
A Molecular Understanding of the Flame Retardant Mechanism of Zinc Stannate/Polypropylene Composites via ReaxFF Simulations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
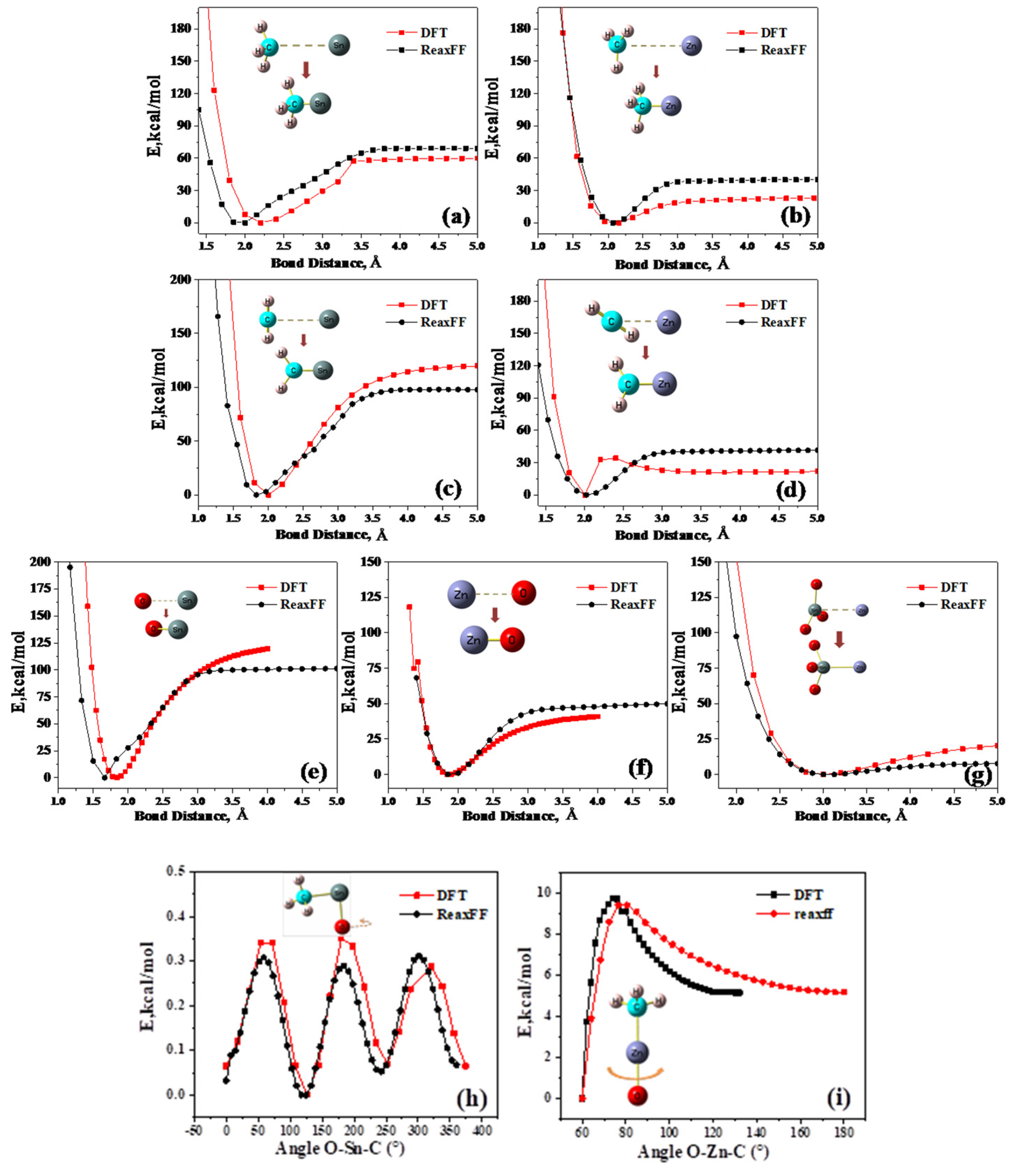
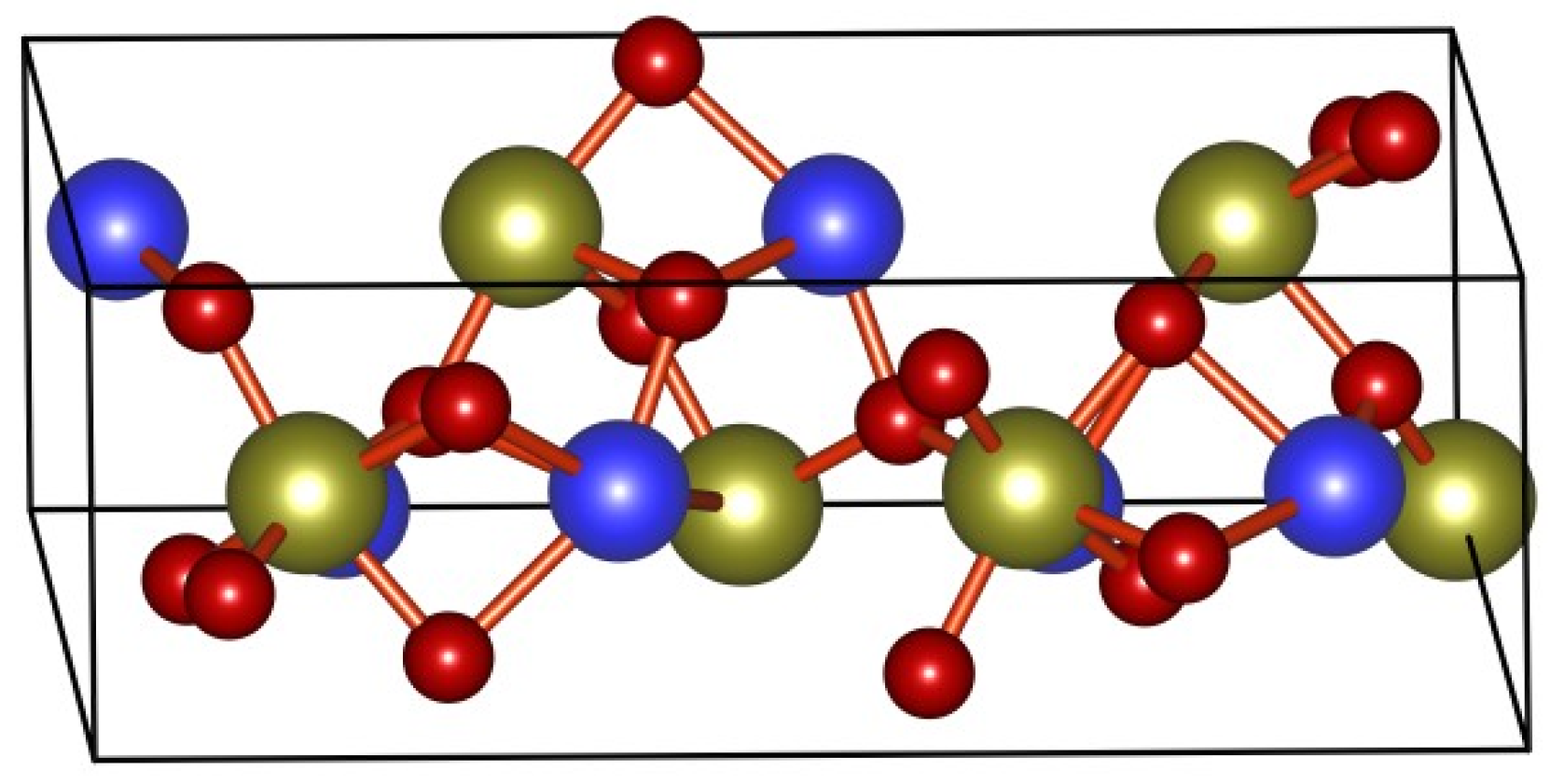
2.1. ReaxFF Force Field Results and Verification
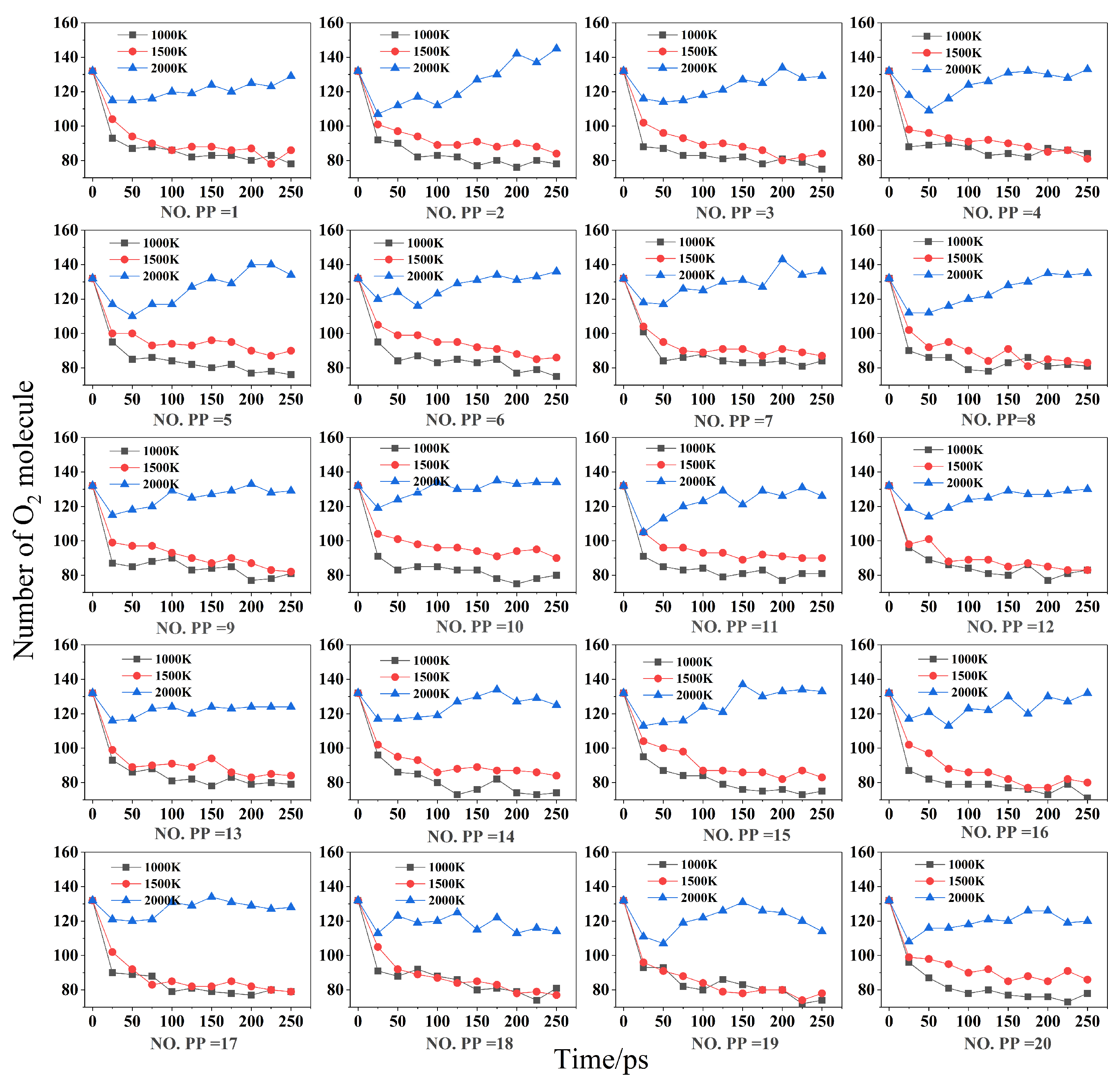
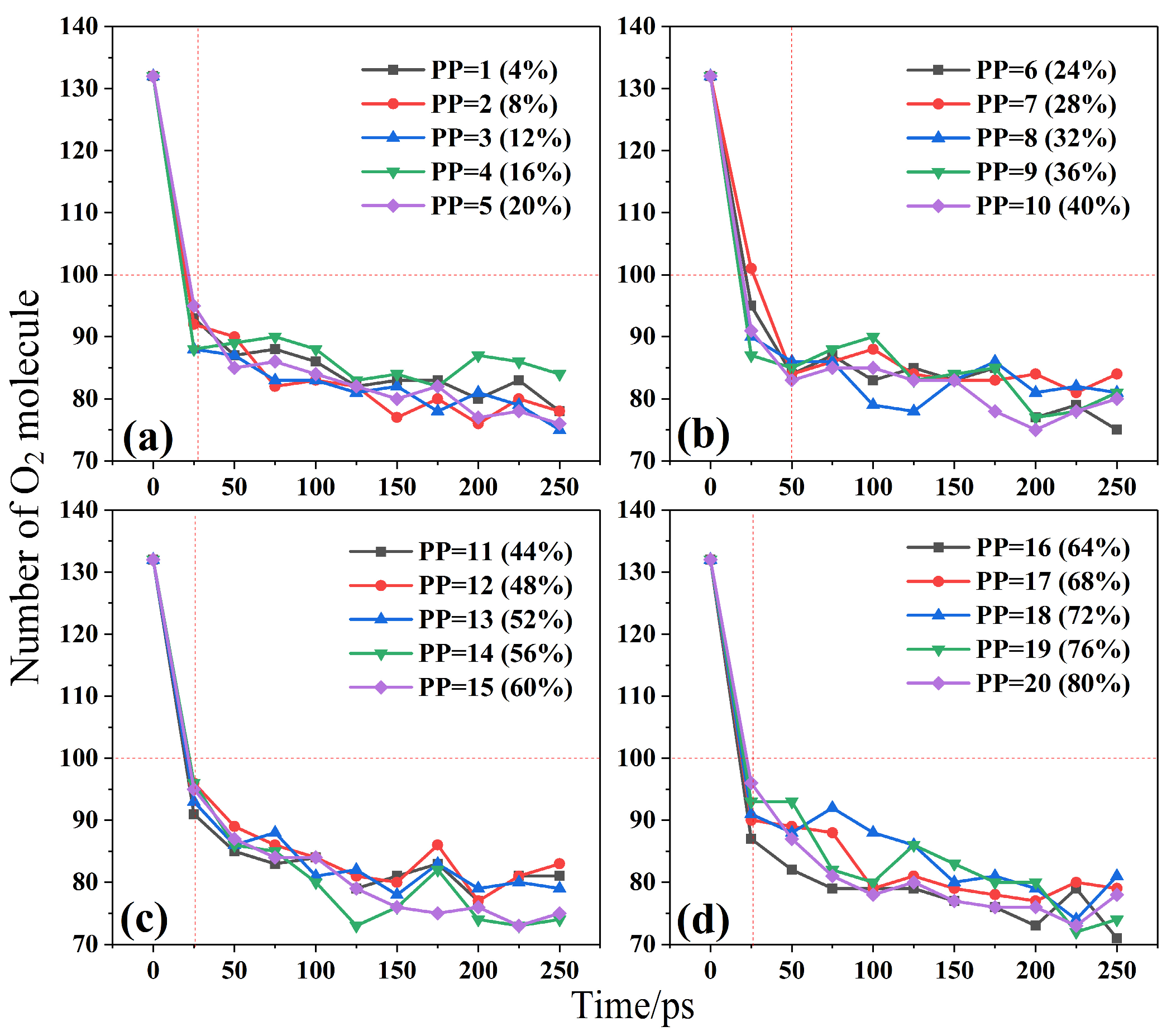
2.2. Effect of Temperature and Composite on Combustion Reactants
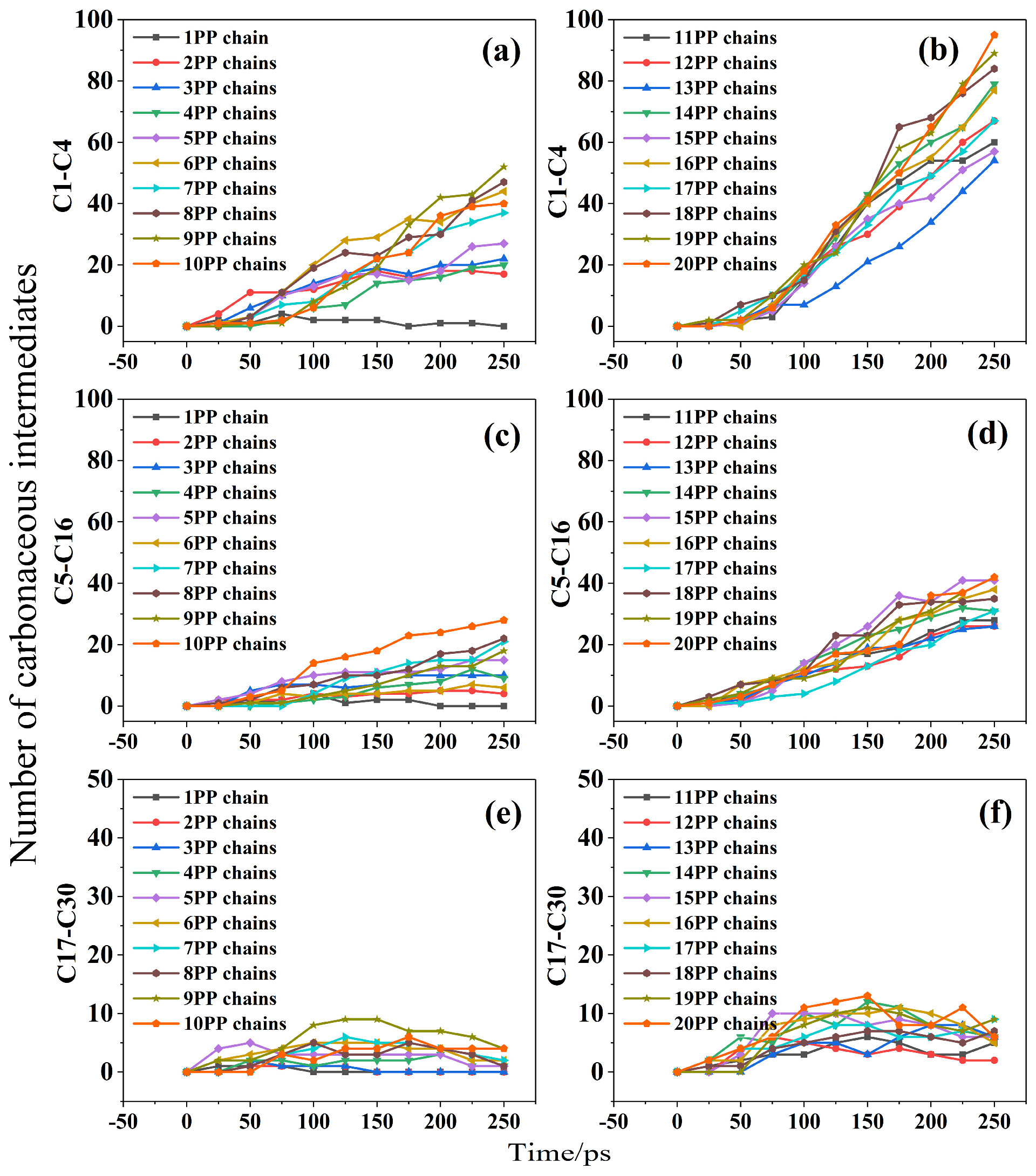
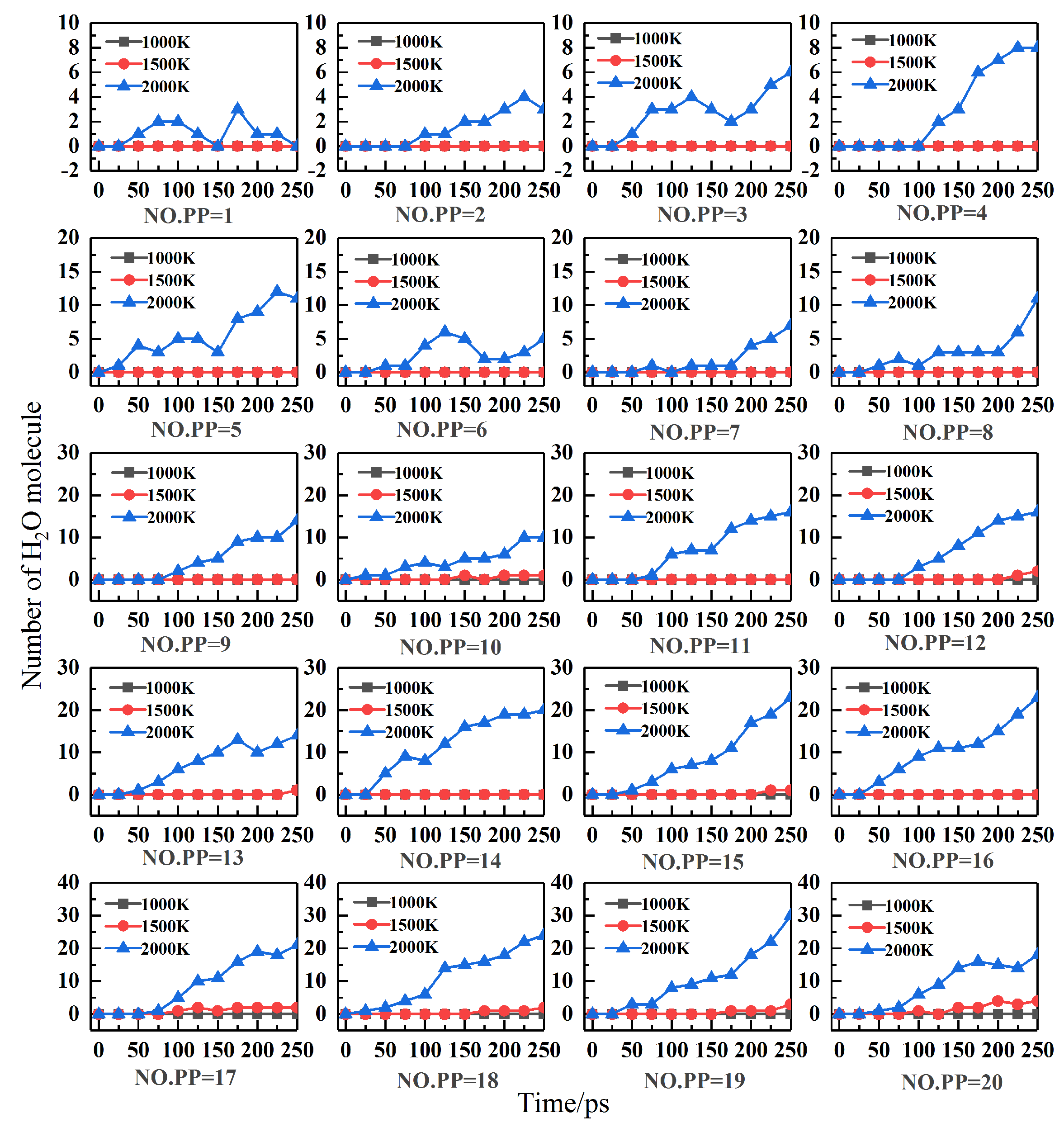
2.3. Effect of Temperature and Content on Combustion Products
3. Models and Simulation Methods
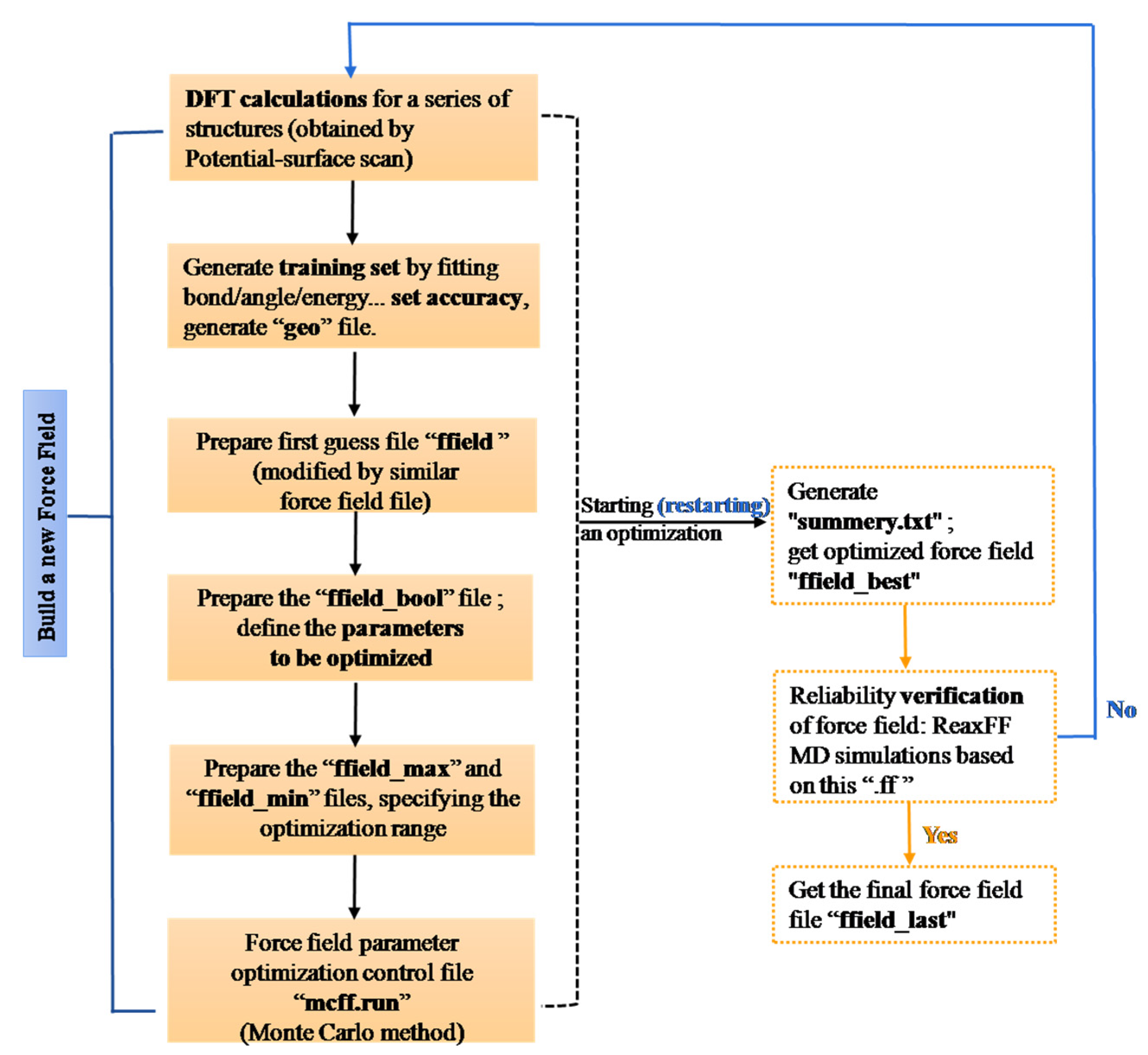
3.1. ReaxFF Force Field
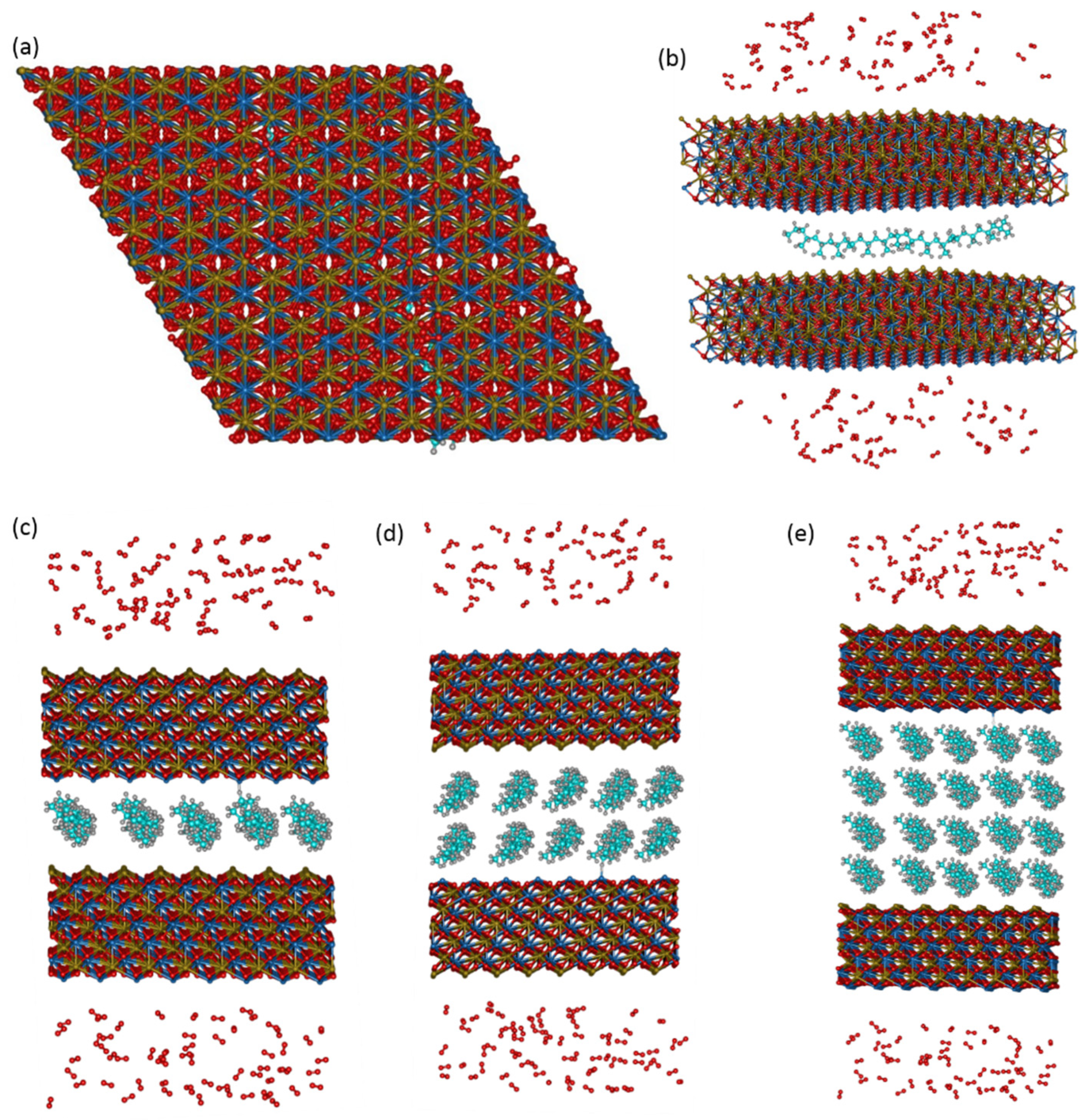
3.2. Details of ReaxFF MD Simulations
3.3. Theoretical Models
4. Conclusions
- (1)
- A ReaxFF force field including Sn/Zn/C/H/O elements was developed for ZS/PP combustion. By determining the essential chemical information in the flame-retardant system, various ground state structures, such as ZS crystals and clusters, were selected to conduct DFT calculations with selected training sets. The accuracy of obtained force field parameters was verified. By comparing DFT and ReaxFF calculations, the dissociation distance error was within 0.15 Å, while the bond angle error was within 2.5°. The results from QM and ReaxFF concur well for each energy proofreading circumstance.
- (2)
- ReaxFF MD simulations were conducted for the combustion reactions of 20 different PP/ZS composites at different temperatures. The results demonstrate that temperature limits the degree of combustion by influencing the molecular oxygen consumption of PP cracking, suggesting the flame retardant feature of ZS. The higher the combustion temperature, the slower the oxygen consumption rate of PP/ZS composites and the lower the total oxygen consumption. When the PP component of composites exceeds 56%, oxygen consumption increases. The rapid oxygen consumption stage of combustion is 25–50 ps, followed by the second slow oxygen consumption stage 50 ps later. The higher the PP percentage in composites, the more oxygen is consumed at the same temperature.
- (3)
- No intermediates smaller than C30 were detected in PP/ZS combustions at 1000 K. The earliest intermediary is C5–C16, while the latest is C1–C4. C17–C30 is the intermediate for PP/ZS composite combustions at 1500 K. The combustion process produces C1–C30 mediators at a temperature of 2000 K. The amount of the C1–C4 mediator was the highest, and C17–C30 was the lowest. The higher the PP percentage, the more C1–C16 fragments are formed during combustion. According to the ReaxFF MD simulations, molecular H2O can form at reaction temperatures beyond 1000 K. The number of H2O in PP/ZS composites increases with PP concentration.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rad, E.R.; Vahabi, H.; de Anda, A.R.; Saeb, M.R.; Thomas, S. Bio-epoxy resins with inherent flame retardancy. Prog. Org. Coat. 2019, 135, 608–612. [Google Scholar] [CrossRef]
- Seidi, F.; Movahedifar, E.; Naderi, G.; Akbari, V.; Ducos, F.; Shamsi, R.; Vahabi, H.; Saeb, M.R. Flame retardant polypropylenes: A review. Polymers 2020, 12, 1701. [Google Scholar] [CrossRef] [PubMed]
- Vahabi, H.; Jouyandeh, M.; Cochez, M.; Khalili, R.; Vagner, C.; Ferriol, M.; Movahedifar, E.; Ramezanzadeh, B.; Rostami, M.; Ranjbar, Z. Short-lasting fire in partially and completely cured epoxy coatings containing expandable graphite and halloysite nanotube additives. Prog. Org. Coat. 2018, 123, 160–167. [Google Scholar] [CrossRef]
- Zhang, Z.X.; Zhang, J.; Lu, B.-X.; Xin, Z.X.; Kang, C.K.; Kim, J.K. Effect of flame retardants on mechanical properties, flammability and foamability of PP/wood–fiber composites. Compos. Part B Eng. 2012, 43, 150–158. [Google Scholar] [CrossRef]
- Renner, G.; Nellessen, A.; Schwiers, A.; Wenzel, M.; Schmidt, T.C.; Schram, J. Data preprocessing & evaluation used in the microplastics identification process: A critical review & practical guide. TrAC Trends Anal. Chem. 2019, 111, 229–238. [Google Scholar]
- Maddah, H.A. Polypropylene as a promising plastic: A review. Am. J. Polym. Sci. 2016, 6, 1–11. [Google Scholar]
- Shen, R.; Quan, Y.; Zhang, Z.; Ma, R.; Wang, Q. Metal-Organic Framework as an Efficient Synergist for Intumescent Flame Retardants against Highly Flammable Polypropylene. Ind. Eng. Chem. Res. 2022, 61, 7292–7302. [Google Scholar] [CrossRef]
- Pan, Y.; Luo, Z.; Wang, B. Synergistic flame retardant effect of piperazine salt and ammonium polyphosphate as intumescent flame retardant system for polypropylene. J. Appl. Polym. Sci. 2021, 138, 49813. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhan, J.; Zhou, K.; Lu, H.; Zeng, W.; Stec, A.A.; Hull, T.R.; Hu, Y.; Gui, Z. The influence of carbon nanotubes on the combustion toxicity of PP/intumescent flame retardant composites. Polym. Degrad. Stab. 2015, 115, 38–44. [Google Scholar] [CrossRef]
- Zhang, S.; Horrocks, A.R. A review of flame retardant polypropylene fibres. Prog. Polym. Sci. 2003, 28, 1517–1538. [Google Scholar] [CrossRef]
- Vahabi, H.; Paran, S.M.R.; Shabanian, M.; Dumazert, L.; Sonnier, R.; Movahedifar, E.; Zarrintaj, P.; Saeb, M.R. Triple-faced polypropylene: Fire retardant, thermally stable, and antioxidative. J. Vinyl Addit. Technol. 2019, 25, 366–376. [Google Scholar] [CrossRef]
- Li, B.; Xu, M. Effect of a novel charring-foaming agent on flame retardancy and thermal degradation of intumescent flame retardant polypropylene. Polym. Degrad. Stab. 2006, 91, 1380–1386. [Google Scholar] [CrossRef]
- Vahabi, H.; Kandola, B.K.; Saeb, M.R. Flame retardancy index for thermoplastic composites. Polymers 2019, 11, 407. [Google Scholar] [CrossRef] [PubMed]
- Vahabi, H.; Dumazert, L.; Khalili, R.; Saeb, M.R.; Cuesta, J.-M.L. Flame retardant PP/PA6 blends: A recipe for recycled wastes. Flame Retard. Therm. Stab. Mater. 2019, 2, 1–8. [Google Scholar] [CrossRef]
- Pani, B.; Sirohi, S.; Singh, D. Studies on the effects of various flame retardants on polypropylene. Am. J. Polym. Sci. 2013, 3, 63–69. [Google Scholar]
- Li, Y.-M.; Hu, S.-L.; Wang, D.-Y. Polymer-based ceramifiable composites for flame retardant applications: A review. Compos. Commun. 2020, 21, 100405. [Google Scholar] [CrossRef]
- Thien, N.D.; Quynh, L.M.; Van Vu, L.; Long, N.N. Phase transformation and photoluminescence of undoped and Eu3+-doped zinc stannate (Zn2SnO4) nanocrystals synthesized by hydrothermal method. J. Mater. Sci. Mater. Electron. 2019, 30, 1813–1820. [Google Scholar] [CrossRef]
- Keles, E.; Yildirim, M.; Öztürk, T.; Yildirim, O.A. Hydrothermally synthesized UV light active zinc stannate:tin oxide (ZTO:SnO2) nanocompositephotocatalysts for photocatalytic applications. Mater. Sci. Semicond. Process. 2020, 110, 104959. [Google Scholar] [CrossRef]
- Levchik, S.V.; Weil, E.D. Combustion and fire retardancy of aliphatic nylons. Polymer International. 2000, 49, 1033–1073. [Google Scholar] [CrossRef]
- Horrocks, A.; Smart, G.; Price, D.; Kandola, B. Zinc stannates as alternative synergists in selected flame retardant systems. J. Fire Sci. 2009, 27, 495–521. [Google Scholar] [CrossRef]
- Zhong, F.; Chen, C.; Yang, X.; Zhou, J.; Zheng, W.; Huang, H.; Wang, P.; Li, W. Self-assembly of zinc hydroxystannate on polyethyleneimine supported boron nitride to improve the flame protection properties of waterborne epoxy coatings. Colloids Surf. A Physicochem. Eng. Asp. 2022, 650, 129557. [Google Scholar] [CrossRef]
- Horrocks, A.; Smart, G.; Kandola, B.; Price, D. Zinc stannate interactions with flame retardants in polyamides; Part 2: Potential synergies with non-halogen-containing flame retardants in polyamide 6 (PA6). Polym. Degrad. Stab. 2012, 97, 645–652. [Google Scholar] [CrossRef]
- Wu, W.; Qu, H.; Li, Z.; Yuan, H. Thermal behavior and flame retardancy of flexible poly (vinyl chloride) treated with zinc hydroxystannate and zinc stannate. J. Vinyl Addit. Technol. 2008, 14, 10–15. [Google Scholar] [CrossRef]
- Morgan, A.B.; Gilman, J.W. An overview of flame retardancy of polymeric materials: Application, technology, and future directions. Fire Mater. 2013, 37, 259–279. [Google Scholar] [CrossRef]
- Hansen, N.; Cool, T.A.; Westmoreland, P.R.; Kohse-Höinghaus, K. Recent contributions of flame-sampling molecular-beam mass spectrometry to a fundamental understanding of combustion chemistry. Prog. Energy Combust. Sci. 2009, 35, 168–191. [Google Scholar] [CrossRef]
- Laoutid, F.; Bonnaud, L.; Alexandre, M.; Lopez-Cuesta, J.M.; Dubois, P. New prospects in flame retardant polymer materials: From fundamentals to nanocomposites. Mater. Sci. Eng. R Rep. 2009, 63, 100–125. [Google Scholar] [CrossRef]
- Altarawneh, M.; Saeed, A.; Al-Harahsheh, M.; Dlugogorski, B.Z. Thermal decomposition of brominated flame retardants (BFRs): Products and mechanisms. Prog. Energy Combust. Sci. 2019, 70, 212–259. [Google Scholar] [CrossRef]
- Sai, T.; Ran, S.; Guo, Z.; Song, P.; Fang, Z. Recent advances in fire-retardant carbon-based polymeric nanocomposites through fighting free radicals. SusMat 2022, 2, 411–434. [Google Scholar] [CrossRef]
- Liu, S.; Wei, L.; Zhou, Q.; Yang, T.; Li, S.; Zhou, Q. Simulation strategies for ReaxFF molecular dynamics in coal pyrolysis applications: A review. J. Anal. Appl. Pyrolysis 2023, 170, 105882. [Google Scholar] [CrossRef]
- Guo, S.T.; Liu, J.; Qian, W.; Zhu, W.H.; Zhang, C.Y. A review of quantum chemical methods for treating energetic molecules. Energetic Mater. Front. 2021, 2, 239–316. [Google Scholar] [CrossRef]
- Han, Y.; Jiang, D.; Zhang, J.; Li, W.; Gan, Z.; Gu, J. Development, applications and challenges of ReaxFF reactive force field in molecular simulations. Front. Chem. Sci. Eng. 2016, 10, 16–38. [Google Scholar] [CrossRef]
- Yuen, A.C.Y.; Chen, T.B.Y.; Li, A.; De Cachinho Cordeiro, I.M.; Liu, L.; Liu, H.; Lo, A.L.P.; Chan, Q.N.; Yeoh, G.H. Evaluating the fire risk associated with cladding panels: An overview of fire incidents, policies, and future perspective in fire standards. Fire Mater. 2021, 45, 663–689. [Google Scholar] [CrossRef]
- Yang, K.; Wu, R.; Shen, L.; Feng, Y.P.; Dai, Y.; Huang, B. Origin of d0 magnetism in II-VI and III-V semiconductors by substitutional doping at anion site. Phys. Rev. B 2010, 81, 125211. [Google Scholar] [CrossRef]
- Shetty, S.; Pal, S.; Kanhere, D.G.; Goursot, A. Structural, Electronic, and Bonding Properties of Zeolite Sn-Beta: A Periodic Density Functional Theory Study. Chem. Eur. J. 2006, 12, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Wong-Ng, W.; Luo, T.; Xie, W.; Tang, W.H.; Kaduk, J.A.; Huang, Q.; Yan, Y.; Chattopadhyay, S.; Tang, X.; Tritt, T. Phase diagram, crystal chemistry and thermoelectric properties of compounds in the Ca–Co–Zn–O system. J. Solid State Chem. 2011, 184, 2159–2166. [Google Scholar] [CrossRef]
- Zhou, B.; Denning, M.S.; Jones, C.; Goicoechea, J.M. Reductive cleavage of Zn–C bonds by group 14 Zintl anions: Synthesis and characterization of [E9ZnR]3− (E = Ge, Sn, Pb; R = Mes, iPr). Dalton Trans. 2009, 9, 1571–1578. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Peng, C.; Li, J.; Wang, J.; Zhang, H. Insight into Sodium Storage Behaviors in Hard Carbon by ReaxFF Molecular Dynamics Simulation. Energy Fuels 2022, 36, 5937–5952. [Google Scholar] [CrossRef]
- Krebs, B.; Henkel, G.; Dartmann, M. Structure of tetramethyltin, Sn(CH3)4. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1989, 45, 1010–1012. [Google Scholar] [CrossRef]
- Wang, Q.; Yan, L.; Meng, L.; Zhang, J.; Li, H.; Geng, C.; Bai, H.; Guo, Q. Mechanism and process of chemical looping depolymerization of polyethylene based on iron-based oxygen carriers. Acta Pet. Sin. (Pet. Process. Sect.) 2023, 39, 358–367. [Google Scholar]
- Chenoweth, K.; Van Duin, A.C.; Goddard, W.A. ReaxFF reactive force field for molecular dynamics simulations of hydrocarbon oxidation. J. Phys. Chem. A 2008, 112, 1040–1053. [Google Scholar] [CrossRef]
- Boes, J.R.; Groenenboom, M.C.; Keith, J.A.; Kitchin, J.R. Neural network and ReaxFF comparison for Au properties. Int. J. Quantum Chem. 2016, 116, 979–987. [Google Scholar] [CrossRef]
- Han, S.S.; Van Duin, A.C.; Goddard, W.A., III; Lee, H.M. Optimization and application of lithium parameters for the reactive force field, ReaxFF. J. Phys. Chem. A 2005, 109, 4575–4582. [Google Scholar] [CrossRef] [PubMed]
- Cavallotti, C.; Moscatelli, D.; Masi, M.; Carrà, S. Accelerated decomposition of gas phase metal organic molecules determined by radical reactions. J. Cryst. Growth 2004, 266, 363–370. [Google Scholar] [CrossRef]
- Abdulsattar, M.A.; Jabbar, R.H.; Abed, H.H. Transition state application to simulate CO gas sensor of pristine and Pt doped tin dioxide clusters. J. Phys. Conf. Ser. 2021, 1963, 012033. [Google Scholar] [CrossRef]
- Senftle, T.P.; Meyer, R.J.; Janik, M.J.; Van Duin, A.C. Development of a ReaxFF potential for Pd/O and application to palladium oxide formation. J. Chem. Phys. 2013, 139, 044109. [Google Scholar] [CrossRef]
- Rahaman, O.; Van Duin, A.C.; Goddard, W.A., III; Doren, D.J. Development of a ReaxFF reactive force field for glycine and application to solvent effect and tautomerization. J. Phys. Chem. B 2011, 115, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Chenoweth, K.; Van Duin, A.C.; Persson, P.; Cheng, M.-J.; Oxgaard, J.; Goddard, W.A., III. Development and application of a ReaxFF reactive force field for oxidative dehydrogenation on vanadium oxide catalysts. J. Phys. Chem. C 2008, 112, 14645–14654. [Google Scholar] [CrossRef]
- Depew, D.D.; Wang, J.; Parmar, S.; Chambreau, S.; Bedrov, D.; van Duin, A.; Vaghjiani, G. AIAA Propulsion and Energy 2019 Forum; American Institute of Aeronautics and Astronautics: Reston, VA, USA, 2019. [Google Scholar]
- Shchygol, G.; Yakovlev, A.; Trnka, T.; Van Duin, A.C.; Verstraelen, T. ReaxFF parameter optimization with Monte-Carlo and evolutionary algorithms: Guidelines and insights. J. Chem. Theory Comput. 2019, 15, 6799–6812. [Google Scholar] [CrossRef]
- Nielson, K.D.; Van Duin, A.C.; Oxgaard, J.; Deng, W.-Q.; Goddard, W.A., III. Development of the ReaxFF reactive force field for describing transition metal catalyzed reactions, with application to the initial stages of the catalytic formation of carbon nanotubes. J. Phys. Chem. A 2005, 109, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Van Duin, A.C.; Bryantsev, V.S.; Diallo, M.S.; Goddard, W.A., III; Rahaman, O.; Doren, D.J.; Raymand, D.; Hermansson, K. Development and validation of a ReaxFF reactive force field for Cu cation/water interactions and copper metal/metal oxide/metal hydroxide condensed phases. J. Phys. Chem. A 2010, 114, 9507–9514. [Google Scholar] [CrossRef]
- Huang, Y.; Wexler, A.S.; Bein, K.J.; Faller, R. Development of a ReaxFF Force Field for Aqueous Phosphoenolpyruvate as a Novel Biomimetic Carbon Capture Absorbent. J. Phys. Chem. C 2022, 126, 9284–9292. [Google Scholar] [CrossRef]
- Zheng, M.; Li, X.; Guo, L. Algorithms of GPU-enabled reactive force field (ReaxFF) molecular dynamics. J. Mol. Graph. Model. 2013, 41, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sengul, M.Y.; Song, Y.; Nayir, N.; Gao, Y.; Hung, Y.; Dasgupta, T.; van Duin, A.C. INDEEDopt: A deep learning-based ReaxFF parameterization framework. Npj Comput. Mater. 2021, 7, 1–9. [Google Scholar] [CrossRef]
- Russo, M.F., Jr.; Van Duin, A.C. Atomistic-scale simulations of chemical reactions: Bridging from quantum chemistry to engineering. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2011, 269, 1549–1554. [Google Scholar] [CrossRef]
- Van Duin, A.C.; Strachan, A.; Stewman, S.; Zhang, Q.; Xu, X.; Goddard, W.A., III. ReaxFF SiO reactive force field for silicon and silicon oxide systems. J. Phys. Chem. A 2003, 107, 3803–3811. [Google Scholar] [CrossRef]
- Li, X.; Zheng, M.; Ren, C.; Guo, L. Reaxff molecular dynamics simulations of thermal reactivity of various fuels in pyrolysis and combustion. Energy Fuels 2021, 35, 11707–11739. [Google Scholar] [CrossRef]
- Mueller, J.E.; van Duin, A.C.T.; Goddard, W.A., III. Application of the Reaxff reactive force field to reactive dynamics of hydrocarbon chemisorption and decomposition. J. Phys. Chem. C 2010, 114, 5675–5685. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Zhu, M.; Geng, C.; Yuan, Y.; Fu, Z.; Yan, S.; Feng, R.; Wang, Y.; Zhou, Y.; Meng, L.; et al. A Molecular Understanding of the Flame Retardant Mechanism of Zinc Stannate/Polypropylene Composites via ReaxFF Simulations. Inorganics 2023, 11, 233. https://doi.org/10.3390/inorganics11060233
Li J, Zhu M, Geng C, Yuan Y, Fu Z, Yan S, Feng R, Wang Y, Zhou Y, Meng L, et al. A Molecular Understanding of the Flame Retardant Mechanism of Zinc Stannate/Polypropylene Composites via ReaxFF Simulations. Inorganics. 2023; 11(6):233. https://doi.org/10.3390/inorganics11060233
Chicago/Turabian StyleLi, Jun, Meilin Zhu, Chang Geng, Yingjie Yuan, Zewei Fu, Shu Yan, Rou Feng, Yingwu Wang, Ying Zhou, Liangliang Meng, and et al. 2023. "A Molecular Understanding of the Flame Retardant Mechanism of Zinc Stannate/Polypropylene Composites via ReaxFF Simulations" Inorganics 11, no. 6: 233. https://doi.org/10.3390/inorganics11060233
APA StyleLi, J., Zhu, M., Geng, C., Yuan, Y., Fu, Z., Yan, S., Feng, R., Wang, Y., Zhou, Y., Meng, L., Zhang, H., & Bai, H. (2023). A Molecular Understanding of the Flame Retardant Mechanism of Zinc Stannate/Polypropylene Composites via ReaxFF Simulations. Inorganics, 11(6), 233. https://doi.org/10.3390/inorganics11060233