








Modulation of Ferrocene–Ferrocene Interactions by Varying Their Reciprocal Positions in L-Dap/Aib Helical Peptides

 , ,
, , 
Abstract
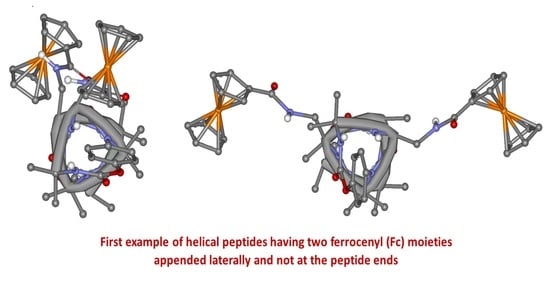
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Peptide Synthesis
2.2. Crystal-State Conformational Analysis
2.3. Solution Conformational Analysis
2.4. CV and DPV Analyses
2.5. Vis-MIR-IR Chemical Oxidation
3. Materials and Methods
3.1. Syntheses and Analyses
3.2. X-ray Diffraction
3.3. Nuclear Magnetic Resonance
3.4. Cyclic Voltammetry
3.5. Differential Pulse Voltammetry
3.6. UV-Vis Analysis
3.7. FT-IR Analysis
3.8. Circular Dichroism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Moriuchi, T. Helical Chirality of Ferrocene Moieties in Cyclic Ferrocene-Peptide Conjugates. Eur. J. Inorg. Chem. 2022, e202100902. [Google Scholar] [CrossRef]
- Tassinari, F.; Jayarathna, D.R.; Kantor-Uriel, N.; Davis, K.L.; Varade, V.; Achim, C.; Naaman, R. Chirality Dependent Charge Transfer Rate in Oligopeptides. Adv. Mater. 2018, 30, 1706423. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Tong, F.; Zhang, W.; Zhou, Y.; He, S.Q.; Xie, R.; Lei, T.; Wang, Y.S.; Peng, S.J.; Li, Z.F.; et al. Self-Delivered Supramolecular Nanomedicine with Transformable Shape for Ferrocene-Amplified Photodynamic Therapy of Breast Cancer and Bone Metastases. Adv. Funct. Mater. 2021, 31, 2104645. [Google Scholar] [CrossRef]
- Gómez, J.; Sierra, D.; Ojeda, C.; Thavalingam, S.; Miller, R.; Guzmán, F.; Metzler-Nolte, N. Solid-phase synthesis and evaluation of linear and cyclic ferrocenoyl/ruthenocenoyl water-soluble hexapeptides as potential antibacterial compounds. J. Biol. Inorg. Chem. 2021, 26, 599–615. [Google Scholar] [CrossRef] [PubMed]
- Gurunarayanan, V.; Ramapanicker, R. Amphiphilic conjugates of ferrocene with amino acids and peptides: Design, synthesis, and studies on their aggregation behavior. J. Pep. Sci. 2021, 27, e3332. [Google Scholar] [CrossRef] [PubMed]
- Costa, N.C.S.; Piccoli, J.P.; Santos-Filho, N.A.; Clementino, L.C.; Fusco-Almeida, A.M.; De Annunzio, S.R.; Fontana, C.R.; Verga, J.B.; Eto, S.F.; Pizauro-Junior, J.M.; et al. Antimicrobial Activity of RP-1 Peptide Conjugate with Ferrocene Group. PLoS ONE 2020, 15, e0228740. [Google Scholar] [CrossRef]
- Peter, S.; Aderibigbe, B.A. Ferrocene-Based Compounds with Antimalaria/Anticancer Activity. Molecules 2019, 24, 3604. [Google Scholar] [CrossRef]
- Falcone, N.; Kraatz, H.-B. Ferrocene Peptide-based Supramolecular Gels: Current Trends and Applications. In Advances in Bioorganometallic Chemistry; Hirao, T., Moriuchi, T., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; Chapter 3; pp. 57–74. [Google Scholar]
- Yu, J.; Horsley, J.R.; Abell, A.D. Peptides as Bio-Inspired Electronic Materials: An Electrochemical and First-Principles Perspective. Acc. Chem. Res. 2018, 51, 2237–2246. [Google Scholar] [CrossRef]
- Albada, B.; Metzler-Nolte, N. Highly Potent Antibacterial Organometallic Peptide Conjugates. Acc. Chem. Res. 2017, 50, 2510–2518. [Google Scholar] [CrossRef]
- Hirao, T.; Moriuchi, M.; Groß, A. Functionalized Redox Systems. Synthetic Reactions and Design of π- and Bio-Conjugates. In Functionalized Redox Systems Chp. 4 Bioconjugates to Induce Chirality Organization; Hirao, T., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; Chapter 4; pp. 111–150. [Google Scholar]
- Adhikariab, B.; Kraatz, H.-B. Redox-triggered changes in the self-assembly of a ferrocene–peptide conjugated. Chem. Commun. 2014, 50, 5551–5553. [Google Scholar] [CrossRef]
- Takahashi, S.; Anzai, J.-I. Recent Progress in Ferrocene-Modified Thin Films and Nanoparticles for Biosensors. Materials 2013, 6, 5742–5762. [Google Scholar] [CrossRef] [PubMed]
- Zuccarello, L.; Barbosa, C.; Todorovic, S.; Silveira, C.M. Electrocatalysis by Heme Enzymes—Applications in Biosensing. Catalysts 2021, 11, 218. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, P.; Jin, X.; Wang, Y.; Yuan, H.; Zhu, X. Enzymatic biofuel cells based on protein engineering: Recent advances and future prospects. Biomater. Sci. 2020, 8, 5230–5524. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.Y.; Myung, N.V. Recent Advances in the Direct Electron Transfer-Enabled Enzymatic Fuel Cells. Front. Chem. 2021, 8, 620153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yagnik, G.; Peng, J.; Wang, J.; Xu, H.H.; Hao, Y.; Liu, Y.-N.; Zhou, F. Kinetic Studies of Inhibition of the Aβ(1–42) Aggregation Using a Ferrocene-tagged β-Sheet Breaker Peptide. Anal Biochem. 2013, 434, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Afrasiabi, R.; Kraatz, H.B. Stimuli-Responsive Supramolecular Gelation in Ferrocene–Peptide Conjugates. Chem. Eur. J. 2013, 19, 17296–17300. [Google Scholar] [CrossRef]
- Martić, S.; Labib, M.; Shipman, P.-O.; Kraatz, H.-B. Ferrocene-peptido conjugates: From synthesis to sensory applications. Dalton Trans. 2011, 40, 7264–7290. [Google Scholar] [CrossRef]
- Moriuchi, T.; Kikushima-Honda, N.; Ohmura, S.D.; Hirao, T. Design and characterization of ferrocene–peptide–oligoaniline conjugates. Tetrahedron Lett. 2010, 51, 4530–4533. [Google Scholar] [CrossRef]
- Okamoto, S.; Morita, T.; Kimura, S. Electron Transfer through a Self-Assembled Monolayer of a Double-Helix Peptide with Linking the Terminals by Ferrocene. Langmuir 2009, 25, 3297–3304. [Google Scholar] [CrossRef]
- Adachi, T.; Kitazumi, Y.; Shirai, O.; Kano, K. Direct Electron Transfer-Type Bioelectrocatalysis of Redox Enzymes at Nanostructured Electrodes. Catalysts 2020, 10, 236. [Google Scholar] [CrossRef]
- Toniolo, C.; Benedetti, E. The polypeptide 310-helix. Trends Biochem. Sci. 1991, 16, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Aravinda, S.; Datta, S.; Shamala, N.; Balaram, P. Hydrogen-Bond Lengths in Polypeptide Helices: No Evidence for Short Hydrogen Bonds. Angew. Chem. Int. Ed. 2004, 43, 6728–6731. [Google Scholar] [CrossRef] [PubMed]
- Donoli, A.; Marcuzzo, V.; Moretto, A.; Toniolo, C.; Cardena, R.; Bisello, A.; Santi, S. Charge Mapping in 310-Helical Peptide Chains by Oxidation of the Terminal Ferrocenyl Group. Org. Lett. 2011, 13, 1282–1285. [Google Scholar] [CrossRef] [PubMed]
- Biondi, B.; Bisello, A.; Cardena, R.; Schiesari, R.; Facci, M.; Cerveson, L.; Rancan, M.; Formaggio, F.; Santi, S. Conformational Analysis and Through-Chain Charge Propagation in Ferrocenyl-Conjugated Homopeptides of 2,3-Diaminopropionic acid (Dap). Eur. J. Inorg. Chem. 2022, e202100966. [Google Scholar] [CrossRef]
- Crisma, M.; Formaggio, F.; Alemán, C.; Torras, J.; Ramakrishnan, C.; Kalmankar, N.; Balaram, P.; Toniolo, C. The fully-extended conformation in peptides and proteins. Pept. Sci. 2018, 110, e23100. [Google Scholar] [CrossRef]
- Biondi, B.; Bisello, A.; Cardena, R.; Schiesari, R.; Cerveson, L.; Facci, M.; Rancan, M.; Formaggio, F.; Santi, S. Flat, Ferrocenyl-Conjugated Peptides: A Combined Electrochemical and Spectroscopic Study. ChemElectroChem 2021, 8, 2693–2700. [Google Scholar] [CrossRef]
- Donoli, A.; Marcuzzo, V.; Moretto, M.; Crisma, M.; Toniolo, C.; Cardena, R.; Bisello, A.; Santi, S. New bis-Ferrocenyl End-Capped Peptides: Synthesis and Charge Transfer Properties. Pept. Sci. 2012, 100, 14–24. [Google Scholar] [CrossRef]
- Santi, S.; Bisello, A.; Cardena, R.; Tomelleri, S.; Schiesari, R.; Biondi, B.; Crisma, M.; Formaggio, F. Flat, Cα,β-Didehydroalanine Foldamers with Ferrocene Pendants: Assessing the Role of α-Peptide Dipolar Moments. ChemPlusChem 2021, 86, 723–730. [Google Scholar] [CrossRef]
- Santi, S.; Biondi, B.; Cardena, R.; Bisello, A.; Schiesari, R.; Tomelleri, S.; Facci, M.; Crisma, F.; Formaggio, F. Helical versus Flat Bis-Ferrocenyl End-Capped Peptides: The Influence of the Molecular Skeleton on Redox Properties. Molecules 2022, 27, 6128. [Google Scholar] [CrossRef]
- Nuskol, M.; Studen, M.; Meden, A.; Kodrin, I.; Cakic Semencic, M. Tight turn in dipeptide bridged ferrocenes: Synthesis, X-ray structural, theoretical and spectroscopic studies. Polyhedron 2019, 161, 137–144. [Google Scholar] [CrossRef]
- Venkatachalam, C.M. Stereochemical criteria for polypeptides and proteins. V. Conformation of a system of three linked peptide units. Biopolymers 1968, 6, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- König, W.; Geiger, R. A new method for synthesis of peptides: Activation of the carboxyl group with dicyclohexylcarbodiimide using 1-hydroxybenzotriazoles as additives. Chem. Ber. 1970, 103, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.E.; Kay, C.M.; Hodges, R.S. Synthetic model proteins: The relative contribution of leucine residues at the nonequivalent positions of the 3-4 hydrophobic repeat to the stability of the two-stranded α-helical coiled-coil. Biochemistry 1992, 31, 5739–5746. [Google Scholar] [CrossRef]
- Wallimann, P.; Kennedy, R.J.; Kemp, D.S. Large Circular Dichroism Ellipticities for N-Templated Helical Polypeptides Are Inconsistent with Currently Accepted Helicity Algorithms. Angew. Chem. Int. Ed. 1999, 38, 1290–1292. [Google Scholar] [CrossRef]
- Metzler-Nolte, N.; Salmain, M. Ferrocenes: Ligands, Materials and Biomolecules; Štepnicka, P., Ed.; Wiley: Chichester, UK, 2008; pp. 499–639. [Google Scholar]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Rance, M.; Sørensen, O.W.; Bodenhausen, G.; Wagner, G.; Ernst, R.R.; Wüthrich, K. Improved spectral resolution in COSY 1H NMR spectra of proteins via double quantum filtering. Biochem. Biophys. Res. Commun. 1983, 117, 479–485. [Google Scholar] [CrossRef]
- Griesinger, C.; Otting, G.; Wüthrich, K.; Ernst, R.R. Clean TOCSY for proton spin system identification in macromolecules. J. Am. Chem. Soc. 1988, 110, 7870–7872. [Google Scholar] [CrossRef]
- Wüthrich, K. NMR of Proteins and Nucleic Acids; Wiley: Chichester, UK, 1991. [Google Scholar]
- Boros, S.; Gáspári, Z.; Batta, G. Accurate NMR Determinations of Proton-Proton Distances. Annu. Rep. NMR Spectro. 2018, 94, 1–39. [Google Scholar]
- Amatore, C.; Lefrou, C.; Pflüger, F. On-line compensation of ohmic drop in submicrosecond time resolved cyclic voltammetry at ultramicroelectrodes. J. Electroanal. Chem. 1989, 270, 43–59. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bisello, A.; Biondi, B.; Cardena, R.; Schiesari, R.; Crisma, M.; Formaggio, F.; Santi, S. Modulation of Ferrocene–Ferrocene Interactions by Varying Their Reciprocal Positions in L-Dap/Aib Helical Peptides. Inorganics 2023, 11, 482. https://doi.org/10.3390/inorganics11120482
Bisello A, Biondi B, Cardena R, Schiesari R, Crisma M, Formaggio F, Santi S. Modulation of Ferrocene–Ferrocene Interactions by Varying Their Reciprocal Positions in L-Dap/Aib Helical Peptides. Inorganics. 2023; 11(12):482. https://doi.org/10.3390/inorganics11120482
Chicago/Turabian StyleBisello, Annalisa, Barbara Biondi, Roberta Cardena, Renato Schiesari, Marco Crisma, Fernando Formaggio, and Saverio Santi. 2023. "Modulation of Ferrocene–Ferrocene Interactions by Varying Their Reciprocal Positions in L-Dap/Aib Helical Peptides" Inorganics 11, no. 12: 482. https://doi.org/10.3390/inorganics11120482
APA StyleBisello, A., Biondi, B., Cardena, R., Schiesari, R., Crisma, M., Formaggio, F., & Santi, S. (2023). Modulation of Ferrocene–Ferrocene Interactions by Varying Their Reciprocal Positions in L-Dap/Aib Helical Peptides. Inorganics, 11(12), 482. https://doi.org/10.3390/inorganics11120482