
Operando CO Infrared Spectroscopy and On-Line Mass Spectrometry for Studying the Active Phase of IrO2 in the Catalytic CO Oxidation Reaction


Abstract
:1. Introduction
2. Experimental Details
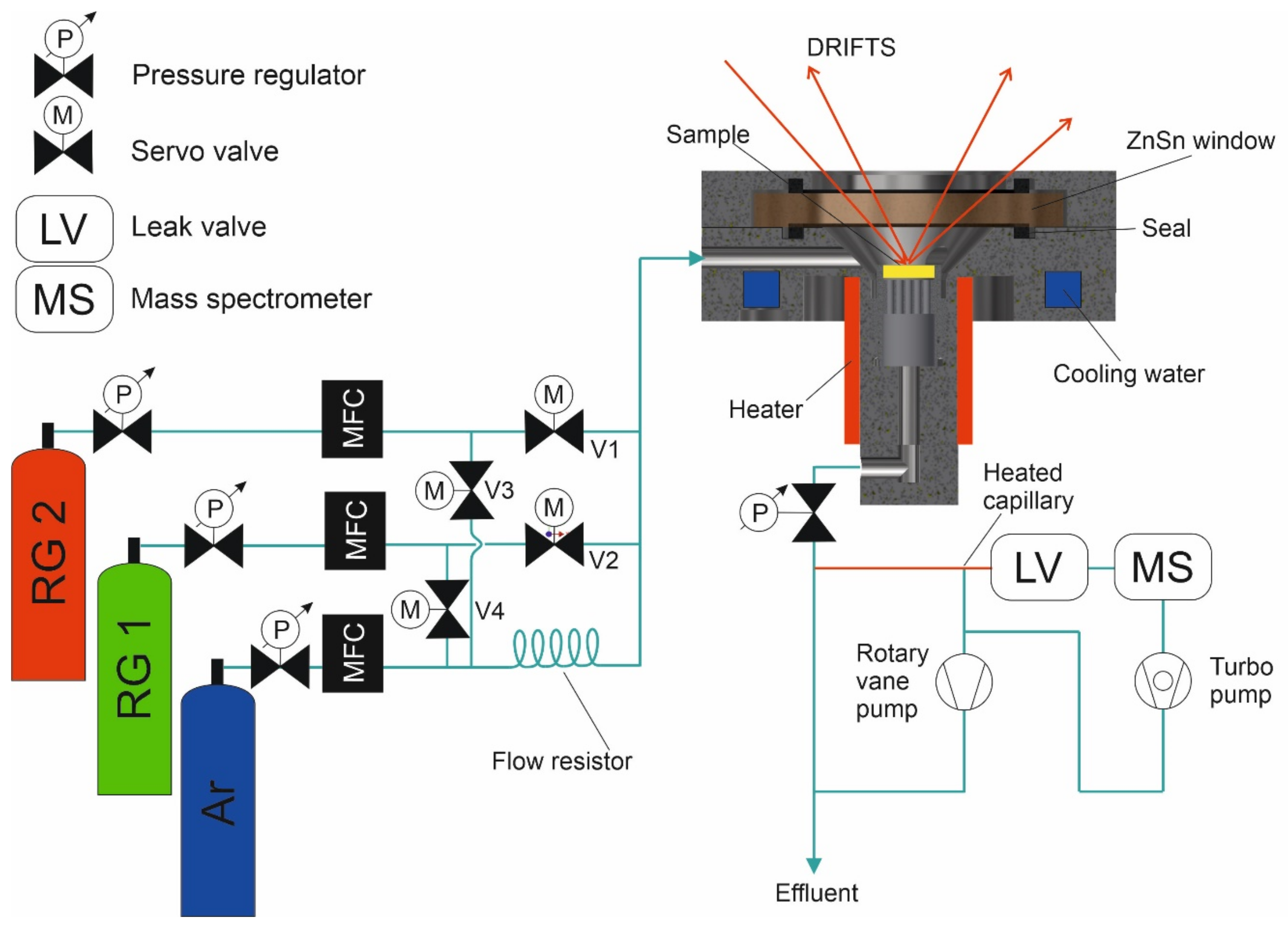
2.1. In Situ DRIFTS Cell
2.2. Sample Preparation and Characterization
2.3. Reaction Conditions
3. Experimental Results
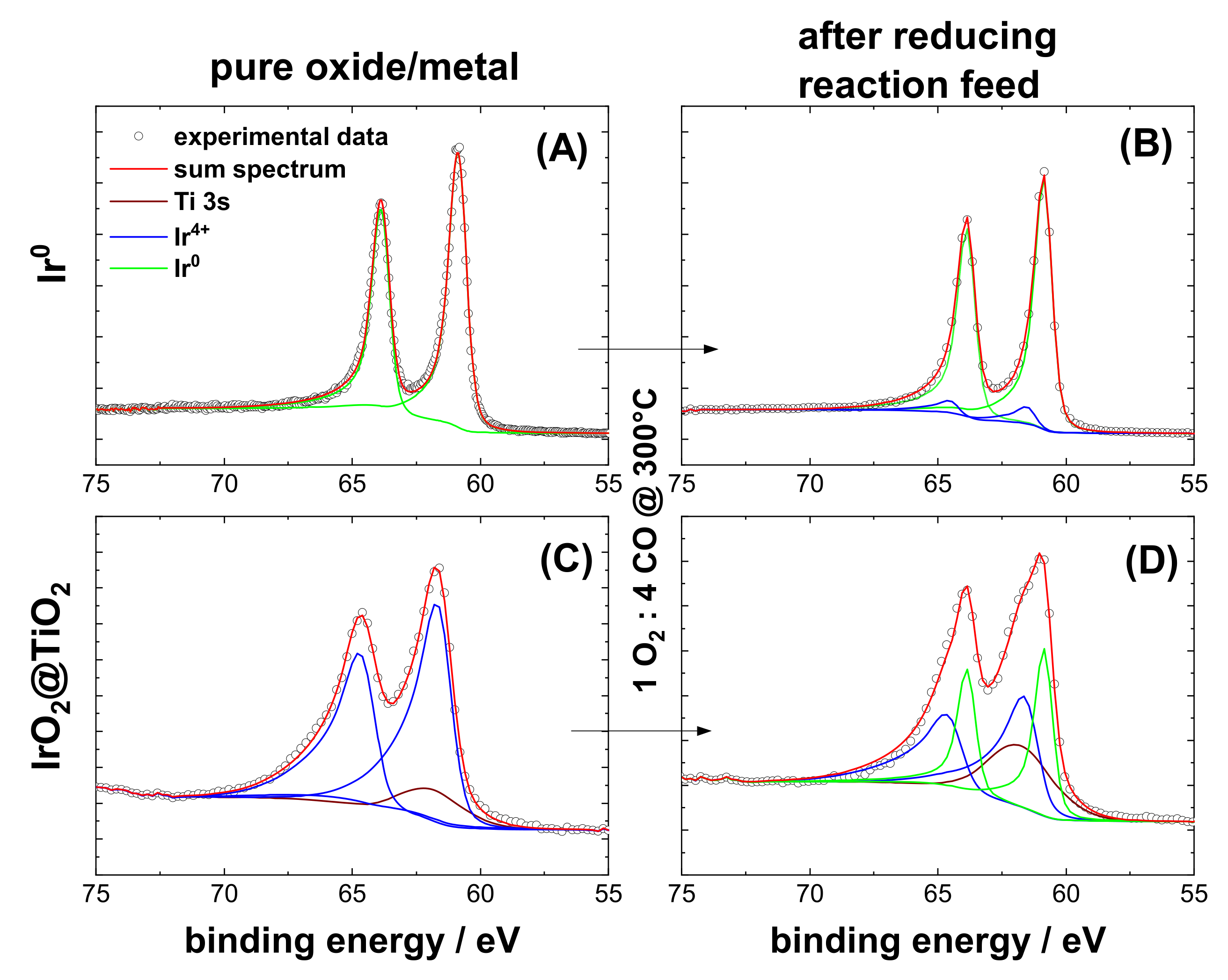
3.1. Characterization of Pre-Oxidized and Pre-Reduced IrO2@TiO2 and Ir0 + TiO2 Samples
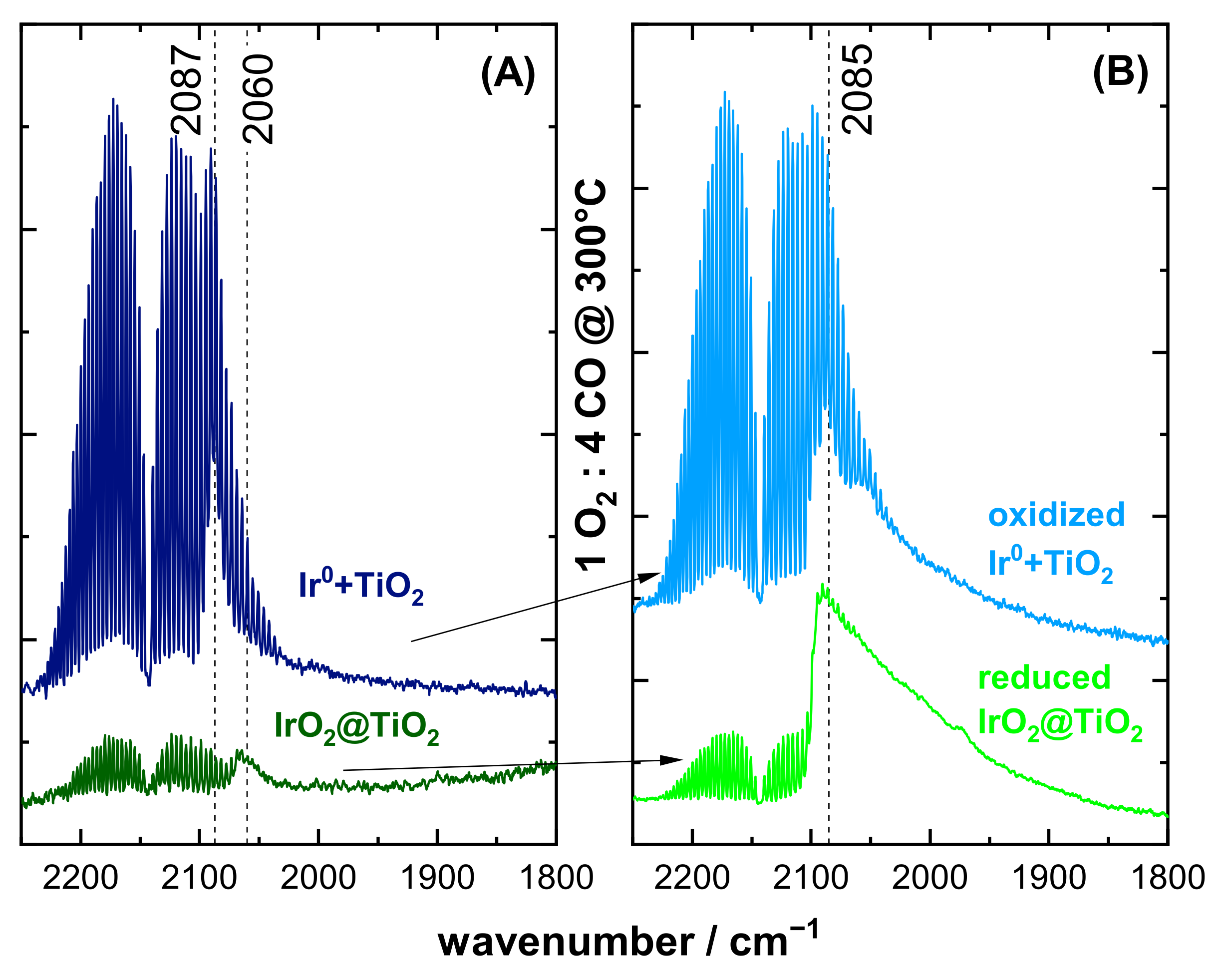
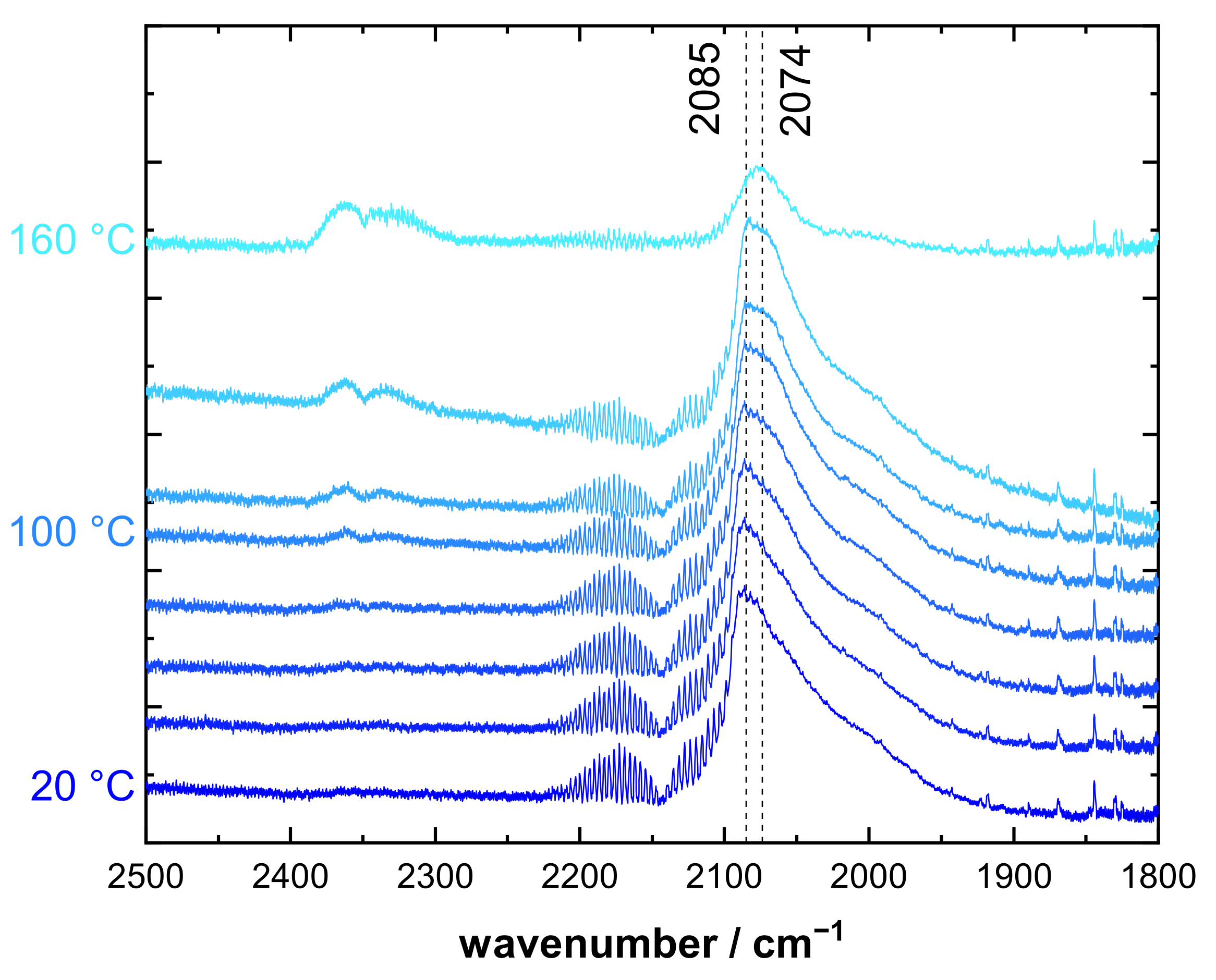
3.2. CO DRIFTS Experiments of Oxidized and Reduced IrO2@TiO2 and Ir0 + TiO2 Samples
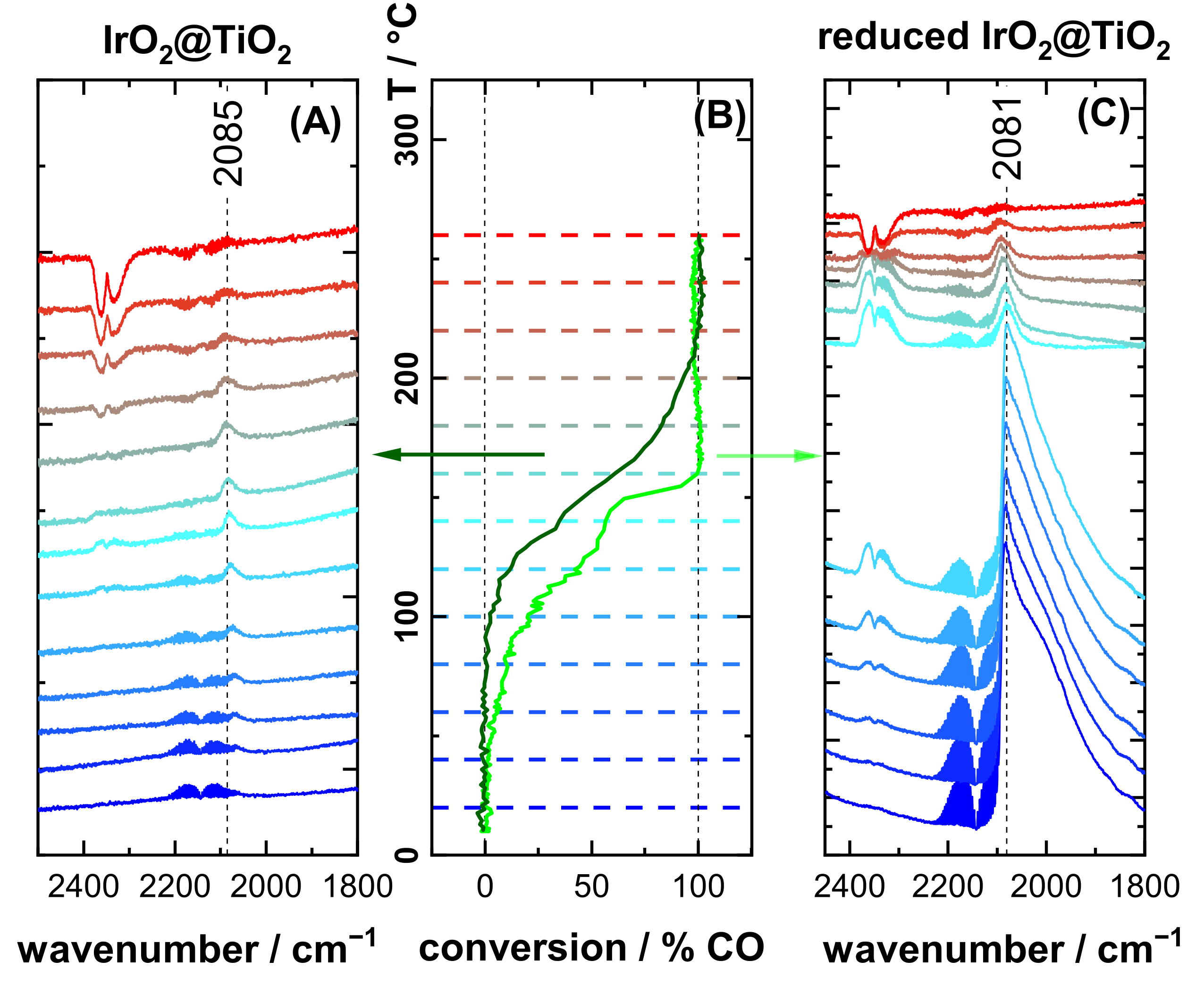
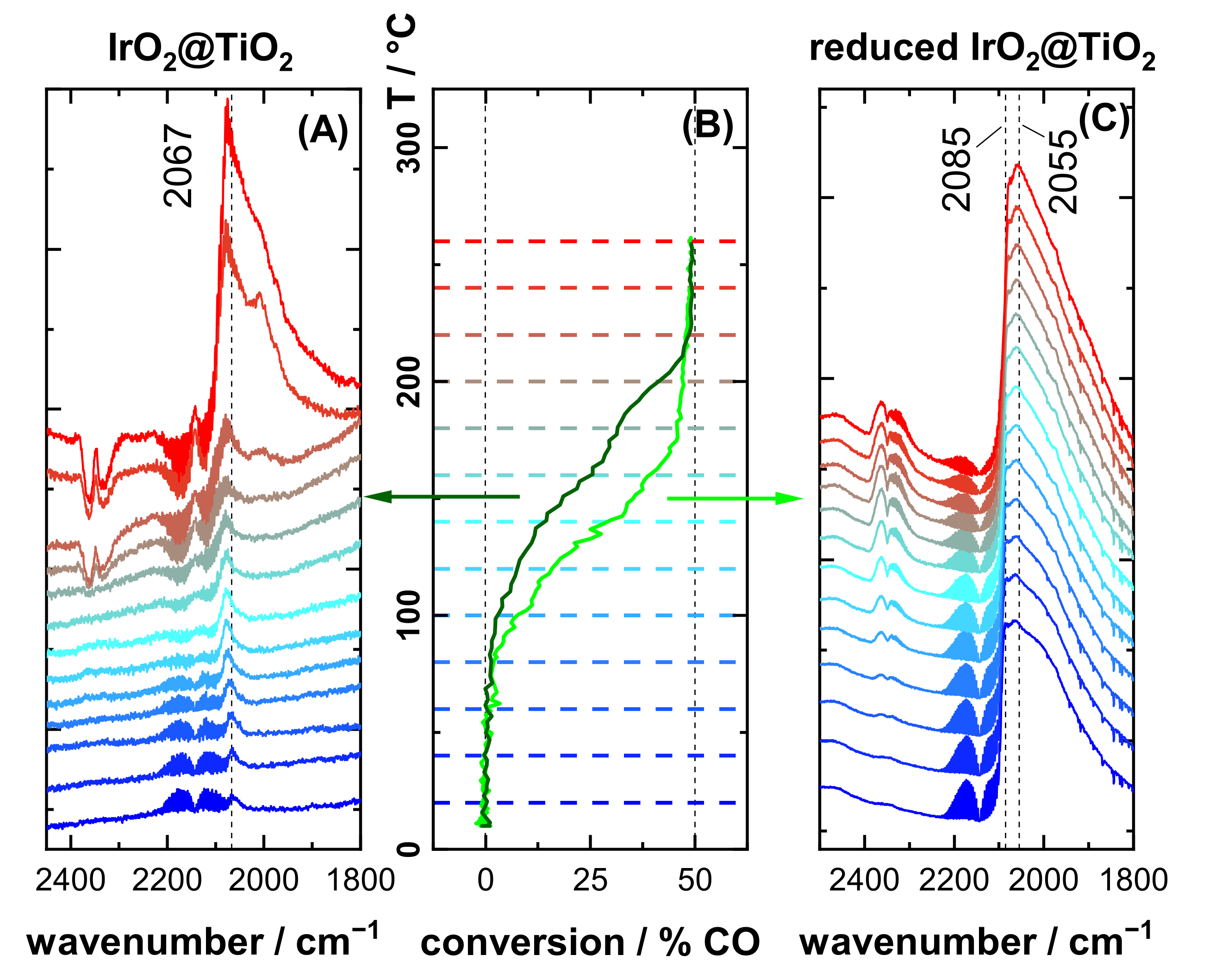
3.3. CO Oxidation Experiments of IrO2@TiO2 Samples
4. Discussion
4.1. CO as a Probe Molecule for the Surface Oxidation State of IrO2
4.2. Case Study: Catalytic CO Oxidation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Over, H.; Balmes, O.; Lundgren, E. Direct Comparison of the Reactivity of the Non-Oxidic Phase of Ru(0001) and the RuO2 Phase in the CO Oxidation Reaction. Surf. Sci. 2009, 603, 298–303. [Google Scholar] [CrossRef]
- Gao, F.; Goodman, D.W. CO Oxidation over Ruthenium: Identification of the Catalytically Active Phases at near-Atmospheric Pressures. Phys. Chem. Chem. Phys. 2012, 14, 6688–6697. [Google Scholar] [CrossRef]
- Gustafson, J.; Balmes, O.; Zhang, C.; Shipilin, M.; Schaefer, A.; Hagman, B.; Merte, L.R.; Martin, N.M.; Carlsson, P.A.; Jankowski, M.; et al. The Role of Oxides in Catalytic CO Oxidation over Rhodium and Palladium. ACS Catal. 2018, 8, 4438–4445. [Google Scholar] [CrossRef]
- Martin, R.; Kim, M.; Lee, C.J.; Mehar, V.; Albertin, S.; Hejral, U.; Merte, L.R.; Asthagiri, A.; Weaver, J.F. Isothermal Reduction of IrO2(110) Films by Methane Investigated Using in Situ x-Ray Photoelectron Spectroscopy. ACS Catal. 2021, 11, 5004–5016. [Google Scholar] [CrossRef]
- Weaver, J.F. Surface Chemistry of Late Transition Metal Oxides. Chem. Rev. 2013, 113, 4164–4215. [Google Scholar] [CrossRef]
- Schlögl, R. Heterogeneous Catalysis. Angew. Chemie Int. Ed. 2015, 54, 3465–3520. [Google Scholar] [CrossRef] [Green Version]
- Weckhuysen, B.M. Determining the Active Site in a Catalytic Process: Operando Spectroscopy Is More than a Buzzword. Phys. Chem. Chem. Phys. 2003, 5, 4351–4360. [Google Scholar] [CrossRef]
- Grunwaldt, J.D.; Baiker, A. In Situ Spectroscopic Investigation of Heterogeneous Catalysts and Reaction Media at High Pressure. Phys. Chem. Chem. Phys. 2005, 7, 3526–3539. [Google Scholar] [CrossRef]
- Ryczkowski, J. IR Spectroscopy in Catalysis. Catal. Today 2001, 68, 263–381. [Google Scholar] [CrossRef]
- Zaera, F. Infrared Absorption Spectroscopy of Adsorbed CO: New Applications in Nanocatalysis for an Old Approach. ChemCatChem 2012, 4, 1525–1533. [Google Scholar] [CrossRef]
- Meunier, F.C. Relevance of IR Spectroscopy of Adsorbed CO for the Characterization of Heterogeneous Catalysts Containing Isolated Atoms. J. Phys. Chem. C 2021, 125, 21810–21823. [Google Scholar] [CrossRef]
- Hoffman, F.M. Infrared Reflection-Absorption Spectroscopy of Adsorbed Molecules. Surf. Sci. Rep. 1983, 3, 107–192. [Google Scholar] [CrossRef]
- Hollins, P.; Pritchard, J. Infrared Studies of Chemisorbed Layers on Single Crystals. Prog. Surf. Sci. 1985, 19, 275–349. [Google Scholar] [CrossRef]
- Villegas, I.; Weaver, M.J. Modeling Electrochemical Interfaces in Ultrahigh Vacuum: Molecular Roles of Solvation in Double-Layer Phenomena. J. Phys. Chem. B 1997, 101, 10166–10177. [Google Scholar] [CrossRef]
- Van Santen, R.A.; Tranca, I.; Hensen, E.J.M. Theory of Surface Chemistry and Reactivity of Reducible Oxides. Catal. Today 2015, 244, 63–84. [Google Scholar] [CrossRef]
- Khalid, O.; Spriewald Luciano, A.; Drazic, G.; Over, H. Mixed RuxIr1−xO2 Supported on Rutile TiO2: Catalytic Methane Combustion, a Model Study. ChemCatChem 2021, 13, 3983–3994. [Google Scholar] [CrossRef]
- Martin, R.; Lee, C.J.; Mehar, V.; Kim, M.; Asthagiri, A.; Weaver, J.F. Catalytic Oxidation of Methane on IrO2 (110) Films Investigated Using Ambient-Pressure X-Ray Photoelectron Spectroscopy. ACS Catal. 2022, 12, 2840–2853. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Khalid, O.; Weber, T.; Luciano, A.S.; Zhan, W.; Smarsly, B.M.; Over, H. Supported RuxIr1-xO2 Mixed Oxides Catalysts for Propane Combustion: Resistance Against Water Poisoning. ChemCatChem 2022, 14, e202200149. [Google Scholar] [CrossRef]
- Assmann, J.; Narkhede, V.; Breuer, N.A.; Muhler, M.; Seitsonen, A.P.; Knapp, M.; Crihan, D.; Farkas, A.; Mellau, G.; Over, H. Heterogeneous Oxidation Catalysis on Ruthenium: Bridging the Pressure and Materials Gaps and Beyond. J. Phys. Condens. Matter 2008, 20, 184017. [Google Scholar] [CrossRef]
- Khalid, O.; Weber, T.; Drazic, G.; Djerdj, I.; Over, H. Mixed RuxIr1-xO2 Oxide Catalyst with Well-Defined and Varying Composition Applied to CO Oxidation. J. Phys. Chem. C 2020, 124, 18670–18683. [Google Scholar] [CrossRef]
- Trasatti, S. Electrocatalysis: Understanding the Success of DSA®. Electrochim. Acta 2000, 45, 2377–2385. [Google Scholar] [CrossRef]
- Kötz, R.; Stucki, S. Stabilization of RuO2 by IrO2 for Anodic Oxygen Evolution in Acid Media. Electrochim. Acta 1986, 31, 1311–1316. [Google Scholar] [CrossRef]
- Over, H. Atomic Scale Insights into Electrochemical versus Gas Phase Oxidation of HCl over RuO2-Based Catalysts: A Comparative Review. Electrochim. Acta 2013, 93, 314–333. [Google Scholar] [CrossRef]
- Over, H. Fundamental Studies of Planar Single-Crystalline Oxide Model Electrodes (RuO2, IrO2) for Acidic Water Splitting. ACS Catal. 2021, 11, 8848–8871. [Google Scholar] [CrossRef]
- Fabbri, E.; Habereder, A.; Waltar, K.; Kötz, R.; Schmidt, T.J. Developments and Perspectives of Oxide-Based Catalysts for the Oxygen Evolution Reaction. Catal. Sci. Technol. 2014, 4, 3800–3821. [Google Scholar] [CrossRef] [Green Version]
- Drochner, A.; Fehlings, M.; Krauß, K.; Vogel, H. A New DRIFTS Cell for the In-Situ Investigation of Heterogeneously Catalyzed Reactions. Chem. Eng. Technol. 2000, 23, 319–322. [Google Scholar] [CrossRef]
- Meunier, F.C. Pitfalls and Benefits of: In Situ and Operando Diffuse Reflectance FT-IR Spectroscopy (DRIFTS) Applied to Catalytic Reactions. React. Chem. Eng. 2016, 1, 134–141. [Google Scholar] [CrossRef]
- Freund, H.J.; Meijer, G.; Scheffler, M.; Schlögl, R.; Wolf, M. CO Oxidation as a Prototypical Reaction for Heterogeneous Processes. Angew. Chemie Int. Ed. 2011, 50, 10064–10094. [Google Scholar] [CrossRef]
- Abb, M.J.S.; Weber, T.; Glatthaar, L.; Over, H. Growth of Ultrathin Single-Crystalline IrO2(110) Films on a TiO2(110) Single Crystal. Langmuir 2019, 35, 7720–7726. [Google Scholar] [CrossRef]
- He, Y.; Langsdorf, D.; Li, L.; Over, H. Versatile Model System for Studying Processes Ranging from Heterogeneous to Photocatalysis: Epitaxial RuO2(110) on TiO2(110). J. Phys. Chem. C 2015, 119, 2692–2702. [Google Scholar] [CrossRef]
- Mamaca, N.; Mayousse, E.; Arrii-Clacens, S.; Napporn, T.W.; Servat, K.; Guillet, N.; Kokoh, K.B. Electrochemical Activity of Ruthenium and Iridium Based Catalysts for Oxygen Evolution Reaction. Appl. Catal. B Environ. 2012, 111–112, 376–380. [Google Scholar] [CrossRef]
- Terezo, A.J.; Pereira, E.C. Preparation and Characterization of Ti/RuO2 Anodes Obtained by Sol-Gel and Conventional Routes. Mater. Lett. 2002, 53, 339–345. [Google Scholar] [CrossRef]
- Reksten, A.; Sunde, S.; Seland, F.; Moradi, F. Iridium-Ruthenium Mixed Oxide for Oxygen Evolution Reaction Prepared By Pechini Synthesis. ECS Meet. Abstr. 2013, MA2013-02, 80. [Google Scholar] [CrossRef]
- Rosario, A.V.; Bulhoões, L.O.S.; Pereira, E.C. Investigation of Pseudocapacitive Properties of RuO2 Film Electrodes Prepared by Polymeric Precursor Method. J. Power Sources 2006, 158, 795–800. [Google Scholar] [CrossRef]
- Freakley, S.J.; Ruiz-Esquius, J.; Morgan, D.J. The X-Ray Photoelectron Spectra of Ir, IrO2 and IrCl3 Revisited. Surf. Interface Anal. 2017, 49, 794–799. [Google Scholar] [CrossRef]
- Abb, M.J.S.; Weber, T.; Langsdorf, D.; Koller, V.; Gericke, S.M.; Pfaff, S.; Busch, M.; Zetterberg, J.; Preobrajenski, A.; Grönbeck, H.; et al. Thermal Stability of Single-Crystalline IrO2(110) Layers: Spectroscopic and Adsorption Studies. J. Phys. Chem. C 2020, 124, 15324–15336. [Google Scholar] [CrossRef]
- Over, H. Surface Chemistry of Ruthenium Dioxide in Heterogeneous Catalysis and Electrocatalysis: From Fundamental to Applied Research. Chem. Rev. 2012, 112, 3356–3426. [Google Scholar] [CrossRef]
- Morgan, D.J. Resolving Ruthenium: XPS Studies of Common Ruthenium Materials. Surf. Interface Anal. 2015, 47, 1072–1079. [Google Scholar] [CrossRef]
- Solymosi, F.; Novák, É.; Molnár, A. Infrared Spectroscopic Study on CO-Induced Structural Changes of Iridium on an Alumina Support. J. Phys. Chem. 1990, 94, 7250–7255. [Google Scholar] [CrossRef]
- Solymosi, F.; Raskó, J. An Infrared Study of CO and NO Adsorption on Alumina-Supported Iridium Catalyst. J. Catal. 1980, 62, 253–263. [Google Scholar] [CrossRef]
- Lauterbach, J.; Boyle, R.W.; Schick, M.; Mitchell, W.J.; Meng, B.; Weinberg, W.H. The Adsorption of CO on Ir(111) Investigated with FT-IRAS. Surf. Sci. 1996, 350, 32–44. [Google Scholar] [CrossRef]
- Fujitani, T.; Nakamura, I.; Kobayashi, Y.; Takahashi, A.; Haneda, M.; Hamada, H. Adsorption and Reactions of NO on Clean and CO-Precovered Ir(111). J. Phys. Chem. B 2005, 109, 17603–17607. [Google Scholar] [CrossRef] [PubMed]
- Farkas, A.; Mellau, G.C.; Over, H. Novel Insight in the CO Oxidation on RuO2(110) by in Situ Reflection-Absorption Infrared Spectroscopy. J. Phys. Chem. C 2009, 113, 14341–14355. [Google Scholar] [CrossRef]
- Farkas, A.; Hess, F.; Over, H. “First-Principles” Kinetic Monte Carlo Simulations Revisited: CO Oxidation over RuO2(110). J. Comput. Chem. 2011, 33, 757–766. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Conditions | Ar/% | O2/% | CO/% |
|---|---|---|---|
| Oxidizing | 96 | 2 | 2 |
| Reducing | 95 | 1 | 4 |
| CO only | 96 | - | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timmer, P.; Weber, T.; Glatthaar, L.; Over, H. Operando CO Infrared Spectroscopy and On-Line Mass Spectrometry for Studying the Active Phase of IrO2 in the Catalytic CO Oxidation Reaction. Inorganics 2023, 11, 102. https://doi.org/10.3390/inorganics11030102
Timmer P, Weber T, Glatthaar L, Over H. Operando CO Infrared Spectroscopy and On-Line Mass Spectrometry for Studying the Active Phase of IrO2 in the Catalytic CO Oxidation Reaction. Inorganics. 2023; 11(3):102. https://doi.org/10.3390/inorganics11030102
Chicago/Turabian StyleTimmer, Phillip, Tim Weber, Lorena Glatthaar, and Herbert Over. 2023. "Operando CO Infrared Spectroscopy and On-Line Mass Spectrometry for Studying the Active Phase of IrO2 in the Catalytic CO Oxidation Reaction" Inorganics 11, no. 3: 102. https://doi.org/10.3390/inorganics11030102
APA StyleTimmer, P., Weber, T., Glatthaar, L., & Over, H. (2023). Operando CO Infrared Spectroscopy and On-Line Mass Spectrometry for Studying the Active Phase of IrO2 in the Catalytic CO Oxidation Reaction. Inorganics, 11(3), 102. https://doi.org/10.3390/inorganics11030102