrossRef]
- Lu, H.; Smirniotis, P.G. Calcium oxide doped sorbents for CO2 uptake in the presence of SO2 at high temperatures. Ind. Eng. Chem. Res. 2009, 48, 5454–5459. [Google Scholar] [CrossRef]
- De, S.; Dokania, A.; Ramirez, A.; Gascon, J. Advances in the Design of Heterogeneous Catalysts and Thermocatalytic Processes for CO2 Utilization. ACS Catal. 2020, 10, 14147–14185. [Google Scholar] [CrossRef]
- Younas, M.; Loong Kong, L.; Bashir, M.J.K.; Nadeem, H.; Shehzad, A.; Sethupathi, S. Recent Advancements, Fundamental Challenges, and Opportunities in Catalytic Methanation of CO2. Energy Fuels 2016, 30, 8815–8831. [Google Scholar] [CrossRef]
- Aziz, M.A.A.; Jalil, A.A.; Triwahyono, S.; Ahmad, A. CO2 methanation over heterogeneous catalysts: Recent progress and future prospects. Green Chem. 2015, 17, 2647–2663. [Google Scholar] [CrossRef]
- Fan, W.K.; Tahir, M. Recent trends in developments of active metals and heterogenous materials for catalytic CO2 hydrogenation to renewable methane: A review. J. Environ. Chem. Eng. 2021, 9, 105460. [Google Scholar] [CrossRef]
- Chen, X.; Chen, Y.; Song, C.; Ji, P.; Wang, N.; Wang, W.; Cui, L. Recent Advances in Supported Metal Catalysts and Oxide Catalysts for the Reverse Water-Gas Shift Reaction. Front. Chem. 2020, 8, 709. [Google Scholar] [CrossRef]
- Daza, Y.A.; Kuhn, J.N. CO2 conversion by reverse water gas shift catalysis: Comparison of catalysts, mechanisms and their consequences for CO2 conversion to liquid fuels. RSC Adv. 2016, 6, 49675–49691. [Google Scholar] [CrossRef]
- Nielsen, D.U.; Hu, X.-M.; Daasbjerg, K.; Skrydstrup, T. Chemically and electrochemically catalysed conversion of CO2 to CO with follow-up utilization to value-added chemicals. Nat. Catal. 2018, 1, 244–254. [Google Scholar] [CrossRef]
- le Saché, E.; Reina, T.R. Analysis of Dry Reforming as direct route for gas phase CO2 conversion. The past, the present and future of catalytic DRM technologies. Prog. Energy Combust. Sci. 2022, 89, 100970. [Google Scholar] [CrossRef]
- Jang, W.-J.; Shim, J.-O.; Kim, H.-M.; Yoo, S.-Y.; Roh, H.-S. A review on dry reforming of methane in aspect of catalytic properties. Catal. Today 2019, 324, 15–26. [Google Scholar] [CrossRef]
- Singh, R.; Dhir, A.; Mohapatra, S.K.; Mahla, S.K. Dry reforming of methane using various catalysts in the process: Review. Biomass Convers. Biorefinery 2020, 10, 567–587. [Google Scholar] [CrossRef]
- Dieterich, V.; Buttler, A.; Hanel, A.; Spliethoff, H.; Fendt, S. Power-to-liquid via synthesis of methanol, DME or Fischer–Tropsch-fuels: A review. Energy Environ. Sci. 2020, 13, 3207–3252. [Google Scholar] [CrossRef]
- Atsbha, T.A.; Yoon, T.; Seongho, P.; Lee, C.-J. A review on the catalytic conversion of CO2 using H2 for synthesis of CO, methanol, and hydrocarbons. J. CO2 Util. 2021, 44, 101413. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, C.; Gao, P.; Wang, H.; Li, X.; Zhong, L.; Wei, W.; Sun, Y. A review of the catalytic hydrogenation of carbon dioxide into value-added hydrocarbons. Catal. Sci. Technol. 2017, 7, 4580–4598. [Google Scholar] [CrossRef]
- Tang, R.; Zhu, Z.; Li, C.; Xiao, M.; Wu, Z.; Zhang, D.; Zhang, C.; Xiao, Y.; Chu, M.; Genest, A.; et al. Ru-Catalyzed Reverse Water Gas Shift Reaction with Near-Unity Selectivity and Superior Stability. ACS Mater. Lett. 2021, 3, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Tawalbeh, M.; Javed, R.M.N.; Al-Othman, A.; Almomani, F.; Ajith, S. Unlocking the potential of CO2 hydrogenation into valuable products using noble metal catalysts: A comprehensive review. Environ. Technol. Innov. 2023, 31, 103217. [Google Scholar] [CrossRef]
- Pakhare, D.; Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 2014, 43, 7813–7837. [Google Scholar] [CrossRef]
- Ojelade, O.A.; Zaman, S.F. A Review on Pd Based Catalysts for CO2 Hydrogenation to Methanol: In-Depth Activity and DRIFTS Mechanistic Study. Catal. Surv. Asia 2020, 24, 11–37. [Google Scholar] [CrossRef]
- Kattel, S.; Liu, P.; Chen, J.G.G. Tuning Selectivity of CO2 Hydrogenation Reactions at the Metal/Oxide Interface. J. Am. Chem. Soc. 2017, 139, 9739–9754. [Google Scholar] [CrossRef]
- Al-Mamoori, A.; Rownaghi, A.A.; Rezaei, F. Combined capture and utilization of CO2 for syngas production over dual-function materials. ACS Sustain. Chem. Eng. 2018, 6, 13551–13561. [Google Scholar] [CrossRef]
- Arellano-Treviño, M.A.; He, Z.; Libby, M.C.; Farrauto, R.J. Catalysts and adsorbents for CO2 capture and conversion with dual function materials: Limitations of Ni-containing DFMs for flue gas applications. J. CO2 Util. 2019, 31, 143–151. [Google Scholar] [CrossRef]
- Duyar, M.S.; Wang, S.; Arellano-Treviño, M.A.; Farrauto, R.J. CO2 utilization with a novel dual function material (DFM) for capture and catalytic conversion to synthetic natural gas: An update. J. CO2 Util. 2016, 15, 65–71. [Google Scholar] [CrossRef]
- Proaño, L.; Arellano-Treviño, M.A.; Farrauto, R.J.; Figueredo, M.; Jeong-Potter, C.; Cobo, M. Mechanistic assessment of dual function materials, composed of Ru-Ni, Na2O/Al2O3 and Pt-Ni, Na2O/Al2O3, for CO2 capture and methanation by in-situ DRIFTS. Appl. Surf. Sci. 2020, 533, 147469. [Google Scholar] [CrossRef]
- Proaño, L.; Tello, E.; Arellano-Trevino, M.A.; Wang, S.; Farrauto, R.J.; Cobo, M. In-situ DRIFTS study of two-step CO2 capture and catalytic methanation over Ru,“Na2O”/Al2O3 Dual Functional Material. Appl. Surf. Sci. 2019, 479, 25–30. [Google Scholar] [CrossRef]
- Bermejo-López, A.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R. Mechanism of the CO2 storage and in situ hydrogenation to CH4. Temperature and adsorbent loading effects over Ru-CaO/Al2O3 and Ru-Na2CO3/Al2O3 catalysts. Appl. Catal. B Environ. 2019, 256, 117845. [Google Scholar] [CrossRef]
- Bobadilla, L.F.; Riesco-García, J.M.; Penelás-Pérez, G.; Urakawa, A. Enabling continuous capture and catalytic conversion of flue gas CO2 to syngas in one process. J. CO2 Util. 2016, 14, 106–111. [Google Scholar] [CrossRef]
- Wang, S.; Farrauto, R.J.; Karp, S.; Jeon, J.H.; Schrunk, E.T. Parametric, cyclic aging and characterization studies for CO2 capture from flue gas and catalytic conversion to synthetic natural gas using a dual functional material (DFM). J. CO2 Util. 2018, 27, 390–397. [Google Scholar] [CrossRef]
- Hu, L.; Urakawa, A. Continuous CO2 capture and reduction in one process: CO2 methanation over unpromoted and promoted Ni/ZrO2. J. CO2 Util. 2018, 25, 323–329. [Google Scholar] [CrossRef]
- Arellano-Treviño, M.A.; Kanani, N.; Jeong-Potter, C.W.; Farrauto, R.J. Bimetallic catalysts for CO2 capture and hydrogenation at simulated flue gas conditions. Chem. Eng. J. 2019, 375, 121953. [Google Scholar] [CrossRef]
- Bermejo-López, A.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R. Ni loading effects on dual function materials for capture and in-situ conversion of CO2 to CH4 using CaO or Na2CO3. J. CO2 Util. 2019, 34, 576–587. [Google Scholar] [CrossRef]
- Sietsma, J.R.A.; Jos van Dillen, A.; de Jongh, P.E.; de Jong, K.P. Application of ordered mesoporous materials as model supports to study catalyst preparation by impregnation and drying. In Studies in Surface Science and Catalysis; Gaigneaux, E.M., Devillers, M., De Vos, D.E., Hermans, S., Jacobs, P.A., Martens, J.A., Ruiz, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 162, pp. 95–102. [Google Scholar]
- Lu, Z.; Kunisch, J.; Gan, Z.; Bunian, M.; Wu, T.; Lei, Y. Gold catalysts synthesized using a modified incipient wetness impregnation method for propylene epoxidation. ChemCatChem 2020, 12, 5993–5999. [Google Scholar] [CrossRef]
- Danks, A.E.; Hall, S.R.; Schnepp, Z. The evolution of ‘sol–gel’ chemistry as a technique for materials synthesis. Mater. Horiz. 2016, 3, 91–112. [Google Scholar] [CrossRef]
- Wu, J.; Zheng, Y.; Fu, J.; Guo, Y.; Yu, J.; Chu, J.; Huang, P.; Zhao, C. Synthetic Ni–CaO–CeO2 dual function materials for integrated CO2 capture and conversion via reverse water–gas shift reaction. Sep. Purif. Technol. 2023, 317, 123916. [Google Scholar] [CrossRef]
- Pechini, M.P. Method of Preparing Lead and Alkaline Earth Titanates and Niobates and Coating Method Using the Same to Form a Capacitor. U.S. Patent US304434A, 11 July 1967. [Google Scholar]
- Sunde, T.O.L.; Grande, T.; Einarsrud, M.-A. Modified pechini synthesis of oxide powders and thin films. In Handbook of Sol-Gel Science and Technology; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Radfarnia, H.R.; Sayari, A. A highly efficient CaO-based CO2 sorbent prepared by a citrate-assisted sol–gel technique. Chem. Eng. J. 2015, 262, 913–920. [Google Scholar] [CrossRef]
- Jin, S.; Bang, G.; Liu, L.; Lee, C.-H. Synthesis of mesoporous MgO–CeO2 composites with enhanced CO2 capture rate via controlled combustion. Microporous Mesoporous Mater. 2019, 288, 109587. [Google Scholar] [CrossRef]
- Ma, X.; Li, X.; Cui, H.; Zhang, W.; Cheng, Z.; Zhou, Z. Metal oxide-doped Ni/CaO dual-function materials for integrated CO2 capture and conversion: Performance and mechanism. AIChE J. 2023, 69, e17520. [Google Scholar] [CrossRef]
- Sun, H.; Wang, J.; Zhao, J.; Shen, B.; Shi, J.; Huang, J.; Wu, C. Dual functional catalytic materials of Ni over Ce-modified CaO sorbents for integrated CO2 capture and conversion. Appl. Catal. B Environ. 2019, 244, 63–75. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, W.; Peng, P.; Zhang, Z.; Du, Q.; Shi, J.; Deng, L. A dual functional sorbent/catalyst material for in-situ CO2 capture and conversion to ethylene production. Fuel 2023, 351, 128701. [Google Scholar] [CrossRef]
- Machida, M.; Uto, M.; Kijima, T. Preparation of large surface area MnOx-ZrO2 for sorptive NOx removal. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 2000; Volume 143, pp. 855–862. [Google Scholar]
- Virji, M.; Stefaniak, A. A Review of Engineered Nanomaterial Manufacturing Processes and Associated Exposures; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Li, C.; Li, M.; van Veen, A.C. Synthesis of Nano-Catalysts in Flow Conditions Using Millimixers. In Advanced Nanomaterials for Catalysis and Energy; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1–28. [Google Scholar]
- Molina-Ramírez, S.; Cortés-Reyes, M.; Herrera, C.; Larrubia, M.; Alemany, L. CO2-SR Cyclic Technology: CO2 Storage and in situ Regeneration with CH4 over a new dual function NiBa unsupported catalyst. J. CO2 Util. 2020, 40, 101201. [Google Scholar] [CrossRef]
- Karami, D.; Mahinpey, N. Study of Al2O3 addition to synthetic Ca-based sorbents for CO2 sorption capacity and stability in cyclic operations. Can. J. Chem. Eng. 2015, 93, 102–110. [Google Scholar] [CrossRef]
- Huang, P.; Chu, J.; Fu, J.; Yu, J.; Li, S.; Guo, Y.; Zhao, C.; Liu, J. Influence of reduction conditions on the structure-activity relationships of NaNO3-promoted Ni/MgO dual function materials for integrated CO2 capture and methanation. Chem. Eng. J. 2023, 467, 143431. [Google Scholar] [CrossRef]
- Wegener Kofoed, M.V.; Jensen, M.B.; Mørck Ottosen, L.D. Chapter 12—Biological upgrading of biogas through CO2 conversion to CH4. In Emerging Technologies and Biological Systems for Biogas Upgrading; Aryal, N., Mørck Ottosen, L.D., Wegener Kofoed, M.V., Pant, D., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 321–362. [Google Scholar] [CrossRef]
- Seemann, M.; Thunman, H. 9—Methane synthesis. In Substitute Natural Gas from Waste; Materazzi, M., Foscolo, P.U., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 221–243. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, Y.; Guan, S.; Huang, J.; Wu, C. Direct and highly selective conversion of captured CO2 into methane through integrated carbon capture and utilization over dual functional materials. J. CO2 Util. 2020, 38, 262–272. [Google Scholar] [CrossRef]
- Kosaka, F.; Liu, Y.; Chen, S.-Y.; Mochizuki, T.; Takagi, H.; Urakawa, A.; Kuramoto, K. Enhanced Activity of Integrated CO2 Capture and Reduction to CH4 under Pressurized Conditions toward Atmospheric CO2 Utilization. ACS Sustain. Chem. Eng. 2021, 9, 3452–3463. [Google Scholar] [CrossRef]
- Porta, A.; Matarrese, R.; Visconti, C.G.; Castoldi, L.; Lietti, L. Storage Material Effects on the Performance of Ru-Based CO2 Capture and Methanation Dual Functioning Materials. Ind. Eng. Chem. Res. 2021, 60, 6706–6718. [Google Scholar] [CrossRef]
- Jeong-Potter, C.; Farrauto, R. Feasibility Study of Combining Direct Air Capture of CO2 and Methanation at Isothermal Conditions with Dual Function Materials. Appl. Catal. B Environ. 2021, 282, 119416. [Google Scholar] [CrossRef]
- Sun, H.; Wang, Y.; Xu, S.; Osman, A.I.; Stenning, G.; Han, J.; Sun, S.; Rooney, D.; Williams, P.T.; Wang, F.; et al. Understanding the interaction between active sites and sorbents during the integrated carbon capture and utilization process. Fuel 2021, 286, 119308. [Google Scholar] [CrossRef]
- Porta, A.; Visconti, C.G.; Castoldi, L.; Matarrese, R.; Jeong-Potter, C.; Farrauto, R.; Lietti, L. Ru-Ba synergistic effect in dual functioning materials for cyclic CO2 capture and methanation. Appl. Catal. B Environ. 2021, 283, 119654. [Google Scholar] [CrossRef]
- Cimino, S.; Russo, R.; Lisi, L. Insights into the cyclic CO2 capture and catalytic methanation over highly performing Li-Ru/Al2O3 dual function materials. Chem. Eng. J. 2022, 428, 131275. [Google Scholar] [CrossRef]
- Onrubia-Calvo, J.A.; Bermejo-López, A.; Pérez-Vázquez, S.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R. Applicability of LaNiO3-derived catalysts as dual function materials for CO2 capture and in-situ conversion to methane. Fuel 2022, 320, 123842. [Google Scholar] [CrossRef]
- Sun, Z.; Shao, B.; Zhang, Y.; Gao, Z.; Wang, M.; Liu, H.; Hu, J. Integrated CO2 capture and methanation from the intermediate-temperature flue gas on dual functional hybrids of AMS/CaMgO||NixCoy. Sep. Purif. Technol. 2023, 307, 122680. [Google Scholar] [CrossRef]
- Faria, A.C.; Trujillano, R.; Rives, V.; Miguel, C.V.; Rodrigues, A.E.; Madeira, L.M. Cyclic operation of CO2 capture and conversion into methane on Ni-hydrotalcite based dual function materials (DFMs). J. CO2 Util. 2023, 72, 102476. [Google Scholar] [CrossRef]
- Onrubia-Calvo, J.A.; Bermejo-López, A.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R. Ca doping effect on the performance of La1−xCaxNiO3/CeO2-derived dual function materials for CO2 capture and hydrogenation to methane. Appl. Catal. B Environ. 2023, 321, 122045. [Google Scholar] [CrossRef]
- Sakai, M.; Imagawa, H.; Baba, N. Layered-double-hydroxide-based Ni catalyst for CO2 capture and methanation. Appl. Catal. A Gen. 2022, 647, 118904. [Google Scholar] [CrossRef]
- Catarina Faria, A.; Miguel, C.V.; Ferreira, A.F.P.; Rodrigues, A.E.; Madeira, L.M. CO2 capture and conversion to methane with Ni-substituted hydrotalcite dual function extrudates. Chem. Eng. J. 2023, 476, 146539. [Google Scholar] [CrossRef]
- Wang, X.; Economides, M. CHAPTER 7—Gas-To-Liquids (GTL). In Advanced Natural Gas Engineering; Wang, X., Economides, M., Eds.; Gulf Publishing Company: Houston, TX, USA, 2009; pp. 243–287. [Google Scholar] [CrossRef]
- Cimino, S.; Boccia, F.; Lisi, L. Effect of alkali promoters (Li, Na, K) on the performance of Ru/Al2O3 catalysts for CO2 capture and hydrogenation to methane. J. CO2 Util. 2020, 37, 195–203. [Google Scholar] [CrossRef]
- Jeong-Potter, C.; Zangiabadi, A.; Farrauto, R. Extended aging of Ru-Ni, Na2O/Al2O3 dual function materials (DFM) for combined capture and subsequent catalytic methanation of CO2 from power plant flue gas. Fuel 2022, 328, 125283. [Google Scholar] [CrossRef]
- Bermejo-López, A.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R. Simulation-based optimization of cycle timing for CO2 capture and hydrogenation with dual function catalyst. Catal. Today 2022, 394–396, 314–324. [Google Scholar] [CrossRef]
- Bermejo-López, A.; Pereda-Ayo, B.; González-Marcos, J.A.; González-Velasco, J.R. Modeling the CO2 capture and in situ conversion to CH4 on dual function Ru-Na2CO3/Al2O3 catalyst. J. CO2 Util. 2020, 42, 101351. [Google Scholar] [CrossRef]
- Garbarino, G.; Bellotti, D.; Finocchio, E.; Magistri, L.; Busca, G. Methanation of carbon dioxide on Ru/Al2O3: Catalytic activity and infrared study. Catal. Today 2016, 277, 21–28. [Google Scholar] [CrossRef]
- Dreyer, J.A.H.; Li, P.; Zhang, L.; Beh, G.K.; Zhang, R.; Sit, P.H.L.; Teoh, W.Y. Influence of the oxide support reducibility on the CO2 methanation over Ru-based catalysts. Appl. Catal. B Environ. 2017, 219, 715–726. [Google Scholar] [CrossRef]
- González-Castaño, M.; Dorneanu, B.; Arellano-García, H. The reverse water gas shift reaction: A process systems engineering perspective. React. Chem. Eng. 2021, 6, 954–976. [Google Scholar] [CrossRef]
- Shao, B.; Hu, G.; Alkebsi, K.A.M.; Ye, G.; Lin, X.; Du, W.; Hu, J.; Wang, M.; Liu, H.; Qian, F. Heterojunction-redox catalysts of FexCoyMg10CaO for high-temperature CO2 capture and in situ conversion in the context of green manufacturing. Energy Environ. Sci. 2021, 14, 2291–2301. [Google Scholar] [CrossRef]
- Sun, S.; He, S.; Wu, C. Ni promoted Fe-CaO dual functional materials for calcium chemical dual looping. Chem. Eng. J. 2022, 441, 135752. [Google Scholar] [CrossRef]
- Sasayama, T.; Kosaka, F.; Liu, Y.Y.; Yamaguchi, T.; Chen, S.Y.; Mochizuki, T.; Urakawa, A.; Kuramoto, K. Integrated CO2 capture and selective conversion to syngas using transition-metal-free Na/Al2O3 dual-function material. J. CO2 Util. 2022, 60, 102049. [Google Scholar] [CrossRef]
- Li, L.; Miyazaki, S.; Yasumura, S.; Ting, K.W.; Toyao, T.; Maeno, Z.; Shimizu, K.-I. Continuous CO2 Capture and Selective Hydrogenation to CO over Na-Promoted Pt Nanoparticles on Al2O3. ACS Catal. 2022, 12, 2639–2650. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, G.; Yu, J.; Huang, P.; Sun, J.; Wang, R.; Wang, T.; Zhao, C. Tailoring the performance of Ni-CaO dual function materials for integrated CO2 capture and conversion by doping transition metal oxides. Sep. Purif. Technol. 2023, 305, 122455. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, C.; Guan, S.; Xu, S.; Williams, P.T.; Wu, C. Ni/support-CaO bifunctional combined materials for integrated CO2 capture and reverse water-gas shift reaction: Influence of different supports. Sep. Purif. Technol. 2022, 298, 121604. [Google Scholar] [CrossRef]
- Bao, Z.; Yu, F. Chapter Two—Catalytic Conversion of Biogas to Syngas via Dry Reforming Process. In Advances in Bioenergy; Li, Y., Ge, X., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 3, pp. 43–76. [Google Scholar]
- Hu, J.; Hongmanorom, P.; Galvita, V.V.; Li, Z.; Kawi, S. Bifunctional Ni-Ca based material for integrated CO2 capture and conversion via calcium-looping dry reforming. Appl. Catal. B Environ. 2021, 284, 119734. [Google Scholar] [CrossRef]
- Kim, S.M.; Abdala, P.M.; Broda, M.; Hosseini, D.; Copéret, C.; Müller, C. Integrated CO2 Capture and Conversion as an Efficient Process for Fuels from Greenhouse Gases. ACS Catal. 2018, 8, 2815–2823. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Jin, B.; Liang, Z. Layered double hydroxide derived bifunctional Ca-Fe-Mg material for integrated CO2 capture and utilization via chemical looping strategy. Chem. Eng. J. 2022, 431, 133826. [Google Scholar] [CrossRef]
- Tian, S.; Yan, F.; Zhang, Z.; Jiang, J. Calcium-looping reforming of methane realizes in situ CO2 utilization with improved energy efficiency. Sci. Adv. 2019, 5, eaav5077. [Google Scholar] [CrossRef]
- Jo, S.B.; Woo, J.H.; Lee, J.H.; Kim, T.Y.; Kang, H.I.; Lee, S.C.; Kim, J.C. CO2 green technologies in CO2 capture and direct utilization processes: Methanation, reverse water-gas shift, and dry reforming of methane. Sustain. Energy Fuels 2020, 4, 5543–5549. [Google Scholar] [CrossRef]
- Hu, J.; Hongmanorom, P.; Chirawatkul, P.; Kawi, S. Efficient integration of CO2 capture and conversion over a Ni supported CeO2-modified CaO microsphere at moderate temperature. Chem. Eng. J. 2021, 426, 130864. [Google Scholar] [CrossRef]
- Merkouri, L.-P.; Ramirez Reina, T.; Duyar, M.S. Feasibility of switchable dual function materials as a flexible technology for CO2 capture and utilisation and evidence of passive direct air capture. Nanoscale 2022, 14, 12620–12637. [Google Scholar] [CrossRef] [PubMed]
- Law, Z.X.; Pan, Y.-T.; Tsai, D.-H. Calcium looping of CO2 capture coupled to syngas production using Ni-CaO-based dual functional material. Fuel 2022, 328, 125202. [Google Scholar] [CrossRef]
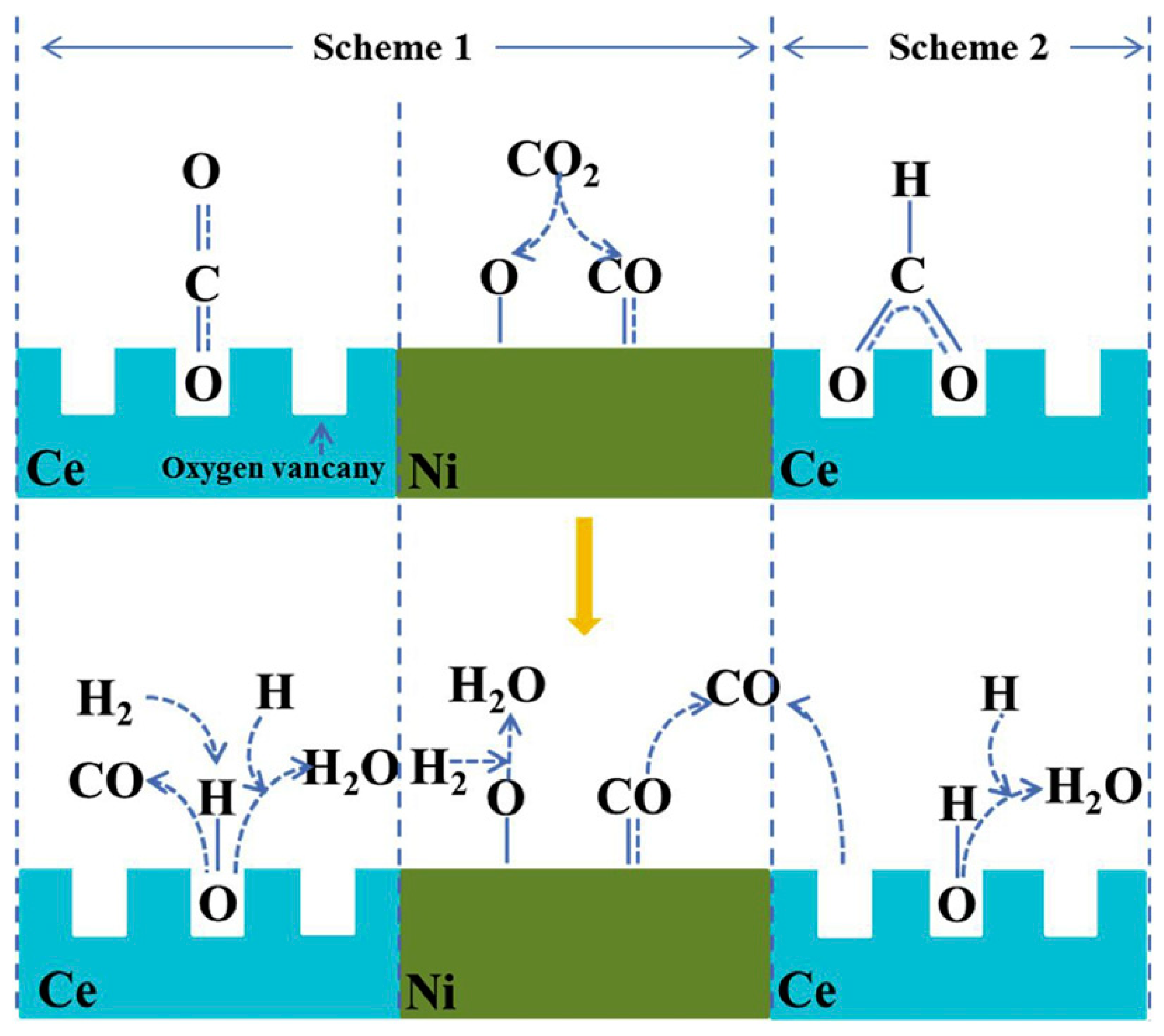
- Hussain, I.; Tanimu, G.; Ahmed, S.; Aniz, C.U.; Alasiri, H.; Alhooshani, K. A review of the indispensable role of oxygen vacancies for enhanced CO2 methanation activity over CeO2-based catalysts: Uncovering, influencing, and tuning strategies. Int. J. Hydrogen Energy 2023, 48, 24663–24696. [Google Scholar] [CrossRef]
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
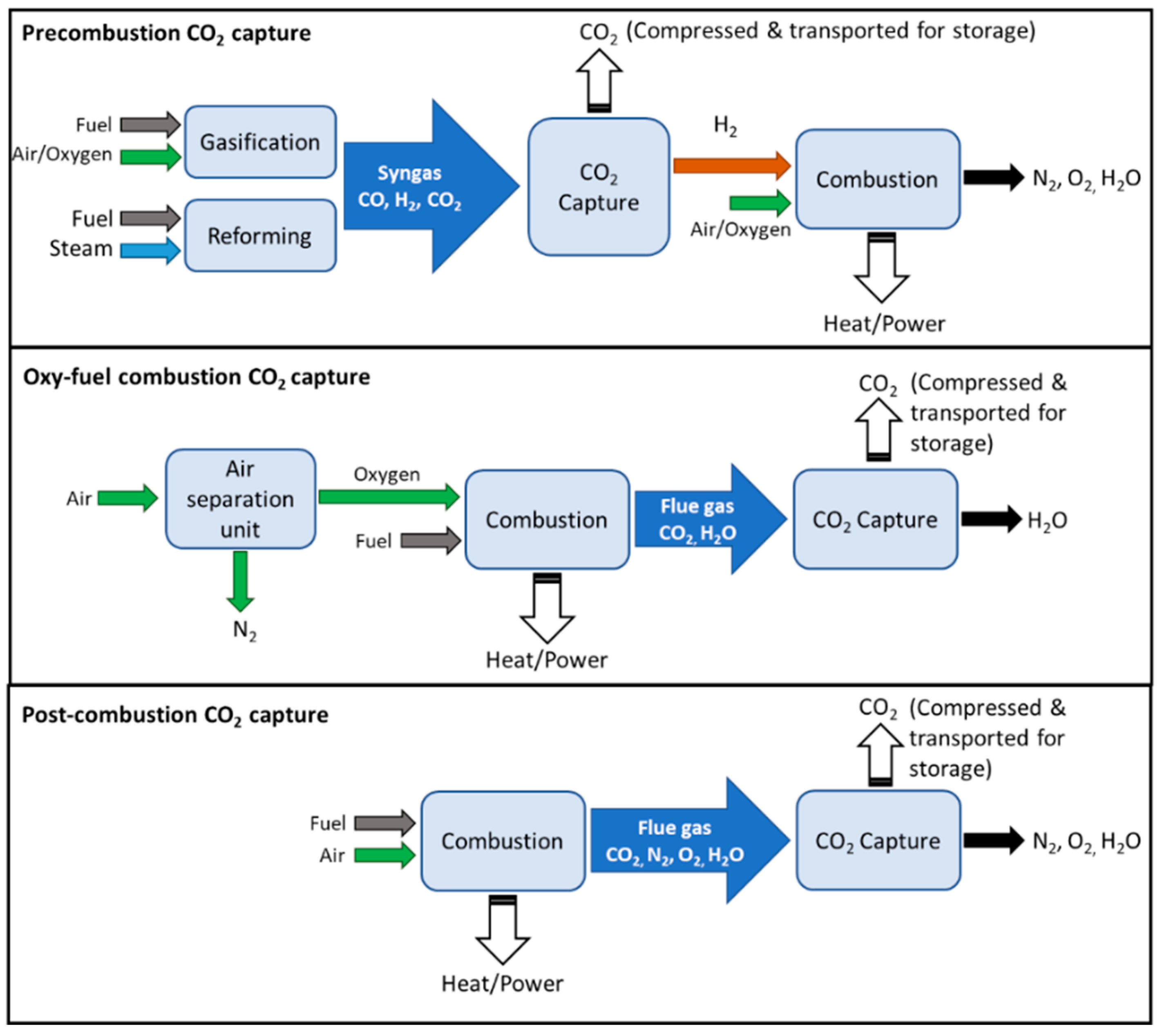
| Category | Pre-Combustion | Oxyfuel Combustion | Post-Combustion | |
|---|---|---|---|---|
| Amine Absorption | Solid Adsorption | |||
| CO2 recovery | 92–93% | 90–94% | 90–98% | 80–95% |
| Energy requirement | low energy | low energy | high regeneration energy | high regeneration energy |
| Costs | less expensive | moderately expensive | expensive | expensive |
| Challenges | increased process complexity | requires air separation unit | corrosion | solid attrition |
| high CO2 concentration | high CO2 concentration | solvent degradation | high pressure drop | |
| high pressure process | high temperature process | low CO2 concentration | easily poisoned by impurities (NOx, SOx) | |
| low CO2 concentration | ||||
| Authors | Year | Brief Description | Reference |
|---|---|---|---|
| Melo Bravo and Debecker | 2019 | Early review of DFMs with focus on CO2 methanation reaction and different types of reactor configurations | [12] |
| Omodolor et al. | 2020 | Overview of oxide- and carbonate-based CO2 adsorbents | [6] |
| Investigation of the active metals, material characteristics, and reaction conditions for Ni-, Ru-, and Rh-based DFMs in CO2 methanation, DRM, and RWGS reactions | |||
| Brief discussion on hydrotalcite-supported Fe-Cr-Cu catalyst as well as promoted Cu supported on alumina for syngas production | |||
| Merkouri et al. | 2021 | Chronological review of advances in DRM, RWGS, and CO2 methanation, as well as evaluation of the reaction mechanism of DFMs, by relating their performances with their physicochemical properties | [13] |
| Sun et al. | 2021 | Investigation of the process parameters and adsorbent–catalyst interaction on high-temperature CO2 capture combined with in situ DRM, RWGS, and methanation reactions | [14] |
| Sabri et al. | 2021 | Environmental and economic evaluation of CO2 capture and utilization process integration, as well as catalyst development for CO2 conversion in various systems, such as photocatalysis, electrocatalysis, and thermocatalysis | [15] |
| Li et al. | 2021 | Discussion on the performances of dual-function oxide particles for CO2 capture integrated with DRM, CO2 hydrogenation, and chemical looping | [16] |
| DFM | Synthesis Method | Carbonation Conditions | Methanation Conditions | CO2 Sorption Capacity (mmol/g) | CO2 Conversion (%) | Methanation Capacity (mmol/g) | Ref. |
|---|---|---|---|---|---|---|---|
| 5% Ru 10% CaO/Al2O3 | IWI | 320 °C, 10%CO2/air | 320 °C, 5%H2/N2 | 0.41 | 76.17% | 0.31 | [11] |
| 5% Ru 10% CaO/Al2O3 | IWI | 320 °C, 10%CO2/N2 | 320 °C, 4%H2/N2 | - | - | 0.5 | [93] |
| 5% Ru 10% K2CO3/Al2O3 | IWI | 320 °C, 10%CO2/N2 | 320 °C, 4%H2/N2 | - | - | 0.91 | [93] |
| 5% Ru 10% Na2CO3/Al2O3 | IWI | 320 °C, 10%CO2/N2 | 320 °C, 4%H2/N2 | - | - | 1.05 | [93] |
| 5% Ru 6.1% Na2O/γ-Al2O3 | IWI | 300 °C, simulated flue gas | 300 °C, 15%H2/N2 | 0.4 (50-cycle average) | 77% | 0.32 | [98] |
| 5% Ru-6.1% “Na2O”/Al2O3 | IWI | 320 °C, 10%CO2/N2 | 320 °C, 10%H2/N2 | 0.651 | 96% | 0.614 | [92] |
| 0.5% Rh-6.1% “Na2O”/Al2O3 | IWI | 320 °C, 10%CO2/N2 | 320 °C, 10%H2/N2 | 0.626 | 69% | 0.422 | [92] |
| 15% Ni 15% CaO/Al2O3 | IWI | 280–520 °C, 10%CO2/Ar | 280–520 °C, 10%H2/Ar | - | - | 0.142 | [101] |
| 15% Ni 10% Na2CO3/Al2O3 | IWI | 280–520 °C, 10%CO2/Ar | 280–520 °C, 10%H2/Ar | - | - | 0.186 | [101] |
| 4% Ru 10% CaO/Al2O3 | IWI | 370 °C, 1.4%CO2/Ar | 370 °C, 10%H2/Ar | 0.253 | - | 0.272 | [96] |
| 4% Ru 10% Na2CO3/Al2O3 | IWI | 370 °C, 1.4%CO2/Ar | 370 °C, 10%H2/Ar | 0.391 | - | 0.398 | [96] |
| 1% Ru, 10% Ni, 6.1% “Na2O”/Al2O3 | IWI | 320 °C, 7.5%CO2-4.5%O2-15%H2O-balance N2 | 320 °C, 15%H2/N2 | 0.52 (3-cycle average) | 81% (3-cycle average) | 0.38 (3-cycle average) | [100] |
| 1% Ru, 10% Ni, 6.1% “Na2O”/Al2O3 | IWI | 320 °C, 7.5%CO2-4.5%O2-15%H2O-balance N2 | 320 °C, 15%H2/N2 | 0.52 (20-cycle average) | 100% (20-cycle average) | 0.38 (20-cycle average) | [100] |
| 1% Pt 10% Ni 6.1% “Na2O”/Al2O3 | IWI | 320 °C, 7.5%CO2-4.5%O2-15%H2O-balance N2 | 320 °C, 15%H2/N2 | 0.35 (3-cycle average) | 87% (3-cycle average) | 0.25 (3-cycle average) | [100] |
| 2.5Ru/CeO2-MgO | Impregnation/physical mixing | 300 °C, 65%CO2/N2 | 300 °C, 5%H2/N2 | - | 60% (1st cycle); 39% (10th cycle) | 5.73 (1st cycle); 1.13 (10th cycle) | [121] |
| 5Ru/CeO2-MgO | Impregnation/physical mixing | 300 °C, 65%CO2/N2 | 300 °C, 5%H2/N2 | - | 74% (1st cycle); 79% (10th cycle) | 6.6 (1st cycle); 3.36 (10th cycle) | [121] |
| 10Ru/CeO2-MgO | Impregnation/physical mixing | 300 °C, 65%CO2/N2 | 300 °C, 5%H2/N2 | - | 89% (1st cycle); 69% (10th cycle) | 7.07 (1st cycle); 2.31 (10th cycle) | [121] |
| Ni/Na-γ-Al2O3 | IWI | 450 °C, 5%CO2/N2 | 450 °C, H2 | 0.209 | 96% | 0.188 | [122] |
| Ni/Na-γ-Al2O3 | IWI | 450 °C, 5%CO2/N2 | 450 °C, H2 | 0.299 | 92% | 0.266 | [122] |
| 0.95% Ru-5% K/Al2O3 | IWI | 350 °C, 2.5%H2O/3%O2/1%CO2/He | 350 °C, 4%H2/He | 0.028 (3rd cycle) | - | 0.028 (3rd cycle) | [123] |
| 0.95% Ru-5.1% Ca/Al2O3 | IWI | 350 °C, 2.5%H2O/3%O2/1%CO2/He | 350 °C, 4%H2/He | 0.116 (3rd cycle) | - | 0.036 (3rd cycle) | [123] |
| 0.84% Ru-16% Ba/Al2O3 | IWI | 350 °C, 2.5%H2O/3%O2/1%CO2/He | 350 °C, 4%H2/He | 0.165 (3rd cycle) | - | 0.080 (3rd cycle) | [123] |
| 0.5% Ru, 6.1% “Na2O”/Al2O3 | IWI | 320 °C, 400 ppm CO2/air | 320 °C, 15% H2/N2 | 0.2 | - | 0.15 | [124] |
| 1%Ni/CeCaO-imp | One-pot citric acid chelation/wet impregnation | 550 °C, 15%CO2/N2 | 550 °C, H2 | 10.6 | 42% | 3.3 | [125] |
| 1%Ni/CeCaCO3-imp | One-pot citric acid chelation/carbonation/wet impregnation | 550 °C, 15%CO2/N2 | 550 °C, H2 | 14.1 | 52% | 6 | [125] |
| 1%Ni/CeO2-CaO-phy | Hydrothermal/one-pot citric acid chelation/physical mixing | 550 °C, 15%CO2/N2 | 550 °C, H2 | 15.3 | 62% | 8 | [125] |
| Ru-BaO/Al2O3 (intimate mixture) | IWI | 350 °C, 1%CO2/He | 350 °C, 4%H2/He | 0.025 | 20% at 291 °C | 0.151 | [126] |
| BaO/Al2O3 + Ru/Al2O3 (mechanical mixture) | IWI | 350 °C, 1%CO2/He | 350 °C, 4%H2/He | 0.055 | 20% at 362 °C | 0.097 | [126] |
| 5Li-Ru/A | IWI | 263 °C, 10%CO2/N2 | 263 °C, 10%H2/N2 | - | 98% | 0.32 | [127] |
| 5Li-Ru/A | IWI | 293 °C, 10%CO2/N2 | 293 °C, 10%H2/N2 | - | 97.4% | 0.34 | [127] |
| 5Li-Ru/A | IWI | 318 °C, 10%CO2/N2 | 318 °C, 10%H2/N2 | - | 95.4% | 0.29 | [127] |
| 30% LaNiO3/CeO2 | Citric acid/impregnation | 400 °C, 1.4%CO2/Ar, | 400 °C, 10%H2/Ar | 0.089 (3-cycle average) | - | 0.08 (3-cycle average) | [128] |
| AMS/CaMgO||Ni1-Co3 | Sol–gel/solvent evaporation | 350 °C, 95%CO2/N2 | 350 °C, 10%H2/N2 | 14.7 (sorbent mass basis) | 76.4% | 5.46 (catalyst mass basis) | [129] |
| AMS/CaMgO||Ni1-Co1 | Sol–gel/solvent evaporation | 350 °C, 95%CO2/N2 | 350 °C, 10%H2/N2 | 14.4 (sorbent mass basis) | 90% | 9.97 (catalyst mass basis) | [129] |
| AMS/CaMgO||Ni3-Co1 | Sol–gel/solvent evaporation | 350 °C, 95%CO2/N2 | 350 °C, 10%H2/N2 | 14.1 (sorbent mass basis) | 75% | 6.66 (catalyst mass basis) | [129] |
| cDFM-2-0.4Ni-10Cs | Co-precipitation/IWI | 350 °C, 15%CO2/N2 | 350 °C, H2 | 0.48 | - | 0.33 | [130] |
| 20% LaNiO3/CeO2 | Citric acid/impregnation | 480 °C, 10%CO2/Ar | 480 °C, 10% H2/Ar | 0.113 | - | 0.075 | [131] |
| 20% La0.5Ca0.5NiO3/CeO2 | Citric acid/impregnation | 480 °C, 10%CO2/Ar | 480 °C, 10% H2/Ar | 0.175 | - | 0.140 | [131] |
| Ni/CaZrO | Sol–gel | 600 °C, 15%CO2/N2 | 600 °C, 66.7%H2/N2 | 9.9–9 (20 cycles) | 86.3–78% (20 cycles) | 6.7 (6th to 20th cycle) | [110] |
| NiO-MgO | Co-precipitation | 320 °C 10%CO2/5%O2/He | 320 °C, 20%H2/He | ~0.3 (10 cycles) | ~96% (10 cycles) | ~0.26 (10 cycles) | [132] |
| Cs-impregnated Ni-MgO-Al2O3 extrudate (EI) | Co-precipitation/impregnation | 350 °C, 15%CO2/N2 | 350 °C, H2 | 0.24 | 75% (250h on-stream) | 0.18 (250h on-stream) | [133] |
| DFM | Synthesis Method | Carbonation Conditions | RWGS Conditions | CO2 Sorption Capacity (mmol/g) | CO2 Conversion (%) | Recycle Study and CO Yield Loss | Ref. |
|---|---|---|---|---|---|---|---|
| Ni-CaO-CeO2 (CP) | Co-precipitation with acetate precursors | 650 °C, 10% CO2/N2 | 650 °C, 10% H2/N2 | 9.81 | 75 | 10 cycles, ~10% loss | [105] |
| Ni-CaO-CeO2 (WM) | Wet mixing with acetate precursors | 650 °C, 10% CO2/N2 | 650 °C, 10% H2/N2 | 8.54 | 84 | 10 cycles, stable | [105] |
| Ni-CaO-CeO2 (SG-A) | Citric acid-based sol–gel with acetate precursors | 650 °C, 10% CO2/N2 | 650 °C, 10% H2/N2 | 15.34 | 92 | 10 cycles, 11.6% loss | [105] |
| Ni-CaO-CeO2 (SG-N) | Citric acid-based sol–gel with nitrate precursors | 650 °C, 10% CO2/N2 | 650 °C, 10% H2/N2 | 13.15 | 96 | 10 cycles, 3.5% loss | [105] |
| Ce-doped Ni/CaO (Ca1Ni0.1Ce0.033) | Citric acid-based sol–gel with nitrate precursors | 650 °C, 15% CO2/N2 | 650 °C, 5% H2/N2 | 14.1 | 51.8 | 20 cycles, stable | [111] |
| Ni-CaO-ZrO2-12 | Citric acid-based sol–gel with nitrate precursors | 650 °C, 10% CO2/N2 | 650 °C, 45% H2/N2 | 16.72 | 63.2 | 12 cycles, 18.6% loss | [146] |
| Ni-CaO-6ZrO2-6CeO2 | Citric acid-based sol–gel with nitrate precursors | 650 °C, 10% CO2/N2 | 650 °C, 45% H2/N2 | 11.88 | 72.1 | 12 cycles, 12% loss | [146] |
| Ni/CeO2-CaO | Physical mixing | 650 °C, 20% CO2/N2 | 650 °C, 5% H2/N2 | 9.58 | 56.1 | 20 cycles, <5% loss | [147] |
| Na/Al2O3 | Wetness impregnation | 500 °C, 5% CO2/N2 | 500 °C, H2 | 0.135 | 76.9 | 50 cycles at 450 °C, stable | [144] |
| Na/Al2O3 | Wetness impregnation | 500 °C, 400 ppm CO2/N2 | 500 °C, H2 | 0.0907 | 77.7 | - | [144] |
| Na-Pt/Al2O3 | Wetness impregnation | 350 °C, 1% CO2/10% O2/N2 | 350 °C, 5% H2/N2 | 0.19 | 89 | 6000 cycles, stable | [145] |
| Ni1Fe9-CaO | Citric acid-based sol–gel with nitrate precursors | 650 °C, 10% CO2/N2 | 650 °C, H2 | 14.78 | 82.5 | 10 cycles, 20.9% loss | [143] |
| Fe5Co5Mg10CaO | Citric acid-based sol–gel with nitrate precursors | 650 °C, 10% CO2/N2 | 650 °C, H2 | 9.2 | 90 | 10 cycles, stable | [142] |
| DFM | Synthesis Method | Carbonation Conditions | Dry Reforming Conditions | CO2 Sorption Capacity | CO2 Conversion | CH4 Conversion | Ref. |
|---|---|---|---|---|---|---|---|
| Ni10-(K-Mg)25/(γ-Al2O3)75 | Sol–gel/wet impregnation | 650 °C, 10%CO2/N2 | 650 °C, 5%C2H6/N2 | 0.22 mmol/g DFM | 14% | 16% (ethane) | [91] |
| Ni10-(Na-Mg)50/(γ-Al2O3)50 | Sol–gel/wet impregnation | 650 °C, 10%CO2/N2 | 650 °C, 5%C2H6/N2 | 0.16 mmol/g DFM | 60% | 47% (ethane) | [91] |
| Ni10-(K-Ca)50/(γ-Al2O3)50 | Sol–gel/wet impregnation | 650 °C, 10%CO2/N2 | 650 °C, 5%C2H6/N2 | 0.99 mmol/g DFM | 65% | 100% (ethane) | [91] |
| Ni10-(Na-Ca)50/(γ-Al2O3)50 | Sol–gel/wet impregnation | 650 °C, 10%CO2/N2 | 650 °C, 5%C2H6/N2 | 0.63 mmol/g DFM | 75% | 100% (ethane) | [91] |
| Ni/Ca-Zr | Precipitation/ammoniacal sol/impregnation | 720 °C, 5%CO2/Ar | 720 °C, 8%CH4/Ar | ~3.64 mmol/g CaO (25 cycles) | - | ~32% | [149] |
| NiCe/Ca-Zr | Precipitation/ammoniacal sol/impregnation | 720 °C, 5%CO2/Ar | 720 °C, 8%CH4/Ar | ~5 mmol/g CaO (25 cycles) | - | ~45% | [149] |
| Ni/MgO-Al2O3 (mixed with CaO) | Co-precipitation | 720 °C, 20%CO2/N2 | 720 °C, 2.4%CH4/N2 | 14.1 mmol/g DFM (initial) | 99.92% | 99.94% | [150] |
| Ca-Fe-Mg oxide | Co-precipitation | 600 °C, 100%CO2 | 900 °C, 4.5%CH4/N2 | 0.64 mmol/g DFM | - | ~50% | [151] |
| DFM | Synthesis Method | Carbonation Conditions | Dry Reforming Conditions | CO2 Sorption Capacity | H2 Yield | CO Yield | Ref. |
|---|---|---|---|---|---|---|---|
| CaO/Ni4 | Sol–gel | 600 °C, 20%CO2/N2 | 800 °C, 20%CH4/N2 | ~5.7 mmol/g CaO (initial) | 5 mmol/g DFM | 4.3 mmol/g DFM | [152] |
| CaO/Ni9 | Sol–gel | 600 °C, 20%CO2/N2 | 800 °C, 20%CH4/N2 | ~5.1 mmol/g CaO (initial) | 5 mmol/g DFM | 4.5 mmol/g DFM | [152] |
| CaO/Ni19 | Sol–gel | 600 °C, 20%CO2/N2 | 800 °C, 20%CH4/N2 | ~4.5 mmol/g CaO (initial) | 3.5 mmol/g DFM | 3.5 mmol/g DFM | [152] |
| Ni-CaO catal-sorbents | Sol–gel | 700 °C, 10%CO2/10%H2O/N2 | 700 °C, 10%CH4/N2 | 14.8 mmol/g DFM | 131.7 mmol/g DFM | 20.2 mmol/g DFM | [153] |
| Ni/Ca85Ce15 | Hydrothermal (sorbent); impregnation (DFM) | 650 °C, 10%CO2/Ar | 650 °C, 6%CH4/Ar | ~13 mmol/g CaO (initial) | - | - | [154] |
| 15% Ni-1% Ru, 10% Na2O/CeO2–Al2O3 | Sequential impregnation | 650 °C, 10%CO2/N2 | 650 °C, 10%CH4/N2 | 0.16 mmol/g DFM | 25.774 mmol/g DFM | 0.153 mmol/g DFM | [155] |
| 15% Ni-1% Ru, 10% K2O/CeO2–Al2O3 | Sequential impregnation | 650 °C, 10%CO2/N2 | 650 °C, 10%CH4/N2 | 0.18 mmol/g DFM | 22.512 mmol/g DFM | 0.239 mmol/g DFM | [155] |
| 15% Ni-1% Ru, 10% CaO/CeO2–Al2O3 | Sequential impregnation | 650 °C, 10%CO2/N2 | 650 °C, 10%CH4/N2 | 0.26 mmol/g DFM | 32.639 mmol/g DFM | 0.338 mmol/g DFM | [155] |
| CaO-0.05Ni-0.05CeO2 | Co-precipitation | 600 °C, 2%CO2/N2 | 800 °C, 2%CH4/N2 | 10.3 mmol/g CaO (initial); 7.9 mmol/g CaO (10th cycle) | 754.4 mmol/g Ni (max, 2nd cycle) | 454.6 mmol/g Ni (max, 2nd cycle) | [156] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, W.J.; Gunawan, P. Integration of CO2 Capture and Conversion by Employing Metal Oxides as Dual Function Materials: Recent Development and Future Outlook. Inorganics 2023, 11, 464. https://doi.org/10.3390/inorganics11120464
Tan WJ, Gunawan P. Integration of CO2 Capture and Conversion by Employing Metal Oxides as Dual Function Materials: Recent Development and Future Outlook. Inorganics. 2023; 11(12):464. https://doi.org/10.3390/inorganics11120464
Chicago/Turabian StyleTan, Wei Jie, and Poernomo Gunawan. 2023. "Integration of CO2 Capture and Conversion by Employing Metal Oxides as Dual Function Materials: Recent Development and Future Outlook" Inorganics 11, no. 12: 464. https://doi.org/10.3390/inorganics11120464
APA StyleTan, W. J., & Gunawan, P. (2023). Integration of CO2 Capture and Conversion by Employing Metal Oxides as Dual Function Materials: Recent Development and Future Outlook. Inorganics, 11(12), 464. https://doi.org/10.3390/inorganics11120464