1. Introduction
Energy plays a crucial role in driving economic prosperity for nations worldwide. With the rapid growth of the population and evolving lifestyles, global energy demands continue to expand. Simultaneously, environmental concerns and the rapid depletion of resources have become alarming challenges. Fossil fuel consumption has notably contributed to environmental degradation, intensifying the need for clean and sustainable energy sources. In this context, hydrogen emerges as a promising solution to meet the growing energy requirements [
1,
2,
3]. As an ecofriendly energy carrier, hydrogen offers a range of advantages, including a high gravimetric energy density, zero emissions, and a renewable nature. Its environmentally benign characteristics position it as a clean energy transporter [
4]. With its immense potential, hydrogen is expected to serve as a highly efficient alternative fuel with diverse applications in the future. However, the development of highly efficient and safe hydrogen transportation and storage technologies remains a subject of discussion and demonstration. Currently, high-pressure compressed hydrogen is the most commonly employed storage technology, although concerns regarding safety persist. Among the three primary techniques for hydrogen storage, namely compressed gas, cryogenic liquid, and solid-state storage, only the former two methods have commercialized. Nevertheless, the cost-effectiveness and safety considerations of hydrogen storage have led to significant research efforts focusing on solid-state storage through the adsorption and/or absorption of various materials and alloys [
5,
6]. The growing development of the new energy automotive sector has spurred numerous studies on secure hydrogen storage. Porous materials, including zeolites, metal-organic frameworks (MOFs), covalent organic frameworks (COFs), and carbon-based materials (such as fullerenes, nanotubes and graphene), have received considerable attention [
7]. However, the low hydrogen storage capacity of zeolites and porous silica, along with the low operating temperature (77K) of MOFs and COFs, pose disadvantages for hydrogen storage [
8,
9]. As a result, 2D carbonaceous materials [
10], particularly graphene [
11,
12], have emerged as a topic of significant interest in the field of hydrogen storage materials. Graphene offers exceptional specific surface area, structural stability, lightweight properties and rapid adsorption kinetics [
13,
14,
15]. Despite the relatively low hydrogen storage capacity of pure graphene (<1 wt% at room temperature), which falls short of the U.S. Department of Energy (DOE) target of 5.5 wt% for onboard applications by 2025 [
16], it remains a subject of exploration and optimization for hydrogen storage purposes. Some researchers confirmed that there is an increment in the hydrogen storage capacity of graphene after being modified by metal atoms, clusters, or nanoparticles. Palladium, platinum, and other precious metals have been widely studied and used to enhance the hydrogen storage capacity of carbon-based materials owing to the hydrogen spillover effect [
17,
18,
19]. However, the high cost has inhibited the large-scale industrial application of these precious metals. The spillover process in hydrogen storage involves the dissociation of hydrogen molecules on the catalyst, migration of hydrogen atoms from the catalyst to the substrate, and diffusion of hydrogen atoms on the substrate surface. However, certain pure transition metal-decorated graphene materials face limitations due to a high energy barrier. For instance, Psofogiannakis et al. [
20] reported a migration barrier of 2.6 eV from the Pt
4 cluster to graphene. This high migration barrier hampers the efficient and thermodynamically favorable transfer of hydrogen atoms from the catalyst to the carbon receptor, thereby impeding the spillover rate of hydrogen.
To reduce the reliance on precious metals such as Pd, the formation of Pd-based alloys has emerged as an effective strategy. The introduction of alloying elements can alter the hydrogen spillover effect and consequently modify hydrogen storage capacity. Guo et al. conducted research on the highest occupied molecular orbital (HOMO) of Penta-Graphene (PG) decorated with Ni
4, Pd
4, and Ni
2Pd
2. Their findings revealed that the orbit of Ni overlaps with that of Pd, resulting in a decrease in the electric field of the Ni
2Pd
2 cluster compared to Ni
4 and Pd
4. This decrease leads to a reduction in hydrogen adsorption energy [
21]. In another study by Wu et al., a systematic comparison of the properties of Pd
6Ni
4 and Pd
4Ni
6 was conducted [
22]. It was observed that the migration of electrons between Pd and Ni caused the D-band center of Pd to shift downward. Our previous research demonstrated that P doping improves the electronic structure of Pd, resulting in a downward shift of the D-band center. This, in turn, weakens the adsorption energy of materials for H
2, lowers the diffusion energy barrier of H atoms, and promotes hydrogen spillover, thereby enhancing hydrogen absorption and desorption capacity [
23]. Based on this understanding, the design and synthesis of Pd–Ni alloys hold promise for improving hydrogen storage performance by adjusting the D-band center.
Moreover, despite extensive research efforts in the field of hydrogen storage, the development of efficient onboard hydrogen storage systems still poses significant scientific and technological challenges. To address this issue, we have conducted a comprehensive investigation, taking into account previous experimental and computational studies towards the development of an economically viable graphene-based hydrogen storage material. In our study, we successfully synthesized the material using a solvothermal method, wherein palladium (Pd) was incorporated to partially replace nickel (Ni). Notably, the utilization of ethylene glycol as the hydrothermal solvent resulted in the coexistence of Ni and Pd in the final material. Concurrently, some Pd atoms infiltrated the Ni lattice, forming a NiPd alloy. Our findings reveal that the NiPd-rGO material exhibits a remarkable maximum hydrogen storage capacity of 2.65 wt% under mild conditions, specifically at 4 MPa and 298 K.
3. Results and Discussion
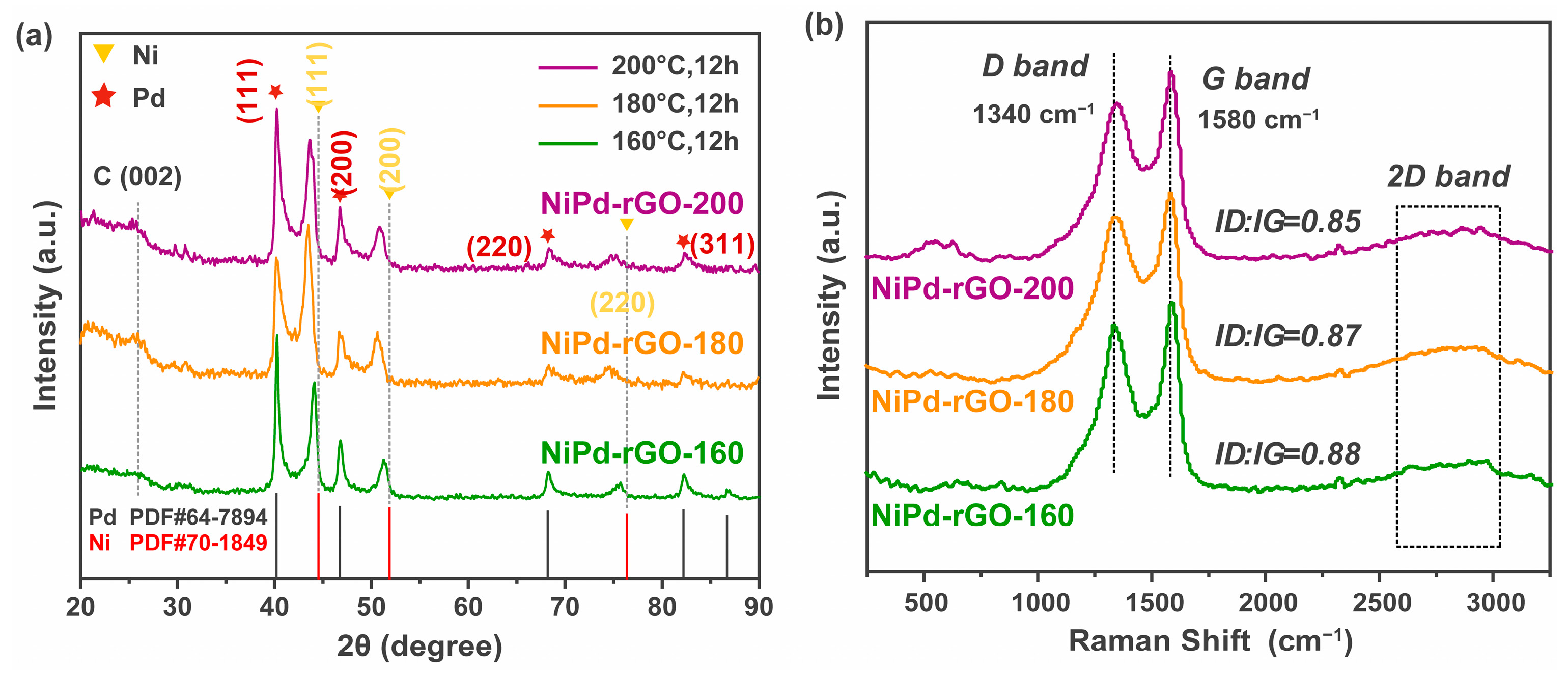
Figure 1 illustrates the X-ray diffraction (XRD) patterns obtained from the NiPd-rGO-X (X = 160, 180, 200 °C) samples, synthesized at different hydrothermal temperatures. A comparison with standard PDF cards allowed for the identification of a biphasic structure within the material, indicating the coexistence of Pd and Ni phases. The peaks observed at 40.2°, 46.8°, 68.3°, 82.4° and 86.9° correspond to Pd (111), Pd (200), Pd (220), Pd (311) and Pd (222), respectively, as per the PDF database. Of particular interest, the peaks associated with Ni (PDF#70-1849) exhibited a noticeable shift towards lower angles, implying the incorporation of Pd atoms into the Ni lattice, thereby forming a NiPd alloy [
24]. This observation is evident from the shift of the Ni (111) diffraction peak towards smaller angles, indicating an expansion of the nickel unit cell due to alloying with palladium. The XRD peaks change with the variation in temperature. At 160 °C, the material predominantly exhibits a split phase composed of palladium and nickel. The XRD analysis indicates that the nickel content within the palladium lattice is relatively low, and correspondingly, the palladium content within the nickel lattice is also relatively small. As the temperature is increased to 180 °C, there is an augmentation in the quantity of palladium integrated into the nickel lattice, leading to the formation of a greater number of palladium–nickel alloys with nickel as the primary phase. Subsequently, at 200 °C, the amount of nickel incorporated into the palladium lattice increases, resulting in the transformation of the material into a palladium–nickel alloy with palladium as the principal phase. Moreover, the shift in the position of the XRD peaks can serve as evidence. At 160 °C, the phase exhibits closer proximity to our palladium and nickel composition. At 180 °C, the palladium content within the nickel lattice notably increases, leading to a greater shift in the peak position. Finally, at 200 °C, the position of the nickel peak shifts in the opposite direction, while the peak shift of palladium becomes more pronounced. This finding suggests a higher proportion of Ni alloyed with Pd in the sample synthesized at 180 °C, indicating that 180 °C may be the optimum hydrothermal temperature for the synthesis of NiPd. The higher proportion of Ni in the NiPd alloy synthesized at 180 °C is particularly significant as Ni is more cost-effective than Pd. Therefore, the increased proportion of Ni offers a promising avenue for achieving a cost-effective hydrogen storage material in the NiPd alloy synthesized at 180 °C.
In order to investigate the presence of defects within the materials, Raman spectroscopy was employed to analyze the NiPd-rGO-X samples (where X represents the hydrothermal temperature of 160, 180, and 200 °C), as illustrated in
Figure 1b. The obtained spectra for all three samples exhibited the presence of the D-band and G-band. The D-band, positioned at 1340 cm
−1, corresponds to the disordered hybridization of sp
2 [
25], while the G-band, observed at 1580 cm
−1, is associated with the stretching of the C-C bond within the graphene material. Furthermore, upon conducting calculations, it was discovered that the NiPd-rGO-160 sample exhibited the highest I
D/I
G ratio, measuring 0.88. It is worth noting that the I
D/I
G ratio tends to decrease as the hydrothermal temperature increases. Consequently, the NiPd-rGO-200 sample displayed the lowest I
D/I
G ratio, amounting to 0.85.
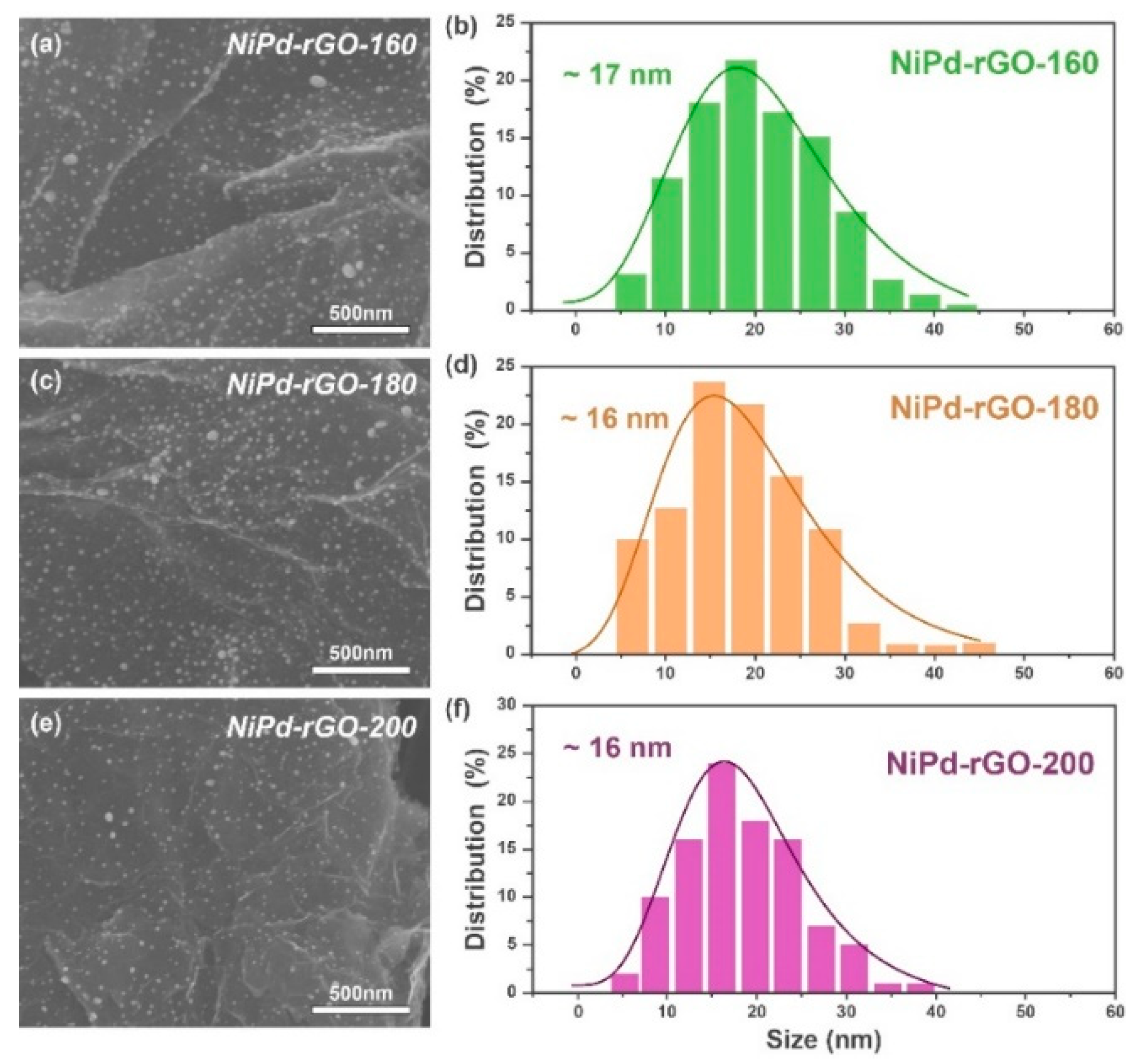
To examine the distribution of nanoparticles on the materials, field emission scanning microscopy (FESEM) was utilized to investigate the morphologies of the NiPd-rGO-X samples. The findings, depicted in
Figure 2, demonstrate a uniform distribution of NiPd nanoparticles on the graphene surface. The grain size of these nanoparticles ranges from 5 to 45 nm, exhibiting a normal distribution pattern. A detailed analysis of
Figure 2 indicates that as the hydrothermal temperature increases, the size of the nanoparticles loaded on the graphene surface tends to decrease. However, it is worth noting that beyond a temperature of 180 °C, the size variations become insignificant, thereby further corroborating that 180 °C represents the optimal hydrothermal temperature. Moreover, an interesting observation is made with the increase in hydrothermal temperature, wherein some larger particles present in the NiPd-rGO-160 sample disappear. In the NiPd-rGO-200 sample, the particle size distribution falls within the range of 5–40 nm, exhibiting a narrower distribution compared to that observed in NiPd-rGO-160.
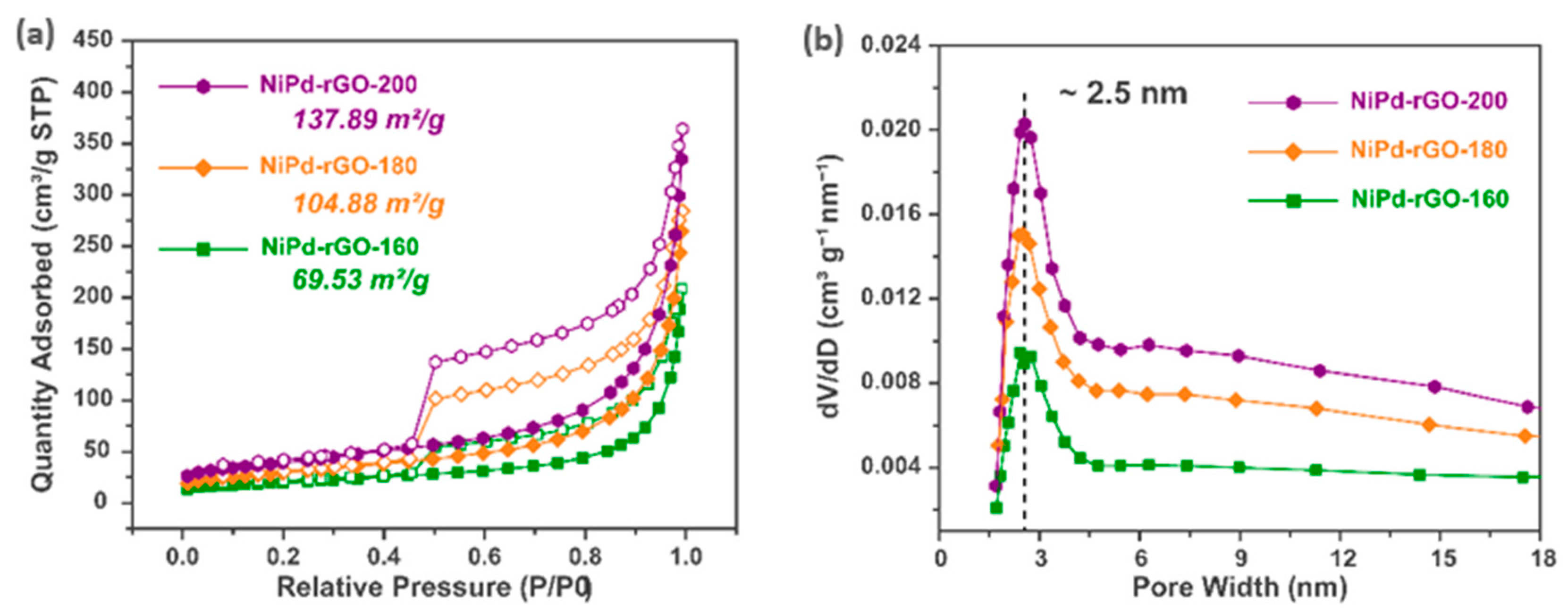
The specific surface area and pore size distribution of the NiPd-rGO-X material were determined through N
2 isothermal adsorption–desorption tests conducted at a temperature of 77K.
Figure 3a,b display the N
2 adsorption–desorption isotherms and pore size distribution curves of NiPd-rGO-X, respectively. It is evident that the N
2 adsorption–desorption isotherms of all three samples exhibit the characteristic features of type-IV adsorption isotherms, suggesting the presence of a mesoporous structure within the material. The observed hysteresis in the isotherms further supports the existence of mesopores. Among the samples, NiPd-rGO-200 exhibited the highest specific surface area, measuring 137.89 m
2/g. It is noteworthy that the specific surface area demonstrated a decreasing trend as the hydrothermal temperature was reduced, which could be attributed to the exfoliation of graphene layers at higher temperatures. The average pore diameter of NiPd-rGO-X was determined to be approximately 2.5 nm, indicating a similarity in the pore structure among the three samples. Nevertheless, the pore volume diminished with decreasing hydrothermal temperature, consistent with the variation in specific surface area, thus suggesting a correlation between pore volume and surface area.
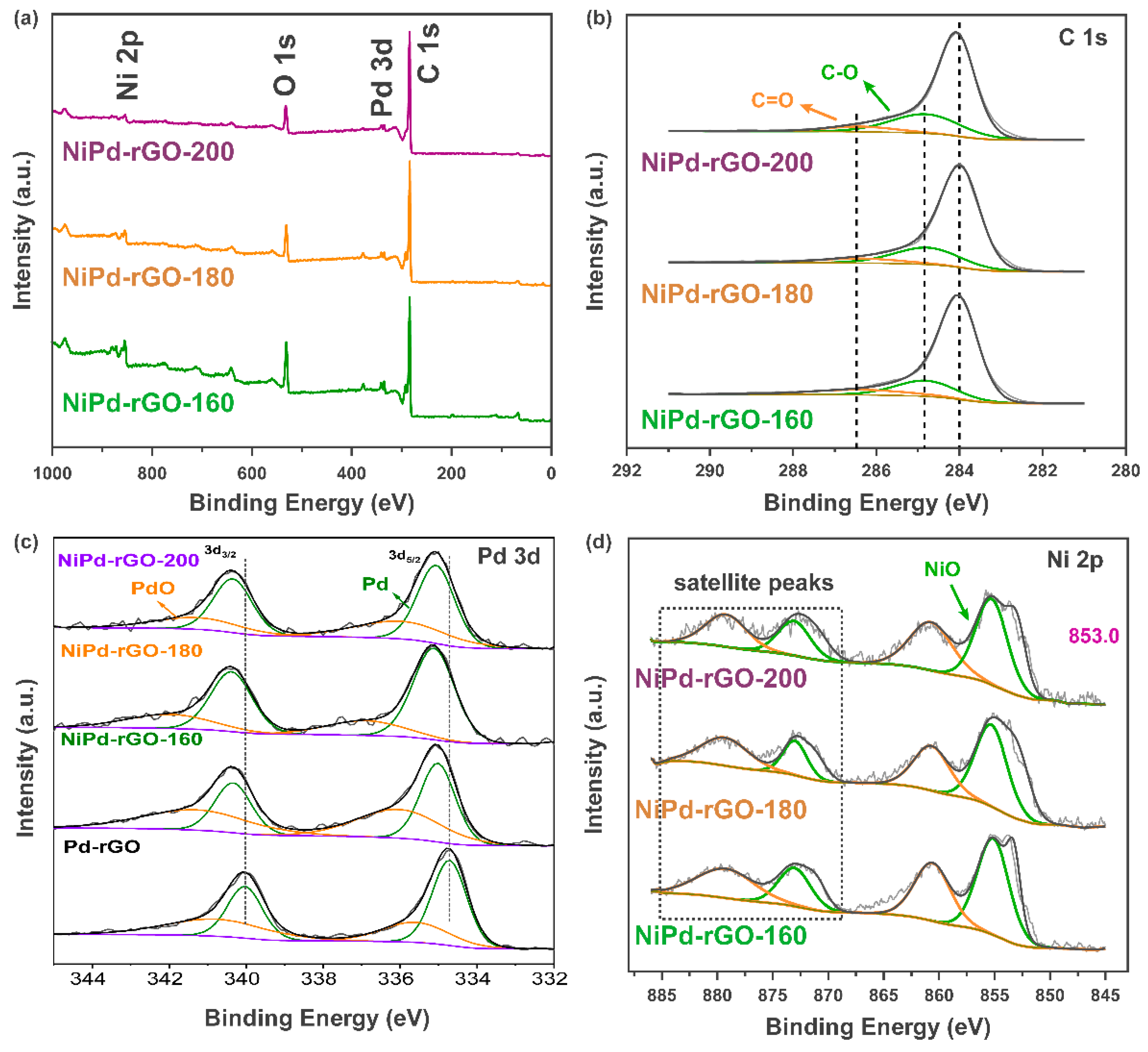
The X-ray photoelectron spectroscopy (XPS) technique was utilized to investigate the surface state of the samples in detail.
Figure 4 showcases the XPS spectrum of NiPd-rGO-X. Upon analyzing the survey spectra, distinct peaks corresponding to C, O, Pd and Ni are observed. In the 1s spectrogram of C, the peaks at 284 eV, 284.8 eV and 286.3 eV correspond to the C=C bond, C-O bond and C=O bond, respectively. These peaks indicate the presence of incompletely reduced oxygen-containing groups in rGO, thus reflecting its surface composition [
26]. Moving on to
Figure 4c, it is observed that the peaks at 335.1 eV and 340.2 eV correspond to the 3d
5/2 orbital and 3d
3/2 orbital of Pd
(0), respectively. Additionally, the peaks at 336 eV and 341.5 eV correspond to the 3d
5/2 orbital and 3d
3/2 orbital of Pd
(II), respectively. This indicates the coexistence of Pd in different oxidation states, suggesting the formation of PdO [
27]. Notably, in comparison to Pd-rGO, the peak of Ni-Pd-rGO shifts towards a higher binding energy, indicating a change in the chemical state of the Pd atom. This forward movement suggests electron transfer from Pd to Ni after alloying, leading to a decrease in the center of the D-band [
28]. The downward shift of the D-band center corresponds to a reduction in the adsorption of H by metals, thereby promoting hydrogen spillover and improving the hydrogen storage performance of the materials. Lastly, as depicted in
Figure 4d, the peaks at 853.0 eV and 871.1 eV are associated with Ni, while the peaks at 855.5 eV and 873.1 eV are attributed to Ni
2+, indicating the oxidation of Ni on the surface of the sample.
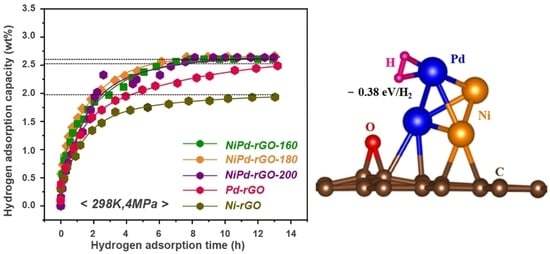
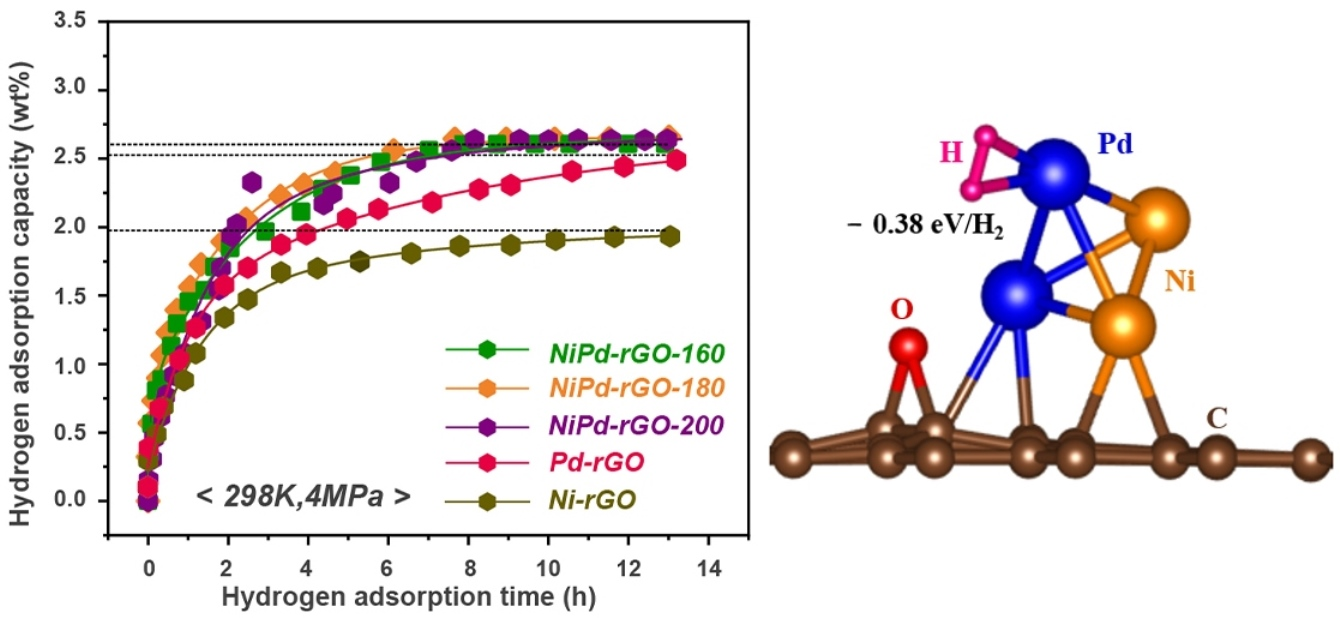
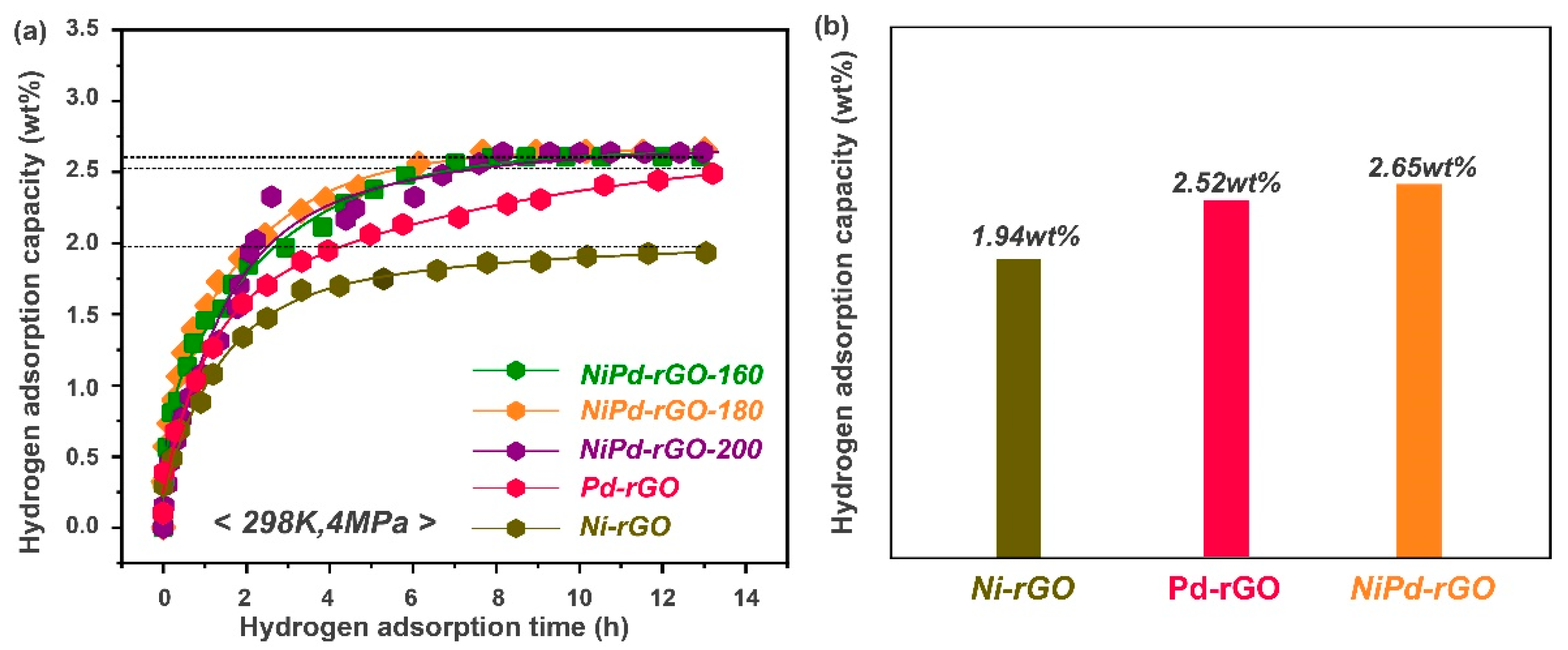
The hydrogen adsorption kinetic curves of NiPd-rGO-X at a pressure of 4 MPa and temperature of 298 K are illustrated in
Figure 4a. The measured hydrogen storage capacities of the NiPd-rGO-160, NiPd-rGO-180, and NiPd-rGO-200 samples are 2.60 wt%, 2.65 wt% and 2.63 wt%, respectively, indicating their close proximity as shown in
Figure 5. The marginally higher hydrogen storage capacity observed in the NiPd-rGO-180 sample can be attributed to the enhanced alloying degree of NiPd, resulting in a more efficient hydrogen spillover effect compared to the Pd and Ni single phases. Notably, the NiPd-rGO-180 material exhibits the highest hydrogen storage capacity (2.65 wt%) among the Ni-rGO, Pd-rGO and NiPd-rGO materials. This remarkable improvement is ascribed to the synergistic effect of rGO and PdNi, which alters the diffusion behavior of hydrogen molecules within the composites. Specifically, the PdNi alloy particles act as activation centers, facilitating the decomposition of hydrogen molecules into hydrogen atoms. These atoms are then stored within the surface defects of graphene, thereby enhancing the overall hydrogen storage capacity of the material. This phenomenon is consistent with the findings of other researchers, who have reported on the higher affinity of defects for hydrogen adsorption from other surface sites [
29,
30].
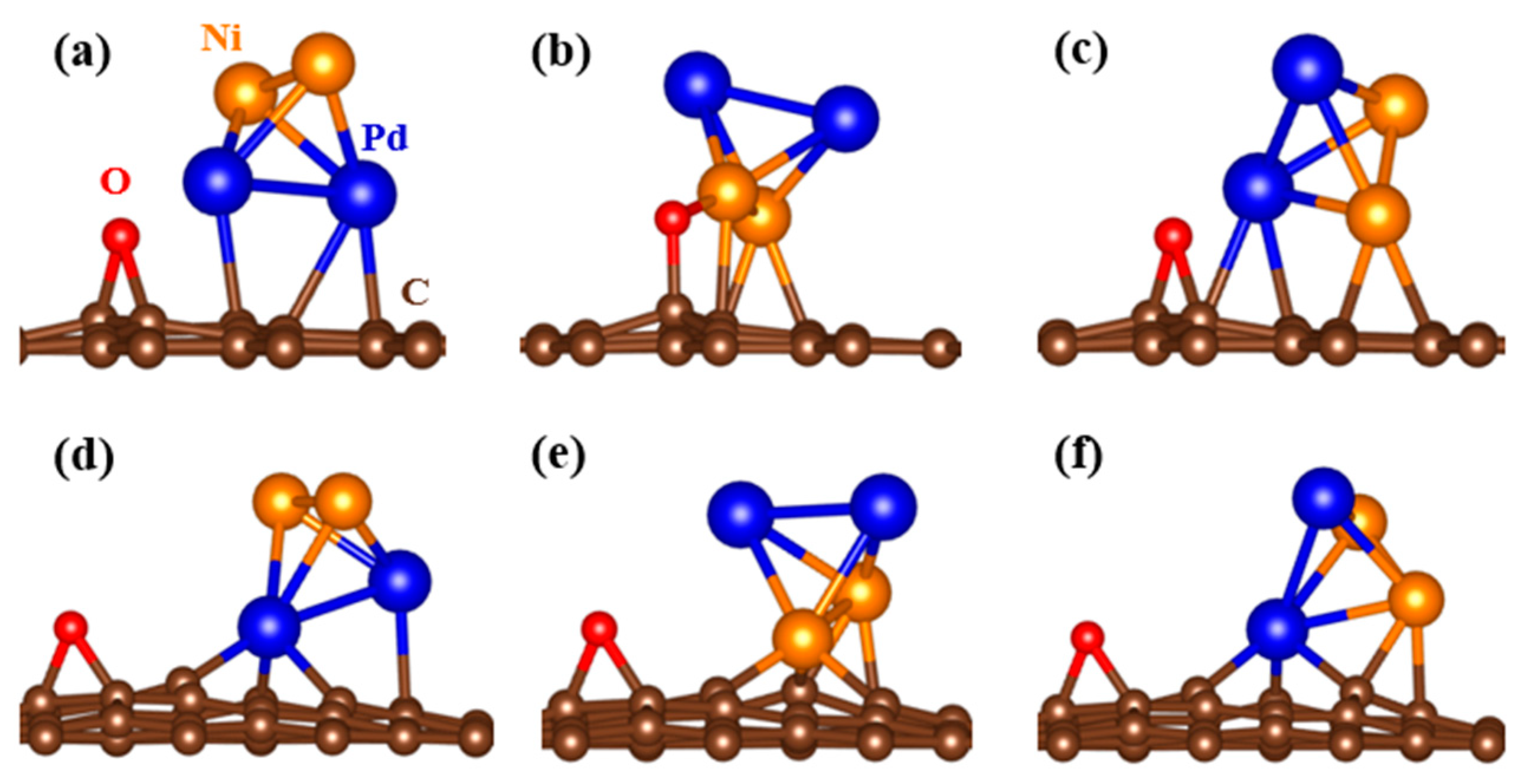
Following the optimization of the graphene slabs, as well as Ni
2Pd
2 clusters, the cluster was placed onto rGO and SVrGO, where the cluster of the Ni
2Pd
2 was tested in different orientations and is presented in
Figure 6. To evaluate the stability of Ni
2Pd
2 on different graphene surfaces, the binding energies were determined using Equation (1), and the findings of our study suggest that the cluster is supported on rGO (with no defects) with three different orientations as shown in the following:
Figure 6a, Pd atoms attached to carbon having a binding energy of −1.03 eV;
Figure 6b, Ni atoms attached to carbon having a binding energy of −2.60 eV, which is higher than the former case due to bonding of the oxygen atom with the Ni atom; and
Figure 6c, Pd and Ni atoms attached to carbon having a binding energy of −1.1 eV, which do not seem to be suitable materials for hydrogen storage. This is attributed to lower binding energy which can cause metal atoms agglomeration which reduces the hydrogen storage capacity. As we know, during metal–metal interaction, metal atoms tend to cluster together, resulting in larger clusters rather than being evenly dispersed on the substrate. This clustering phenomenon is much stronger than the cluster–substrate interaction, which minimizes the efficiency of hydrogen molecule interaction.
Moreover, the desorption of metal–hydrogen complexes competes with the desorption of H
2 from pure graphene, further hindering the overall hydrogen storage process [
31]. To address these issues, defect modifications have been employed as techniques to enhance the interaction between graphene and hydrogen molecules. Conversely, the binding energies of Ni
2Pd
2 supported on SVrGO with three different orientations are shown in the following:
Figure 6d, Pd atoms attached to carbon vacancy having a binding energy of −4.72 eV;
Figure 6e, Ni atoms attached to carbon having a binding energy of −6.61 eV; and, finally,
Figure 6f, Pd and Ni atoms attached to carbon having a binding energy of −5.17 eV. Therefore, these substrates are not conducive to clustering. The results of our study also highlight the significance of defects in the graphene slab, as they significantly increase the binding energy between the transition metal cluster and the substrate. Our calculations indicate that the binding energy is approximately four times higher with the introduction of defects, which is consistent with previous findings reported by Kim et al. [
32]. This increase in binding energy not only enhances the hydrogen storage capacity of the system but also addresses the challenge of complex hydride desorption during hydrogen molecule release [
31]. By effectively increasing the binding energy, the presence of defects in the graphene structure offers a promising solution for improving hydrogen storage capabilities and overcoming the issue of complex hydride desorption.
In order to calculate the adsorption energy of the H
2 molecule, Equation (2) was applied which considers the vibration effect only for the gas phase since it is almost negligible for a solid system. The optimized structures and corresponding calculated adsorption energies of single H
2 molecules adsorbed on Ni
2Pd
2-rGO and Ni
2Pd
2-SVrGO at different positions within the orientation of the cluster are illustrated in
Figure 7. The adsorption energies with negative values in
Figure 7 indicate favorable adsorption, as they signify the attraction of H
2 molecules toward the cluster. Upon structural optimization, the H-H bond underwent relaxation, resulting in bond lengths ranging from 0.83 Å to 0.88 Å, and adsorbed H
2 molecules exhibited an increase in bond length on all materials as compared to the free H
2 bond length of 0.75 Å. However, when compared to an isolated H
2 molecule, the stretching of the H-H bond length was found to be minimal, with a difference of less than 0.3 Å [
33], indicating non-dissociative chemisorption or “molecular” adsorption of H
2, with the association between the H
2 molecule and the Ni and Pd atoms resulting from Kubas-type interaction. Additionally, the bond length between Ni and H varies between 1.56 Å and 1.64 Å, and the bond length of Pd and H varies between 1.72 Å and 1.79 Å. As shown in
Figure 7, it is observed that the adsorption energies are different from each other, which means that the orientation of the cluster and defects in the slabs alter the adsorption energy of the H
2 molecule. Successful hydrogen storage under ambient conditions requires an adsorption mechanism that falls between physical and chemical adsorption, with previous studies recommending an adsorption energy range of −0.20 to −0.60 eV [
34], which is consistent with the adsorption energies observed in this study. All materials fall within this range except
Figure 7(e, g), indicating their potential for hydrogen storage.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






