

Structural, Spectroscopic, and Thermal Decomposition Features of [Carbonatotetraamminecobalt(III)] Iodide—Insight into the Simultaneous Solid-Phase Quasi-Intramolecular Redox Reactions

 , , , , , and
, , , , , and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Preparation and Properties of Compound 1
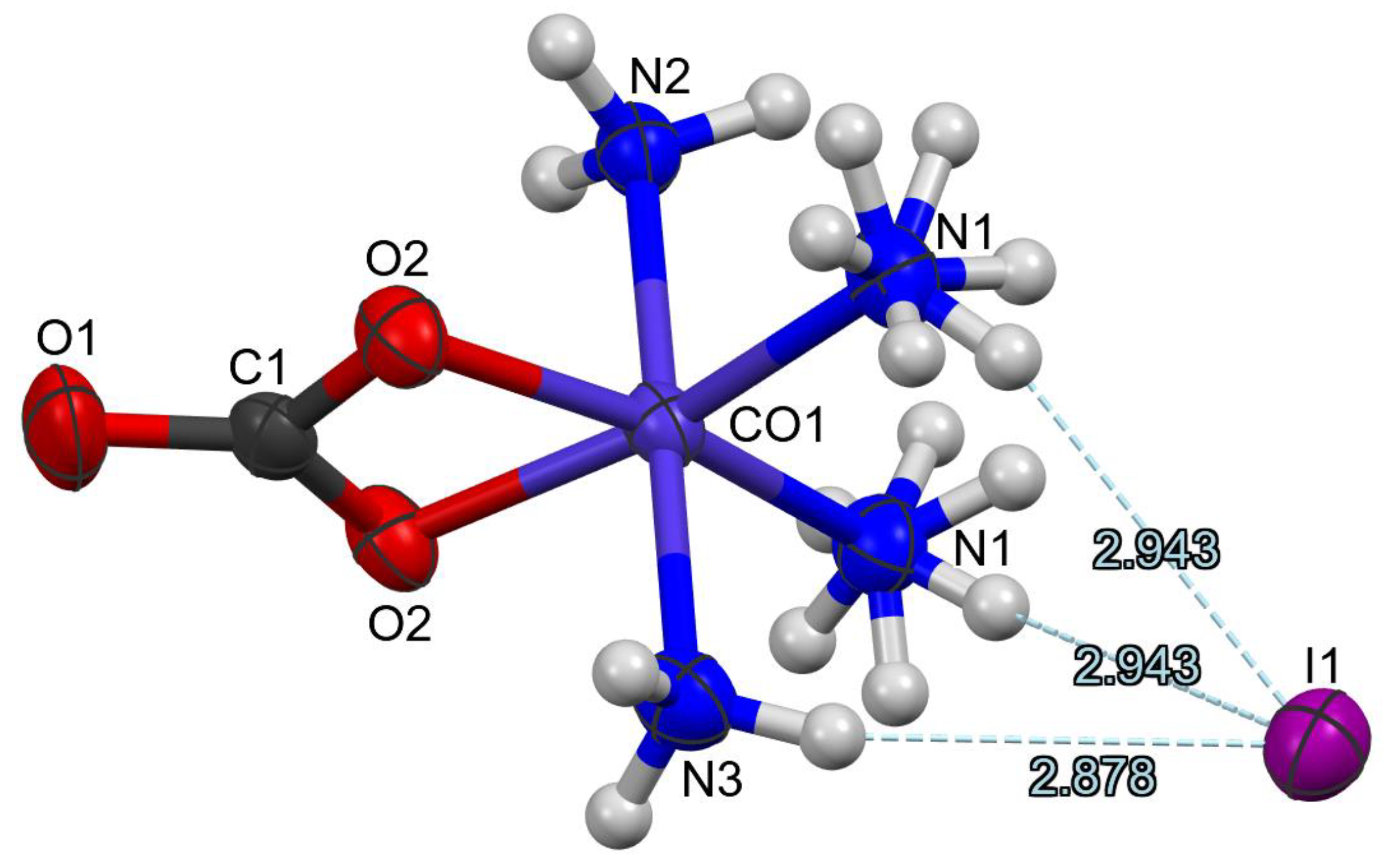
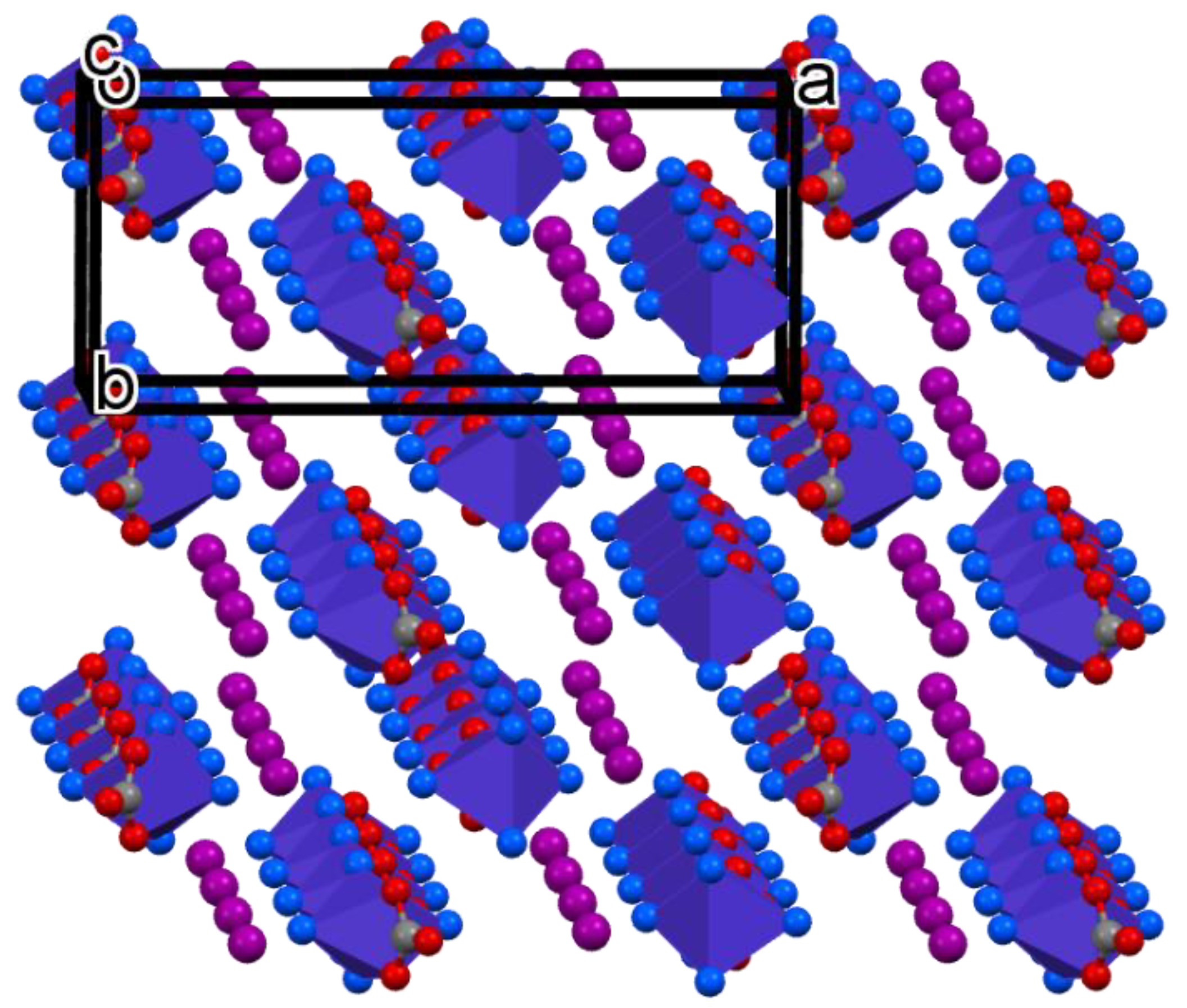
2.2. Crystal Structure Features of Compound 1
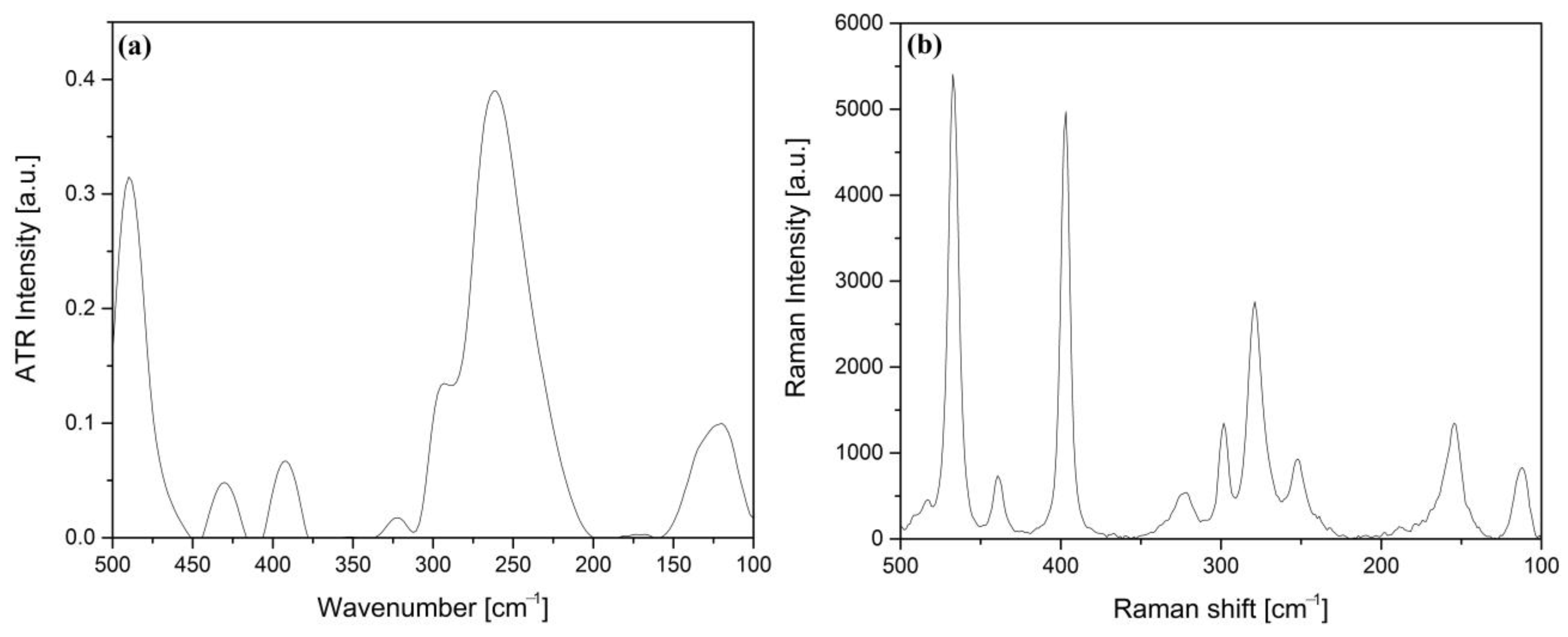
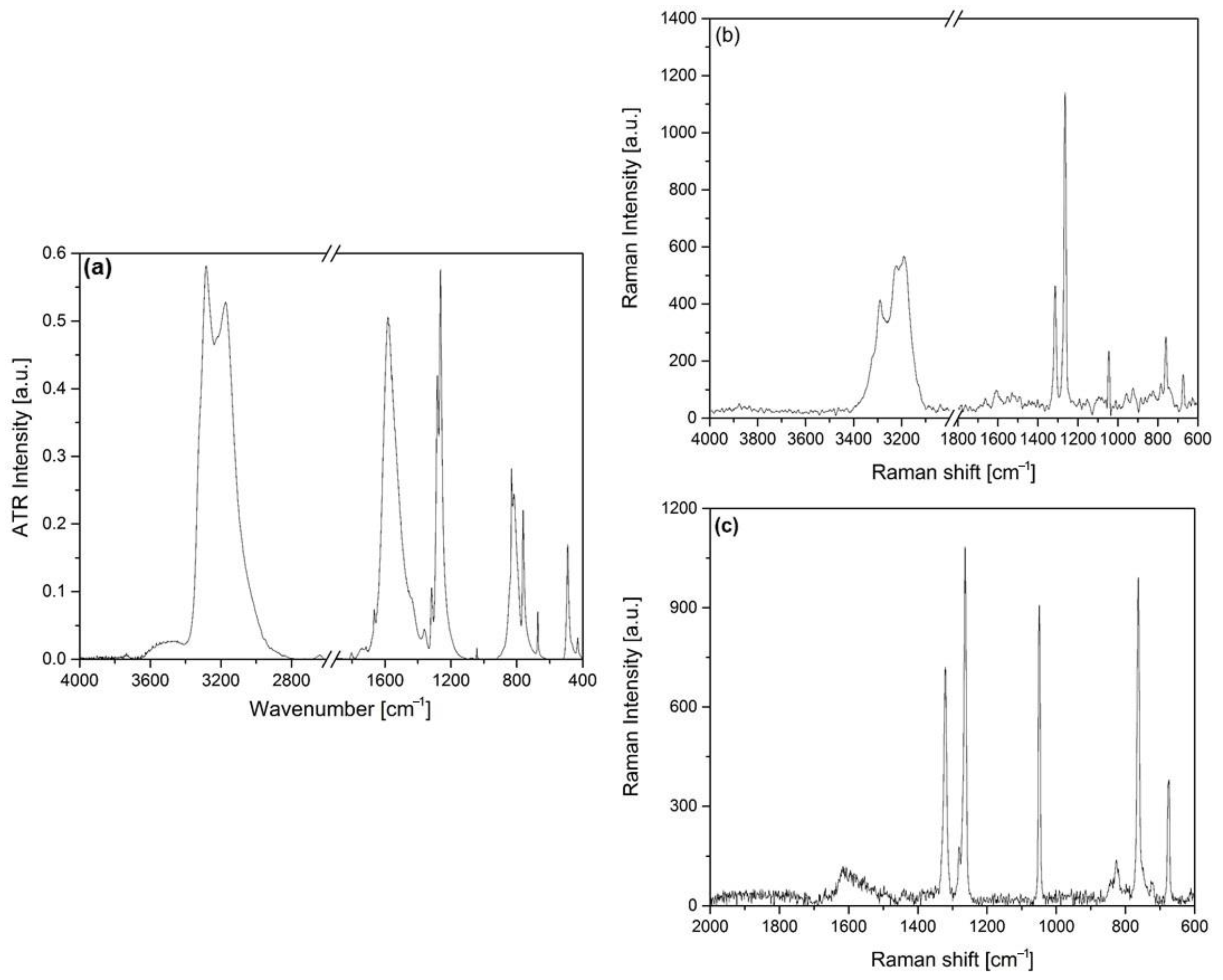
2.3. IR, Raman and UV Spectroscopic Characterization of Compound 1
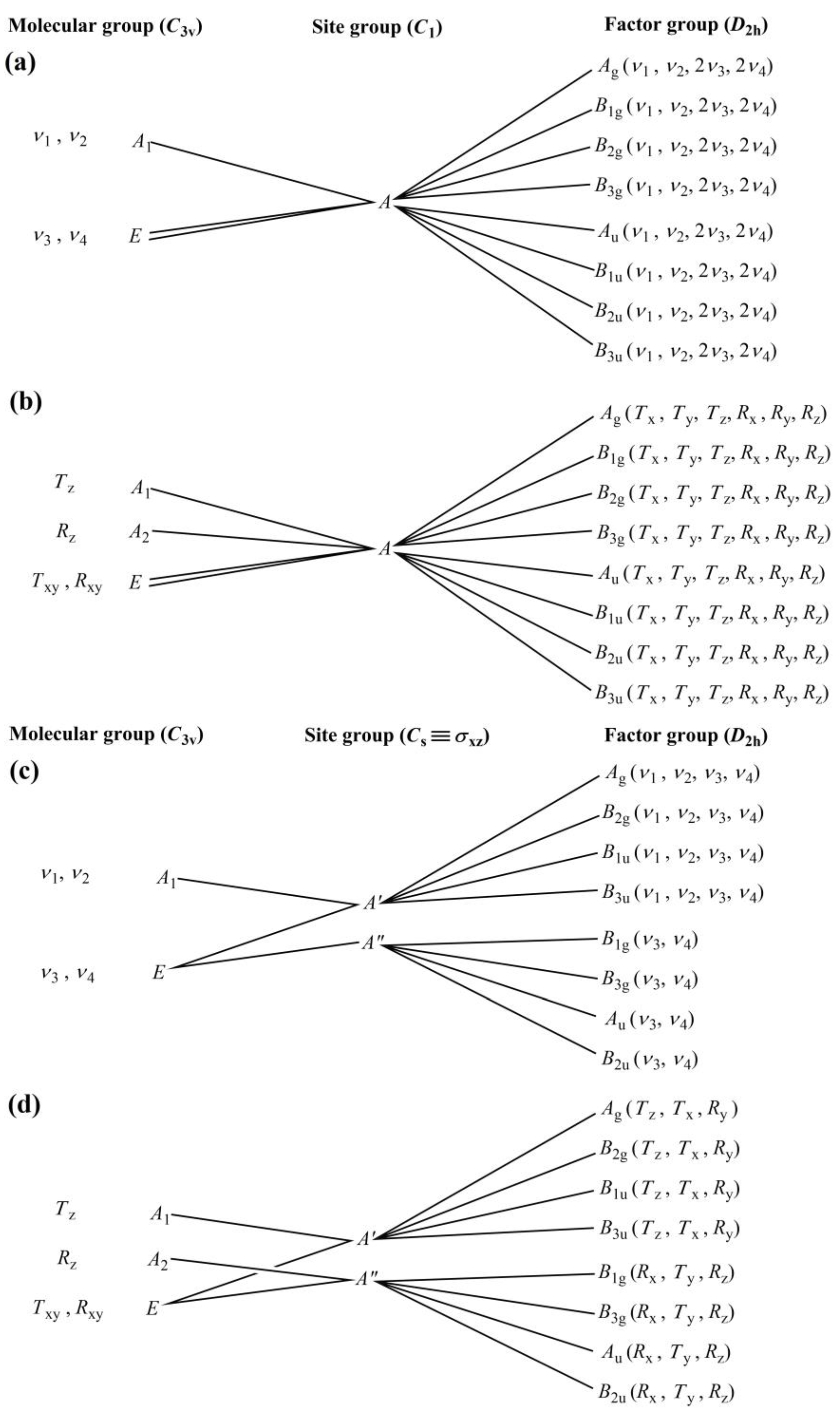
2.3.1. Correlation Analysis of Compound 1
2.3.2. The Ammonia Ligands in Compound 1
2.3.3. The CoIII and I− ions in Compound 1
2.3.4. The Vibrational Modes of the Ammonia and Carbonate Ion Ligands in Compound 1
2.3.5. UV Spectroscopy
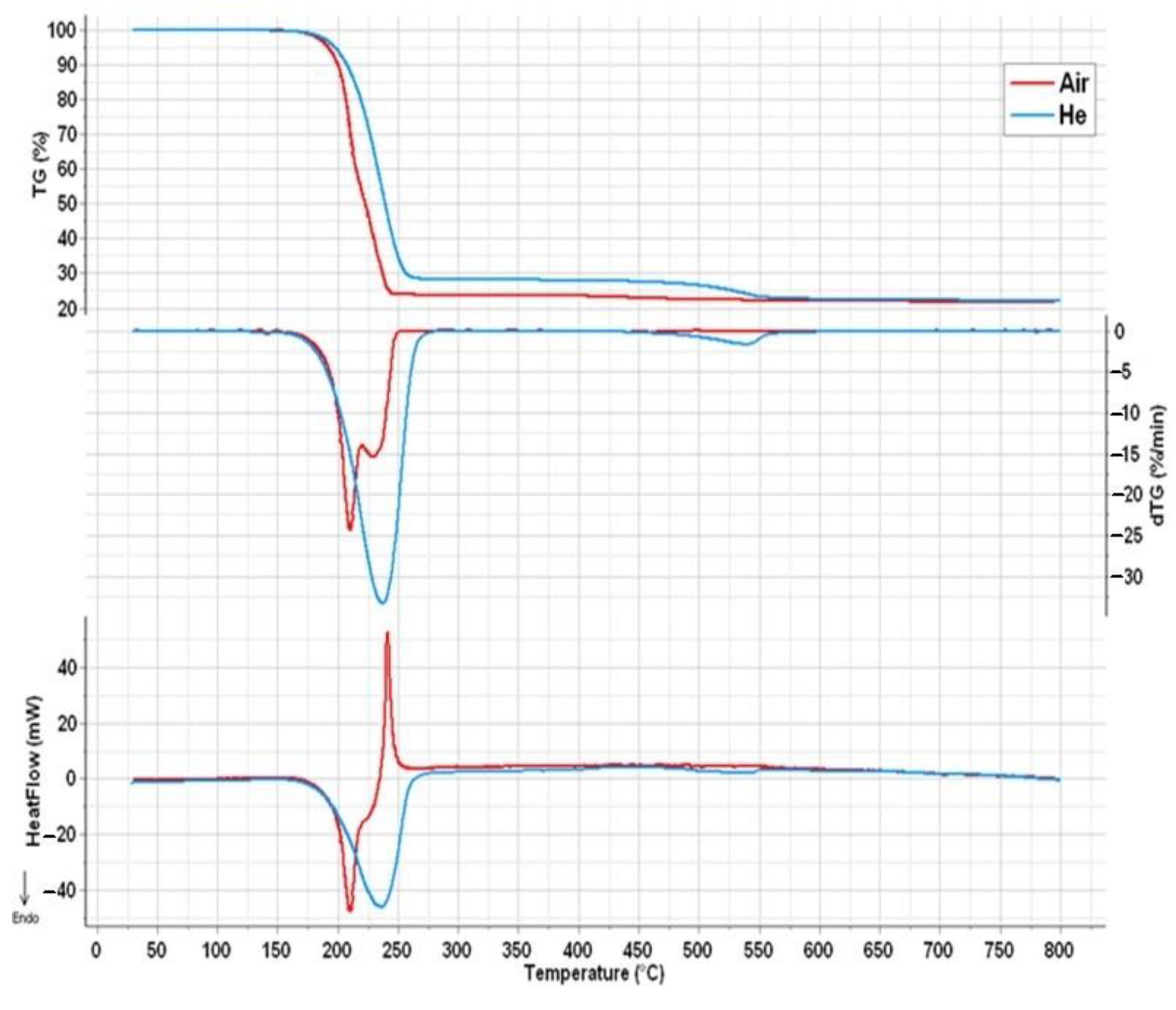
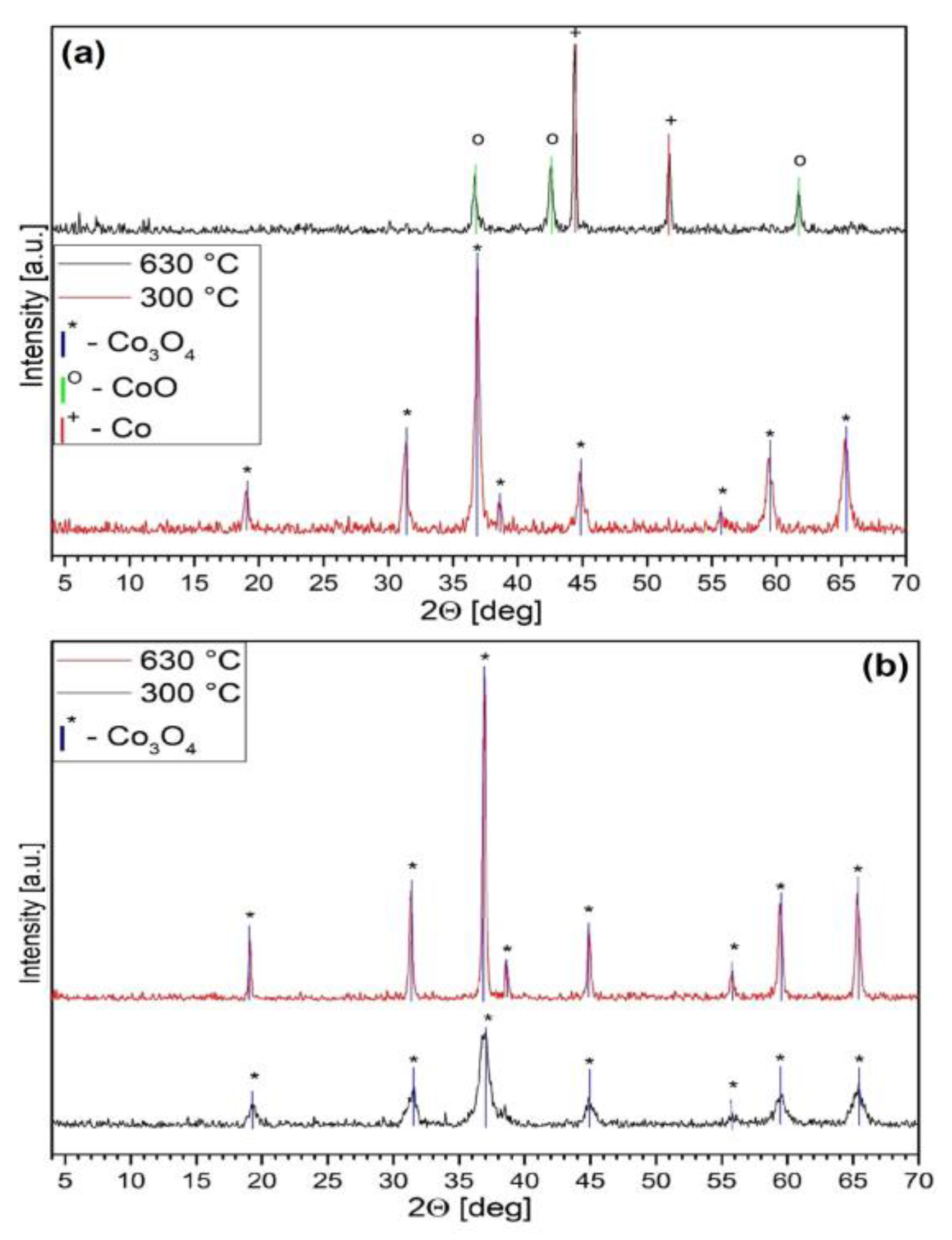
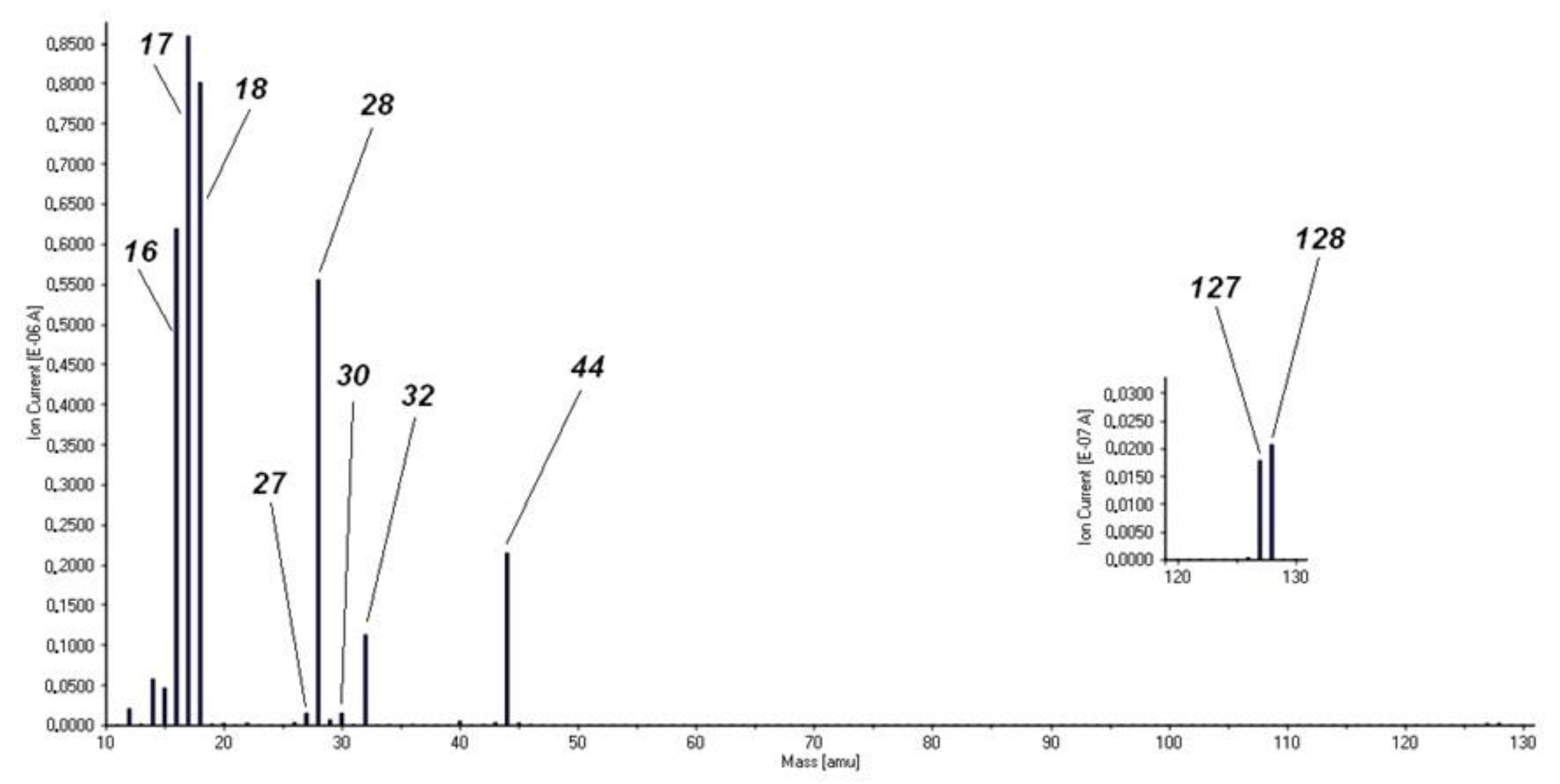
2.4. Thermal Analysis of Compound 1
- (1)
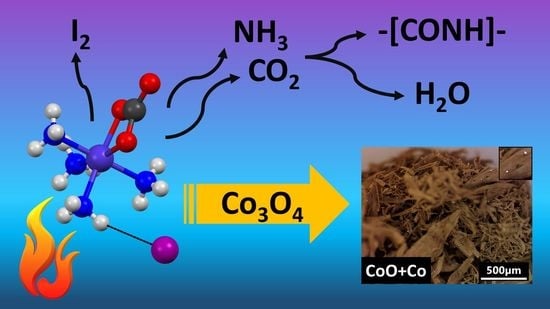
- The initial thermal decomposition step of compound 1 proceeded the same way both in inert atmosphere and in air (Figure 6) and consisted of the solid-phase quasi-intramolecular oxidation of iodide ion with CoIII:The character of the mass loss observed [35] may be attributed to the sublimation of elementary iodine, and it became more intensive only around the boiling point (184.3 °C) of iodine. This could explain why Wendland found a lack of regular relationship between the mass loss and the CoII content during the thermal decomposition of compound 1, since CoIII did not oxidize bromide and chloride ions in compounds 2 and 3 into elementary halogens, but the oxidation of ammonia by CoIII was accompanied by Co2OX2 (X = Br, Cl) formation, and the CoII content was found to be proportional to the mass loss in these reactions [33];[Co(NH3)4CO3]I(s) = [Co(NH3)4CO3](s) + 1/2I2 (s)
- (2)
- The subsequent decomposition of the intermediate [Co(NH3)4CO3] was not the same in inert and in oxygen-containing atmosphere. Cobalt(II) carbonate does not form [Co(NH3)4CO3] or other complex in an NH3 stream [55], and the only known ammine complex of cobalt(II) carbonate, Co(NH3)3CO3·4H2O prepared in aq. ammonia solution is very unstable even at room temperature. Thus, [Co(NH3)4CO3] was expected to be a metastable compound at 200 °C. In the absence of air, ligand loss reactions and interactions between the NH3 and CO2 (carbonate) components resulted in the formation of C-N bonds containing intermediates.Since the oxidation number of cobalt in [CoII(NH3)4CO3] increased during the Co3O4 formation (CoIICoIII2O4) in the oxygen-free environment, 25% of the oxygen content of Co3O4 had to originate formally from carbon dioxide [CoII(NH3)4CO3 = CoIIO + 4NH3 + CO2]. This oxygen transfer should be accompanied by the formation of carbon compounds with reduced oxygen content (the O/C ratio should be <2, because the O/C ratio is 2 in CO2). Based on the TG-MS, XRD and IR results (Figures S7–S9) the intermediate CoCO3 may be decomposed into Co3O4 and CO [56], or a quasi-intramolecular redox reaction of [Co(NH3)4CO3] may result in the formation of the C-N bond containing intermediates as urea, biuret or other amides, which in situ transforms into HNCO/HOCN type decomposition products at ~200 °C (the decomposition points of urea and biuret are 132.7 °C and 190 °C, respectively). The amount of organic compounds formed in inert atmosphere is enough to reduce Co3O4 into CoO and partly into metallic Co (Scheme 1).
- (3)
- In the sample prepared at 200 °C in air, we could not detect crystalline CoCO3 (Figure S9); only badly crystallized Co3O4 was found. IR studies, however, showed the presence of other X-ray amorphous components (ESI Figure S8). The strongly overlapped bands may belong to C(=O)-NH, ammonium ion and carbonate ion containing materials, and the aqueous leaching left a water-insoluble residue showing carbonate ion and coordinated hydroxide ion bands (νOH = ~1055 cm−1). The IR spectrum of this residue was similar to the IR spectrum of basic cobalt carbonate [56], and based on the intensity ratios of the carbonate (~1400 cm−1) and hydroxide ion signals (~1050 cm−1), a hydroxide-ion rich basic cobalt carbonate formed from the decomposition intermediate prepared at 200 °C. The aqueous leaching resulted in removing the water-soluble compounds that showed coinciding bands with the carbonate ion.
3. Materials and Methods
3.1. Vibrational Spectroscopy
3.2. UV−Vis Spectroscopy
3.3. Scanning Electron Microscopy
3.4. Powder X-ray Diffractometry
3.5. Single Crystal X-ray
3.6. Thermogravimetric Analysis
3.7. [Carbonatotetraamminecobalt(III)] Nitrate Hemihydrate
3.8. [Carbonatotetraamminecobalt(III)] Iodide (Compound 1)
3.8.1. Procedure 1
3.8.2. Procedure 2
4. Conclusions
- [κ2-O,O′-Carbonatotetraamminecobalt(III)] iodide, ([Co(NH3)4CO3]I), compound 1 was prepared with 89% yield. The correlation analysis showed three kinds of spectroscopically different ammonia ligands in compound 1. All normal modes in the vibrational spectra (IR and Raman) and the UV bands of compound 1 were assigned;
- Compound 1 was orthorhombic, and isomorphous with the analogous bromide. The distorted octahedral complex cation contained four ammonia ligands and a bidentate-coordinated carbonate anion. The carbonate ion formed a four-membered symmetric planar chelate ring. There was no observed trans-effect for the carbonate ion on the equatorial Co-N distances. The complex cations and iodide ions were arranged into ion pairs and each cation bound its iodide pair through three hydrogen bonds. The complex cations were bound to each other by N-H···O hydrogen bonds and formed zigzag sheets via an extended 2D hydrogen bond network;
- The thermal decomposition of compound 1 started with the solid-phase quasi-intramolecular oxidation of the iodide ion by CoIII with the formation of [CoII(NH3)4CO3] and I2. The intermediate CoII-complex in situ decomposed into Co3O4 and C-N containing intermediates. The ammonia ligand loss resulted in CoCO3, which in situ decomposed into Co3O4 and carbon monoxide. A quasi-intramolecular solid-phase redox reaction of [Co(NH3)4CO3] might result in C-N bond-containing compounds with a sub-stoichiometric release of ammonia and CO2 from compound 1. In an inert atmosphere, the C-N containing compounds reduced Co3O4 into CoO and Co. In oxygen-containing atmosphere, the end-product was Co3O4, and the endothermic ligand loss reaction coincided with the consecutive exothermic oxidation processes. The carbonate ion remaining in the decomposition residues formed at 200 °C transformed into a coordinated hydroxide-ion rich basic cobalt(II) carbonate during aqueous leaching.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bereczki, L.; Petruševski, V.M.; Franguelli, F.P.; Béres, K.A.; Farkas, A.; Holló, B.B.; Czégény, Z.; Szilágyi, I.M.; Kótai, L. [Hexaamminecobalt(III)] Dichloride Permanganate—Structural Features and Heat-Induced Transformations into (CoII,MnII)(CoIII,MnIII)2O4 Spinels. Inorganics 2022, 10, 252. [Google Scholar] [CrossRef]
- Béres, K.A.; Homonnay, Z.; Kvitek, L.; Dürvanger, Z.; Kubikova, M.; Harmat, V.; Szilágyi, F.; Czégény, Z.; Németh, P.; Bereczki, L.; et al. Thermally Induced Solid−Phase Quasi−Intramolecular Redox Reactions of [Hexakis(urea−O)iron(III)] Permanganate: An Easy Reaction Route to Prepare Potential (Fe,Mn)Ox Catalysts for CO2 Hydrogenation. Inorg. Chem. 2022, 61, 14403–14418. [Google Scholar] [CrossRef] [PubMed]
- Lagunova, V.I.; Filatov, E.Y.; Plyusnin, P.E.; Korenev, S.V. In Situ and Ex Situ Studies of Tetrammineplatinum(II) Chromate Thermolysis. Russ. J. Inorg. Chem. 2020, 65, 1566–1570. [Google Scholar] [CrossRef]
- Fogaca, L.A.; Kováts, É.; Németh, G.; Kamarás, K.; Berés, K.A.; Németh, P.; Petruševski, V.; Bereczki, L.; Holló, B.B.; Sajó, I.E.; et al. Solid−Phase Quasi−Intramolecular Redox Reaction of [Ag(NH3)2]MnO4: An Easy Way to Prepare Pure AgMnO2. Inorg. Chem. 2021, 60, 3749–3760. [Google Scholar] [CrossRef] [PubMed]
- Serebrennikova, P.S.; Komarov, V.Y.; Sukhikh, A.S.; Khranenko, S.P.; Zadesenets, A.V.; Gromilov, S.A.; Yusenko, K.V. [Nien3](MoO4)0.5(WO4)0.5 co-crystals as single−source precursors for ternary refractory Ni–Mo–W alloys. Nanomaterials 2021, 11, 3272–3284. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.G.; Mogk, J.; Conrad, M.; Kraus, F. Octaammine EuII and YbII Azides and Their Thermal Decompositions to the Nitrides. Eur. J. Inorg. Chem. 2016, 26, 4162–4169. [Google Scholar] [CrossRef]
- Solt, H.E.; Németh, P.; Mohai, M.; Sajó, I.E.; Klébert, S.; Franguelli, F.P.; Fogaca, L.A.; Pawar, R.P.; Kótai, L. Temperature−Limited Synthesis of Copper Manganites along the Borderline of the Amorphous/Crystalline State and Their Catalytic Activity in CO Oxidation. ACS Omega 2021, 6, 1523–1533. [Google Scholar] [CrossRef]
- Kótai, L.; Petruševski, V.M.; Bereczki, L.; Béres, K.A. Catalytic Properties of the Spinel-Like CuxMn3−xO4 Copper Manganese Oxides—An Overview. Catalysts 2023, 13, 129. [Google Scholar] [CrossRef]
- Bereczki, L.; Fogaca, L.A.; Durvanger, Z.; Harmat, V.K.; Németh, G.; Holló, B.B.; Petruševski, V.M.; Bódis, E.; Farkas, A.; Szilágyi, I.M.; et al. Dynamic disorder in the high−temperature polymorph of bis[diamminesilver(I)] sulfate–reasons and consequences of simultaneous ammonia release from two different polymorphs. J. Coord. Chem. 2021, 74, 2144–2162. [Google Scholar] [CrossRef]
- Domonov, D.P.; Pechenyuk, S.I.; Semushina, Y.P.; Yusenko, K.V. Solid−state transformations in inner coordination sphere of [Co(NH3)6][Fe(C2O4)3]·3H2O as a route to access catalytically active Co−Fe materials. Materials 2019, 12, 221. [Google Scholar] [CrossRef] [Green Version]
- Yusenko, K.V.; Pechenyuk, S.I.; Vikulova, E.S.; Semushina, Y.P.; Baidina, I.A.; Filatov, E.Y. Isostructurality and Thermal Properties in the Series of Double Complex Salts [M1(NH3)6][M2(C2O4)3]·3H2O (M1=Co, Ir, M2 = Fe, Cr). J. Struct. Chem. 2019, 60, 1062–1071. [Google Scholar] [CrossRef]
- Farhadi, S.; Pourzare, K.; Bazgir, S. Co3O4 nanoplates: Synthesis, characterization and study of optical and magnetic properties. J. Alloys Compd. 2014, 587, 632–637. [Google Scholar] [CrossRef]
- Béres, K.A.; Sajó, I.E.; Lendvay, G.; Trif, L.; Petruševski, V.M.; Holló, B.B.; Korecz, L.; Franguelli, F.P.; László, K.; Szilágyi, I.M.; et al. Solid−Phase “Self−Hydrolysis” of [Zn(NH3)4MoO4@2H2O] Involving Enclathrated Water—An Easy Route to a Layered Basic Ammonium Zinc Molybdate Coordination Polymer. Molecules 2021, 26, 4022–4041. [Google Scholar] [CrossRef]
- Franguelli, F.P.; Béres, K.A.; Kótai, L. Pyridinesilver Tetraoxometallate Complexes: Overview of the Synthesis, Structure and Properties of Pyridine Complexed AgXO4 (X = Cl, Mn, Re) Compounds. Inorganics 2021, 9, 79–92. [Google Scholar] [CrossRef]
- Garkul’, I.A.; Zadesenets, A.V.; Plyusnin, P.E.; Filatov, E.Y.; Asanova, T.I.; Kozlov, D.V.; Korenev, S.V. Zinc(II) and Manganese(II) Oxalatopalladates as Precursors of Bimetallic Nanomaterials. Russ. J. Inorg. Chem. 2020, 65, 1571–1576. [Google Scholar] [CrossRef]
- Ilyushin, M.A.; Shugalei, I.V.; Tverjanovich, A.S.; Smirnov, A.V. Influence of the Mechanism of the Initial Stages of the Ligand Decomposition on the Initiating Ability of Cobalt(III) Ammine Tetrazolate Complexes. Russ. J. Inorg. Chem. 2020, 90, 640–647. [Google Scholar] [CrossRef]
- Franguelli, F.P.; Kováts, É.; Czégény, Z.; Bereczki, L.; Petruševski, V.M.; Holló, B.B.; Béres, K.A.; Farkas, A.; Szilágyi, I.M.; Kótai, L. Multi−Centered Solid−Phase Quasi−Intramolecular Redox Reactions of [(Chlorido)Pentaamminecobalt(III)] Permanganate—An Easy Route to Prepare Phase Pure CoMn2O4 Spinel. Inorganics 2022, 10, 18–40. [Google Scholar] [CrossRef]
- Béres, K.A.; Homonnay, Z.; Holló, B.; Gracheva, M.; Petruševski, V.M.; Farkas, A.; Dürvanger, Z.; Kótai, L. Synthesis, structure, and Mössbauer spectroscopic studies on the heat-induced solid-phase redox reactions of hexakis(urea-O)iron(III) peroxodisulfate. J. Mater. Res. 2022. [Google Scholar] [CrossRef]
- Mohan, S.C.; Jenniefer, S.J.; Muthiah, P.T.; Jothivenkatachalam, K. Tetraammine(carbonato-κ2O,O’)-cobalt(III) perchlorate. Acta Cryst. 2013, 69, 45–46. [Google Scholar] [CrossRef]
- Gosteva, A.; Kossoy, A.; Tsvetov, N.; Semushina, Y.; Kozerozhets, I. Dynamics of Thermal Decomposition of the Double Complex Salt [Cr(ur)6][Co(CN)6]⋅4H2O. Russ. J. Inorg. Chem. 2022, 67, 1257–1263. [Google Scholar] [CrossRef]
- Mansouri, M.; Atashi, H.; Tabrizi, F.F.; Mirzaei, A.A.; Mansouri, G. Kinetics studies of nano−structured cobalt−manganese oxide catalysts in Fischer-Tropsch synthesis. J. Ind. Eng. Chem. 2013, 19, 1177–1183. [Google Scholar] [CrossRef]
- Mansouri, G.; Mansouri, M. Synthesis and characterization of Co−Mn nanocatalyst prepared by thermal decomposition for Fischer-Tropsch reaction. Iran. J. Chem. Eng. 2018, 37, 1–9. [Google Scholar]
- Macikenas, D.; Hazell, R.G.; Christensen, A.N. X-ray Crystallographic Study of Tetrammine-CarbonatocobaIt(III) Sulfate Trihydratef [Co(NH3)4CO3]2SO4.3H2O. Acta Chem. Scand. 1995, 49, 636–639. [Google Scholar] [CrossRef]
- Franguelli, F.P.; Holló, B.B.; Petruševski, V.M.; Sajó, I.E.; Klébert, S.; Farkas, A.; Bódis, E.; Szilágyi, I.M.; Pawar, R.P.; Kótai, L. Thermal decomposition and spectral characterization of di[carbonatotetraamminecobalt(III)] sulfate trihydrate and the nature of its thermal decomposition products. J. Therm. Anal. Calorim. 2021, 145, 2907–2923. [Google Scholar] [CrossRef]
- Iman, K.; Naqi Ahamad, M.; Ansari, M.A.; Saleh, H.A.M.; Khan, M.S.; Ahmad, M.; Haque, R.A.; Shahid, M. How to identify a smoker: A salient crystallographic approach to detect thiocyanate content. RSC Adv. 2021, 11, 16881–16891. [Google Scholar] [CrossRef] [PubMed]
- Haagensen, C.O.; Rasmussen, S.E. Accuracy of a Crystal Structure Determination. The Structure of Carbonato-Tetrammine-Cobalt (III) Bromide. Acta Chem. Scand. 1963, 17, 1630–1634. [Google Scholar] [CrossRef]
- Barclay, G.A.; Hoskins, B.F. The crystal structure of carbonatotetra-amminecobalt(III) bromide. J. Chem. Soc. 1962, 586–591. [Google Scholar] [CrossRef]
- Junk, P.C.; Steed, J.W. Syntheses and X-ray crystal structures of [Co(η2-CO3)(NH3)4](NO3)·0.5H2O and [(NH3)3Co(μ2-OH)(μ-CO3)Co(NH3)3][NO3]2·H2O. Polyhedron 1999, 18, 3593–3597. [Google Scholar] [CrossRef]
- Bernal, I.; Cetrullo, J. The Phenomenon of Conglomerate Crystallization. XVIII. Clavic Dissymmetry in Coordination Compounds. XVI. The Crystal Structure of Racemic {[Co(NH3)4(CO3)]NO3}2·H2O. Struct. Chem. 1990, 1, 227–234. [Google Scholar] [CrossRef]
- Le Bail, A. Tetraammine(carbonato-κ2-O,O′)-cobalt(III) nitrate: A powder X-ray diffraction study. Acta Cryst. 2013, 69, i42–i43. [Google Scholar]
- Miolati, A. Preparazione del bicarbonate di carbonatotetramincobalto. Rendiconti Atti Accad. Lincei 1897, 5, 344–348. [Google Scholar]
- Jörgensen, S.M. Constitution of the cobalt, chromium and rhodium bases IV. Z. Anorg. Allgem. Chem. 1892, 2, 279–300. [Google Scholar] [CrossRef]
- Wendlandt, W.W.; Woodlock, J.H. The thermal dissociation of [Co(NH3)4CO3]X type complexes. J. Inorg. Nucl. Chem. 1966, 28, 1485–1488. [Google Scholar] [CrossRef]
- Miyokawa, K.; Masuda, H.; Masuda, I. Thermal Decomposition Reactions of Analogous Pentaamminenitrosylcobalt(III) Complexes. Bull. Chem. Sot. Jpn. 1980, 53, 3573–3576. [Google Scholar] [CrossRef]
- Simmons, E.L.; Wendlandt, W.W. The thermal decomposition of metal complexes—XVIII Intermediates formed in the decomposition of hexamminecobalt (III) chloride. J. Inorg. Nucl. Chem. 1966, 28, 2187–2191. [Google Scholar] [CrossRef]
- Zhuang, J.; Zhang, W.; Zheng, L.; Xin, X. Studies on the solid-state reactions of coordination compounds. LVII. Effect of anions on the thermal decomposition of [Co(NH3)4CO3]Cl. Wuji Huaxue Xuebao 1993, 9, 260–265. [Google Scholar]
- Li-Min, Z.; Wei-Hua, Z.; Xin-Quan, X. The effect of KI on the thermal decomposition of cobalt (III)-ammine complexes. Thermochim. Acta 1993, 222, 273–277. [Google Scholar] [CrossRef]
- Siebert, H. Ultrarotspektren von Kobalt(lll)-Komplexen mit Ammoniak und Resten von Sauerstoffsaeuren als Liganden. Z. Anorg. Allgem. Chem. 1959, 298, 51–63. [Google Scholar] [CrossRef]
- Goldsmith, J.A.; Ross, S.D. Factors affecting the infra-red spectra of planar anions with D3h symmetry—IV The vibrational spectra of some complex carbonates in the region 4000–400 cm−1. Spectrochim Acta A 1968, 24, 993–998. [Google Scholar] [CrossRef]
- Wu, H.; Zhu, J.; Ma, L.; Shen, X.; Shi, G. Powder diffraction of [Co(NH3)5CO3]NO3·½H2O and [Co(NH3)4CO3]NO3·½H2O. Powder Diffr. 1998, 13, 32–34. [Google Scholar] [CrossRef]
- Nakai, H.; Kushi, Y.; Kurotani, T. Crystal structure of carbonatopentamminecobal(III) iodide monohydrate. Nippon. Kagaku Zasshi 1967, 88, 1126–1127. [Google Scholar] [CrossRef]
- Barnet, M.T.; Craven, B.M.; Freeman, H.C.; Kime, N.E.; Ibers, J.A. The Co–N bond lengths in CoII and CoIII hexammines. Chem. Commun. 1966, 10, 307–308. [Google Scholar]
- Goldsmith, J.A.; Hezel, A.; Ross, S.D. The skeletal vibrations of some cobalt (III) carbonato-, phosphato-and sulphato-complexes. Spectrochim. Acta A Mol. Biomol. Spectrosc. 1968, 24, 1139–1147. [Google Scholar] [CrossRef]
- Fujita, J.; Martell, A.E.; Nakamoto, K. Infrared spectra of metal chelate compounds. VIII. Infrared spectra of Co(III) carbonato complexes. J. Chem. Phys. 1962, 36, 339–345. [Google Scholar] [CrossRef]
- Fujita, J.; Nakamoto, K.; Kobayashi, M. Infrared Spectra of Metallic Complexes. I. The Effect of Coordination on the Infrared Spectra of Ammine, Rhodanato and Azido Complexes. J. Am. Chem. Soc. 1956, 78, 3295–3297. [Google Scholar] [CrossRef]
- Carassiti, V.; Martelli, R. Absorption spectra of solutions of complex salts. II. Cobalt tetra- and pentaammines. Ann. Chim. 1957, 47, 402–409. [Google Scholar]
- Sastri, V.S.; Langford, C.H. Electronic spectra of carbonato and oxalato ammine complexes of cobalt(III); average field model for singlet, triplet, and charge transfer bands. Can. J. Chem. 1969, 47, 34237–34240. [Google Scholar] [CrossRef]
- Sastri, V.S. Studies on the Disposition of Carbonato Group in Cobalt(III) Complexes. Inorganica Chim. Acta 1972, 6, 264–266. [Google Scholar] [CrossRef]
- Kotai, L.; Argay, G.; Holly, S.; Keszler, A.; Pukanszky, B.; Banerji, K.K. Study on the Existence of Hydrogen Bonds in Ammonium Permanganate. Z. Anorg. Allg. Chem. 2001, 627, 114–118. [Google Scholar] [CrossRef]
- Simmons, E.L.; Wendlandt, W.W. Intermediates formed in the thermal decomposition of hexammiaecobalt (III) bromide. J. Inorg. Nucl. Chem. 1966, 28, 2437–2439. [Google Scholar] [CrossRef]
- Tang, C.W.; Wang, C.B.; Chien, S.H. Characterization of cobalt oxides studied by FT-IR, Raman, TPR and TG-MS. Thermochim. Acta 2008, 473, 68–73. [Google Scholar] [CrossRef]
- Kocsis, T.; Magyari, J.; Sajó, I.E.; Pasinszki, T.; Homonnay, Z.; Szilágyi, I.M.; Farkas, A.; May, Z.; Effenberger, H.; Szakáll, S.; et al. Evidence of quasi-intramolecular redox reactions during thermal decomposition of ammonium hydroxodisulfitoferriate (III), (NH4)2[Fe(OH)(SO3)2]· H2O. J. Therm. Anal. Calorim. 2018, 132, 493–502. [Google Scholar] [CrossRef]
- Li, Y.; Qiu, W.; Qin, F.; Fang, H.; Hadjiev, V.G.; Litvinov, D.; Bao, J. Identification of Cobalt Oxides with Raman Scattering and Fourier Transform Infrared Spectroscopy. J. Phys. Chem. C 2016, 120, 4511–4516. [Google Scholar] [CrossRef]
- Christoskova, S.G.; Stoyanova, M.; Georgieva, M.; Mehandjiev, D. Preparation and characterization of a higher cobalt oxide. Mater. Chem. Phys. 1999, 60, 39–43. [Google Scholar] [CrossRef]
- Clark, G.L.; Buckner, H.K. The properties of subsidiary valence groups. III. the preparation, properties and molecular volume relationships of the hydrates and ammines of cobalt fluoride, bromide, iodide, nitrate, carbonate and citrate. J. Am. Chem. Soc. 1922, 44, 230–244. [Google Scholar] [CrossRef]
- Yang, J.J.; Frost, R.L. Synthesis and characterisation of cobalt hydroxy carbonate Co2CO3(OH)2 Nanomaterials. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2011, 78, 420–428. [Google Scholar] [CrossRef] [Green Version]
- Martiz, A.; Károly, Z.; Domján, A.; Mohai, M.; Bereczki, L.; Trif, L.; Farkas, A.; László, K.; Menyhárd, A.; Kótai, L. Nano-ZrO2@C, Nano-(ZrC, ZrO2)@C and Nano-ZrC@C Composites Prepared by Plasma-Assisted Carbonization of Zr-Loaded Iminodiacetate-Functionalized Styrene-Divinylbenzene Copolymers. Inorganics 2022, 10, 77–104. [Google Scholar] [CrossRef]
- Martiz, A.; Károly, Z.; Trif, L.; Mohai, M.; Bereczki, L.; Németh, P.; Molnár, Z.; Menyhárd, A.; Pawar, R.P.; Tekale, S.; et al. Plasma-assisted preparation of nano-(ZrC, ZrO2)@ carbon composites from Zr-loaded sulfonated styrene–divinylbenzene copolymers. J. Therm. Anal. Calorim. 2022, 147, 9353–9365. [Google Scholar] [CrossRef]
- Martiz, A.; Farkas, A.; Karoly, Z.; Franguelli, F.P.; Samaniego, S.K.; Menyhard, A.; Kotai, L. Raman studies on carbon-containing phases in nanosized-ZrO2/C and nanosized-(ZrC; ZrO2)/C composites. Nano Stud. 2022, 10, 77. [Google Scholar]
- Sheldrick, G.M. SHELXT– Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Nassar, M.Y.; Ahmed, I.S. Hydrothermal synthesis of cobalt carbonates using different counter ions: An efficient precursor to nano-sized cobalt oxide (Co3O4). Polyhedron 2011, 30, 2431–2437. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Béres, K.A.; Szilágyi, F.; Homonnay, Z.; Dürvanger, Z.; Bereczki, L.; Trif, L.; Petruševski, V.M.; Farkas, A.; Bayat, N.; Kótai, L. Structural, Spectroscopic, and Thermal Decomposition Features of [Carbonatotetraamminecobalt(III)] Iodide—Insight into the Simultaneous Solid-Phase Quasi-Intramolecular Redox Reactions. Inorganics 2023, 11, 68. https://doi.org/10.3390/inorganics11020068
Béres KA, Szilágyi F, Homonnay Z, Dürvanger Z, Bereczki L, Trif L, Petruševski VM, Farkas A, Bayat N, Kótai L. Structural, Spectroscopic, and Thermal Decomposition Features of [Carbonatotetraamminecobalt(III)] Iodide—Insight into the Simultaneous Solid-Phase Quasi-Intramolecular Redox Reactions. Inorganics. 2023; 11(2):68. https://doi.org/10.3390/inorganics11020068
Chicago/Turabian StyleBéres, Kende Attila, Fanni Szilágyi, Zoltán Homonnay, Zsolt Dürvanger, Laura Bereczki, László Trif, Vladimir M. Petruševski, Attila Farkas, Niloofar Bayat, and László Kótai. 2023. "Structural, Spectroscopic, and Thermal Decomposition Features of [Carbonatotetraamminecobalt(III)] Iodide—Insight into the Simultaneous Solid-Phase Quasi-Intramolecular Redox Reactions" Inorganics 11, no. 2: 68. https://doi.org/10.3390/inorganics11020068
APA StyleBéres, K. A., Szilágyi, F., Homonnay, Z., Dürvanger, Z., Bereczki, L., Trif, L., Petruševski, V. M., Farkas, A., Bayat, N., & Kótai, L. (2023). Structural, Spectroscopic, and Thermal Decomposition Features of [Carbonatotetraamminecobalt(III)] Iodide—Insight into the Simultaneous Solid-Phase Quasi-Intramolecular Redox Reactions. Inorganics, 11(2), 68. https://doi.org/10.3390/inorganics11020068