
H2O·HF@C70: Encapsulation Energetics and Thermodynamics





Abstract
:1. Introduction
2. Calculations
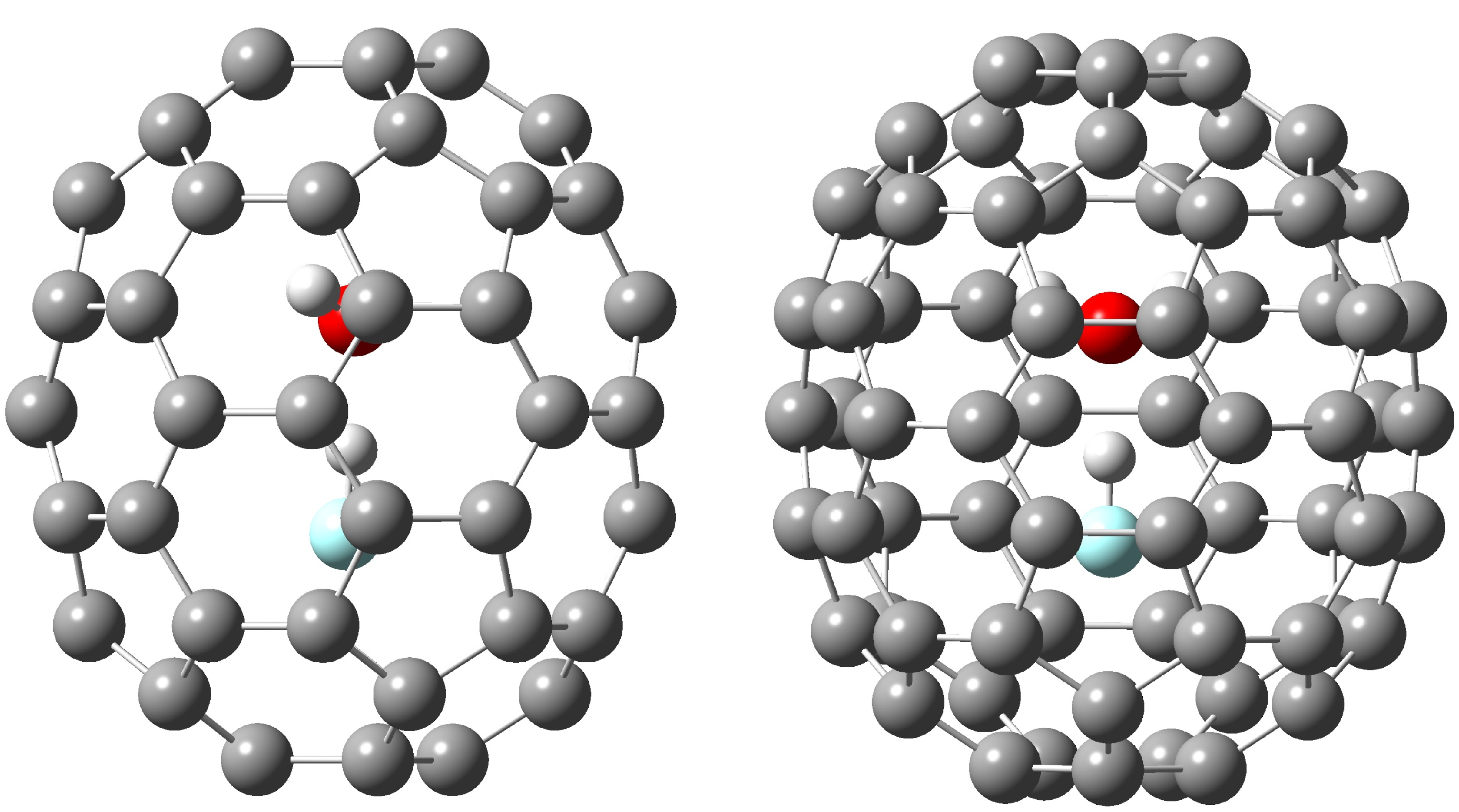
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
| Calc. Level | 6-31++G** | 6-311++G** |
|---|---|---|
| M06-2X | ||
| B2PLYPD | ||
| (kcal/mol) | (kcal/mol) | (kcal/mol) | (atm) | |
|---|---|---|---|---|
| B2PLYPD/6-311++G** | 1.346 × 10 |
References
- Dresselhaus, M.S.; Dresselhaus, G.; Eklund, P.C. Science of Fullerenes and Carbon Nanotubes; Academic Press: San Diego, CA, USA, 1996; p. 316. [Google Scholar]
- Peres, T.; Cao, B.P.; Cui, W.D.; Khong, A.; Cross, R.J.; Saunders, M.; Lifshitz, C. Some new diatomic molecule containing endohedral fullerenes. Int. J. Mass Spectr. 2001, 210/211, 241–247. [Google Scholar] [CrossRef]
- Suetsuna, T.; Dragoe, N.; Harneit, W.; Weidinger, A.; Shimotani, H.; Ito, S.; Takagi, H.; Kitazawa, K. Separation of N2@C60 and N@C60. Chem. Eur. J. 2002, 8, 5079–5083. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.A.; Pawlik, T.; Weidinger, A.; Höhne, M.; Alcala, R.; Spaeth, J.-M. Observation of atomlike nitrogen in nitrogen-implanted solid C60. Phys. Rev. Lett. 1996, 77, 1075–1078. [Google Scholar] [CrossRef] [PubMed]
- Knapp, C.; Dinse, K.-P.; Pietzak, B.; Waiblinger, M.; Weidinger, A. Fourier transform EPR study of N@C60 in solution. Chem. Phys. Lett. 1997, 272, 433–437. [Google Scholar] [CrossRef]
- Pietzak, B.; Waiblinger, M.; Murphy, T.A.; Weidinger, A.; Höhne, M.; Dietel, E.; Hirsch, A. Buckminsterfullerene C60: A chemical Faraday cage for atomic nitrogen. Chem. Phys. Lett. 1997, 279, 259–263. [Google Scholar] [CrossRef]
- Cao, B.P.; Peres, T.; Cross, R.J.; Saunders, M.; Lifshitz, C. Do nitrogen-atom-containing endohedral fullerenes undergo the shrink-wrap mechanism? J. Phys. Chem. A 2001, 105, 2142–2146. [Google Scholar] [CrossRef]
- Kobayashi, K.; Nagase, S.; Dinse, K.-P. A theoretical study of spin density distributions and isotropic hyperfine couplings of N and P atoms in N@C60, P@C60, N@C70, N@C60(CH2)6, and N@C60(SiH2)6. Chem. Phys. Lett. 2003, 377, 93–98. [Google Scholar] [CrossRef]
- Wakahara, T.; Matsunaga, Y.; Katayama, A.; Maeda, Y.; Kako, M.; Akasaka, T.; Okamura, M.; Kato, T.; Choe, Y.K.; Kobayashi, K.; et al. A comparison of the photochemical reactivity of N@C60 and C60: Photolysis with disilirane. Chem. Commun. 2003, 39, 2940–2941. [Google Scholar] [CrossRef]
- Saunders, M.; Jiménez-Vázquez, H.A.; Cross, R.J.; Poreda, R.J. Stable compounds of helium and neon: He@C60 and Ne@C60. Science 1993, 259, 1428–1430. [Google Scholar] [CrossRef]
- Saunders, M.; Jiménez-Vázquez, H.A.; Cross, R.J.; Mroczkowski, S.; Freedberg, D.I.; Anet, F.A.L. Probing the interior of fullerenes by 3He NMR spectroscopy of endohedral 3He@C60 and 3He@C70. Nature 1994, 367, 256–258. [Google Scholar] [CrossRef]
- Cross, R.J.; Saunders, M.; Prinzbach, H. Putting helium inside dodecahedrane. Org. Lett. 1999, 1, 1479–1481. [Google Scholar] [CrossRef]
- Cross, R.J.; Saunders, M. Catalyzed incorporation of nobel gases in fullerenes. In Recent Advances in the Chemistry and Physics of Fullerenes and Related Materials, Volume 11—Fullerenes for the New Millennium; Kadish, K.M., Kamat, P.V., Guldi, D., Eds.; The Electrochemical Society: Pennington, NJ, USA, 2001; pp. 298–300. [Google Scholar]
- Rubin, Y.; Jarrosson, T.; Wang, G.-W.; Bartberger, M.D.; Houk, K.N.; Schick, G.; Saunders, M.; Cross, R.J. Insertion of helium and molecular hydrogen through the orifice of an open fullerene. Angew. Chem., Int. Ed. Engl. 2001, 40, 1543–1546. [Google Scholar] [CrossRef]
- Carravetta, M.; Murata, Y.; Murata, M.; Heinmaa, I.; Stern, R.; Tontcheva, A.; Samoson, A.; Rubin, Y.; Komatsu, K.; Levitt, M.H. Solid-state NMR spectroscopy of molecular hydrogen trapped inside an open-cage fullerene. J. Am. Chem. Soc. 2004, 126, 4092–4093. [Google Scholar] [CrossRef] [Green Version]
- Iwamatsu, S.-I.; Uozaki, T.; Kobayashi, K.; Re, S.; Nagase, S.; Murata, S. A bowl-shaped fullerene encapsulates a water into the cage. J. Am. Chem. Soc. 2004, 126, 2668–2669. [Google Scholar] [CrossRef]
- Komatsu, K.; Murata, M.; Murata, Y. Encapsulation of molecular hydrogen in fullerene C60 by organic synthesis. Science 2005, 307, 238–240. [Google Scholar] [CrossRef]
- Kurotobi, K.; Murata, Y. A single molecule of water encapsulated in fullerene C60. Science 2011, 333, 613–616. [Google Scholar] [CrossRef]
- Zhang, R.; Murata, M.; Aharen, T.; Wakamiya, A.; Shimoaka, T.; Hasegawa, T.; Murata, Y. Synthesis of a distinct water dimer inside fullerene C70. Nature Chem. 2016, 8, 435–441. [Google Scholar] [CrossRef]
- Iwamatsu, S.; Stanisky, C.M.; Cross, R.J.; Saunders, M.; Mizorogi, N.; Nagase, S.; Murata, S. Carbon monoxide inside an open-cage fullerene. Angew. Chem. Intl. Ed. 2006, 45, 5337–5340. [Google Scholar] [CrossRef]
- Shi, L.J.; Yang, D.Z.; Colombo, F.; Yu, Y.M.; Zhang, W.X.; Gan, L.B. Punching a carbon atom of C60 into its own cavity to form an endohedral complex CO@C59O6 under mild conditions. Chem. Eur. J. 2013, 19, 16545–16549. [Google Scholar] [CrossRef]
- Li, Y.; Lou, N.; Xu, D.; Pan, C.; Lu, X.; Gan, L. Oxygen-delivery materials: Synthesis of an open-cage fullerene derivative suitable for encapsulation of H2O2 and O2. Angew. Chem. Int. Ed. 2018, 57, 14144–14148. [Google Scholar] [CrossRef]
- Gan, L. Molecular containers derived from [60]fullerene through peroxide chemistry. Acc. Chem. Res. 2019, 52, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Cioslowski, J. Endohedral chemistry: Electronic structures of molecules trapped inside the C60 cage. J. Am. Chem. Soc. 1991, 113, 4139–4141. [Google Scholar] [CrossRef]
- Charkin, O.P.; Klimenko, N.M.; Charkin, D.O.; Mebel, A.M. Theoretical study of host-guest interaction in model endohedral fullerenes with tetrahedral molecules and ions of MH4 hydrides inside the C60H36, C60H24, C84, and C60 cages. Russ. J. Inorg. Chem. 2004, 49, 868–880. [Google Scholar]
- Slanina, Z.; Uhlík, F.; Adamowicz, L.; Nagase, S. Computing fullerene encapsulation of non-metallic molecules: N2@C60 and NH3@C60. Mol. Simul. 2005, 31, 801–806. [Google Scholar] [CrossRef]
- Ramachandran, C.N.; Sathyamurthy, N. Water clusters in a confined nonpolar environment. Chem. Phys. Let. 2005, 410, 348–351. [Google Scholar] [CrossRef]
- Slanina, Z.; Nagase, S. A computational characterization of N2@C60. Mol. Phys. 2006, 104, 3167–3171. [Google Scholar] [CrossRef]
- Shameema, O.; Ramachandran, C.N.; Sathyamurthy, N. Blue shift in X-H stretching frequency of molecules due to confinement. J. Phys. Chem. A 2006, 110, 2–4. [Google Scholar] [CrossRef]
- Slanina, Z.; Pulay, P.; Nagase, S. H2, Ne, and N2 energies of encapsulation into C60 evaluated with the MPWB1K functional. J. Chem. Theory Comput. 2006, 2, 782–785. [Google Scholar] [CrossRef]
- Mazurek, A.P.; Sadlej-Sosnowska, N. Is fullerene C60 large enough to host an aromatic molecule? Int. J. Quantum Chem. 2011, 111, 2398–2405. [Google Scholar] [CrossRef]
- Rodríguez-Fortea, A.; Balch, A.L.; Poblet, J.M. Endohedral metallofullerenes: A unique host-guest association. Chem. Soc. Rev. 2011, 40, 3551–3563. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R. Can a single molecule of water be completely isolated within the subnano-space inside the fullerene C60 cage? A Quantum chemical prospective. Chem. Eur. J. 2012, 18, 15345–15360. [Google Scholar] [CrossRef]
- Farimani, A.B.; Wu, Y.B.; Aluru, N.R. Rotational motion of a single water molecule in a buckyball. Phys. Chem. Chem. Phys. 2013, 15, 17993–18000. [Google Scholar] [CrossRef]
- Popov, A.A.; Yang, S.; Dunsch, L. Endohedral fullerenes. Chem. Rev. 2013, 113, 5989–6113. [Google Scholar] [CrossRef]
- Uhlík, F.; Slanina, Z.; Lee, S.-L.; Wang, B.-C.; Adamowicz, L.; Nagase, S. Water-dimer stability and its fullerene encapsulations. J. Comput. Theor. Nanosci. 2015, 12, 959–964. [Google Scholar] [CrossRef]
- Slanina, Z.; Uhlík, F.; Lu, X.; Akasaka, T.; Lemke, K.H.; Seward, T.M.; Nagase, S.; Adamowicz, L. Calculations of the water-dimer encapsulations into C84. Fullerenes Nanotub. Carbon Nanostruct. 2016, 24, 1–7. [Google Scholar] [CrossRef]
- Slanina, Z.; Uhlík, F.; Nagase, S.; Lu, X.; Akasaka, T.; Adamowicz, L. Computed relative populations of D2(22)-C84 endohedrals with encapsulated monomeric and dimeric water. ChemPhysChem 2016, 17, 1109–1111. [Google Scholar] [CrossRef] [Green Version]
- Slanina, Z.; Uhlík, F.; Nagase, S.; Akasaka, T.; Adamowicz, L.; Lu, X. Computational comparison of the water-dimer encapsulations into D2(22)-C84 and D2d(23)-C84. ECS J. Solid State Sci. Technol. 2017, 6, M3113–M3115. [Google Scholar] [CrossRef]
- Slanina, Z.; Uhlík, F.; Nagase, S.; Akasaka, T.; Adamowicz, L.; Lu, X. A computational characterization of CO@C60. Fullerenes Nanotub. Carbon Nanostruct. 2017, 25, 624–629. [Google Scholar] [CrossRef]
- Slanina, Z.; Uhlík, F.; Nagase, S.; Akasaka, T.; Lu, X.; Adamowicz, L. Cyclic water-trimer encapsulation into D2(22)-C84 fullerene. Chem. Phys. Lett. 2018, 695, 245–248. [Google Scholar] [CrossRef]
- Slanina, Z.; Uhlík, F.; Adamowicz, L.; Pan, C.; Lu, X. A computational characterization of H2O2@C60. Fullerenes Nanotub. Carbon Nanostruct. 2022, 30, 258–262. [Google Scholar] [CrossRef]
- Zhang, R.; Murata, M.; Wakamiya, A.; Shimoaka, T.; Hasegawa, T.; Murata, Y. Isolation of the simplest hydrated acid. Sci. Adv. 2017, 3, e1602833-1–e1602833-6. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Schwabe, T.; Grimme, S. Double-hybrid density functionals with long-range dispersion corrections: Higher accuracy and extended applicability. Phys. Chem. Chem. Phys. 2007, 9, 3397–3406. [Google Scholar] [CrossRef] [PubMed]
- Moller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the difference of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Slanina, Z.; Lee, S.-L.; Adamowicz, L.; Uhlík, F.; Nagase, S. Computed atructure and energetics of La@C60. Int. J. Quantum Chem. 2005, 104, 272–277. [Google Scholar] [CrossRef]
- Basiuk, V.A.; Basiuk, E.V. Noncovalent complexes of Ih-C80 fullerene with phthalocyanines. Fulleren. Nanotub. Carb. Nanostruct. 2018, 26, 69–75. [Google Scholar] [CrossRef]
- Basiuk, V.A.; Tahuilan-Anguiano, D.E. Complexation of free-base and 3d transition metal(II) phthalocyanines with endohedral fullerene Sc3N@C80. Chem. Phys. Lett. 2019, 722, 146–152. [Google Scholar] [CrossRef]
- Tahuilan-Anguiano, D.E.; Basiuk, V.A. Complexation of free-base and 3d transition metal(II) phthalocyanines with endohedral fullerenes H@C60, H2@C60 and He@C60: The effect of encapsulated species. Diam. Rel. Mat. 2021, 118, 108510-1–108510-5. [Google Scholar] [CrossRef]
- Slanina, Z.; Uhlík, F.; Lee, S.-L.; Adamowicz, L.; Akasaka, T.; Nagase, S. Computed stabilities in metallofullerene series: Al@C82, Sc@C82, Y@C82, and La@C82. Int. J. Quant. Chem. 2011, 111, 2712–2718. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Rev. D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Slanina, Z.; Uhlík, F.; Lee, S.-L.; Wang, B.-C.; Adamowicz, L.; Suzuki, M.; Haranaka, M.; Feng, L.; Lu, X.; Nagase, S.; et al. Towards relative populations of non-isomeric metallofullerenes: La@C76(Td) vs. La2@C76(Cs,17490). Fullerenes Nanotub. Carbon Nanostruct. 2014, 22, 299–306. [Google Scholar] [CrossRef]
- Simon, S.; Bertran, J.; Sodupe, M. Effect of counterpoise correction on the geometries and vibrational frequencies of hydrogen bonded systems. J. Phys. Chem. A 2001, 105, 4359–4364. [Google Scholar] [CrossRef]
- Legon, A.C.; Millen, D.J.; North, H.M. Experimental determination of the dissociation energies D0 and De of H2O...HF. Chem. Phys. Let. 1987, 135, 303–306. [Google Scholar] [CrossRef]
- Slanina, Z. Contemporary Theory of Chemical Isomerism; Academia: Prague, Czech Republic; D. Reidel Publ. Comp.: Dordrecht, The Netherlands, 1986; pp. 22–23, 160–164. [Google Scholar]
- Slanina, Z.; Uhlík, F.; Lee, S.-L.; Adamowicz, L.; Nagase, S. Computations of endohedral fullerenes: The Gibbs energy treatment. J. Comput. Meth. Sci. Engn. 2006, 6, 243–250. [Google Scholar] [CrossRef] [Green Version]
- Slanina, Z.; Uhlík, F.; Adamowicz, L. Theoretical predictions of fullerene stabilities. In Handbook of Fullerene Science and Technology; Lu, X., Akasaka, T., Slanina, Z., Eds.; Springer: Singapore, 2022; pp. 111–179. [Google Scholar]
- Akasaka, T.; Nagase, S.; Kobayashi, K.; Walchli, M.; Yamamoto, K.; Funasaka, H.; Kako, M.; Hoshino, T.; Erata, T. 13C and 139La NMR studies of La2@C80: First evidence for circular motion of metal atoms in endohedral dimetallofullerenes. Angew. Chem. Int. Ed. 1997, 36, 1643–1645. [Google Scholar] [CrossRef]
- Slanina, Z.; Uhlík, F.; Feng, L.; Akasaka, T.; Lu, X.; Adamowicz, L. Calculations of the Lu3N@C80 Two-Isomer Equilibrium. Fullerenes Nanotub. Carbon Nanostruct. 2019, 27, 382–386. [Google Scholar] [CrossRef]
- Slanina, Z. Some aspects of mathematical chemistry of equilibrium and rate processes: Steps towards a completely non-empirical computer design of syntheses. J. Mol. Struct. (Theochem) 1989, 185, 217–228. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Raghavachari, V.; Redfern, P.C.; Rassolov, V.; Pople, J.A. Gaussian-3 (G3) theory for molecules containing first and second-row atoms. J. Chem. Phys. 1998, 109, 7764–7776. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Slanina, Z.; Uhlík, F.; Lu, X.; Akasaka, T.; Adamowicz, L. H2O·HF@C70: Encapsulation Energetics and Thermodynamics. Inorganics 2023, 11, 123. https://doi.org/10.3390/inorganics11030123
Slanina Z, Uhlík F, Lu X, Akasaka T, Adamowicz L. H2O·HF@C70: Encapsulation Energetics and Thermodynamics. Inorganics. 2023; 11(3):123. https://doi.org/10.3390/inorganics11030123
Chicago/Turabian StyleSlanina, Zdeněk, Filip Uhlík, Xing Lu, Takeshi Akasaka, and Ludwik Adamowicz. 2023. "H2O·HF@C70: Encapsulation Energetics and Thermodynamics" Inorganics 11, no. 3: 123. https://doi.org/10.3390/inorganics11030123
APA StyleSlanina, Z., Uhlík, F., Lu, X., Akasaka, T., & Adamowicz, L. (2023). H2O·HF@C70: Encapsulation Energetics and Thermodynamics. Inorganics, 11(3), 123. https://doi.org/10.3390/inorganics11030123