
Iron(III)-Complexes with N-Phenylpyrazole-Based Ligands

 , ,
, , 
Abstract
:
1. Introduction
2. Results
2.1. Synthesis and Characterization
2.2. Cyclic Voltammetry and Optical Spectroscopy
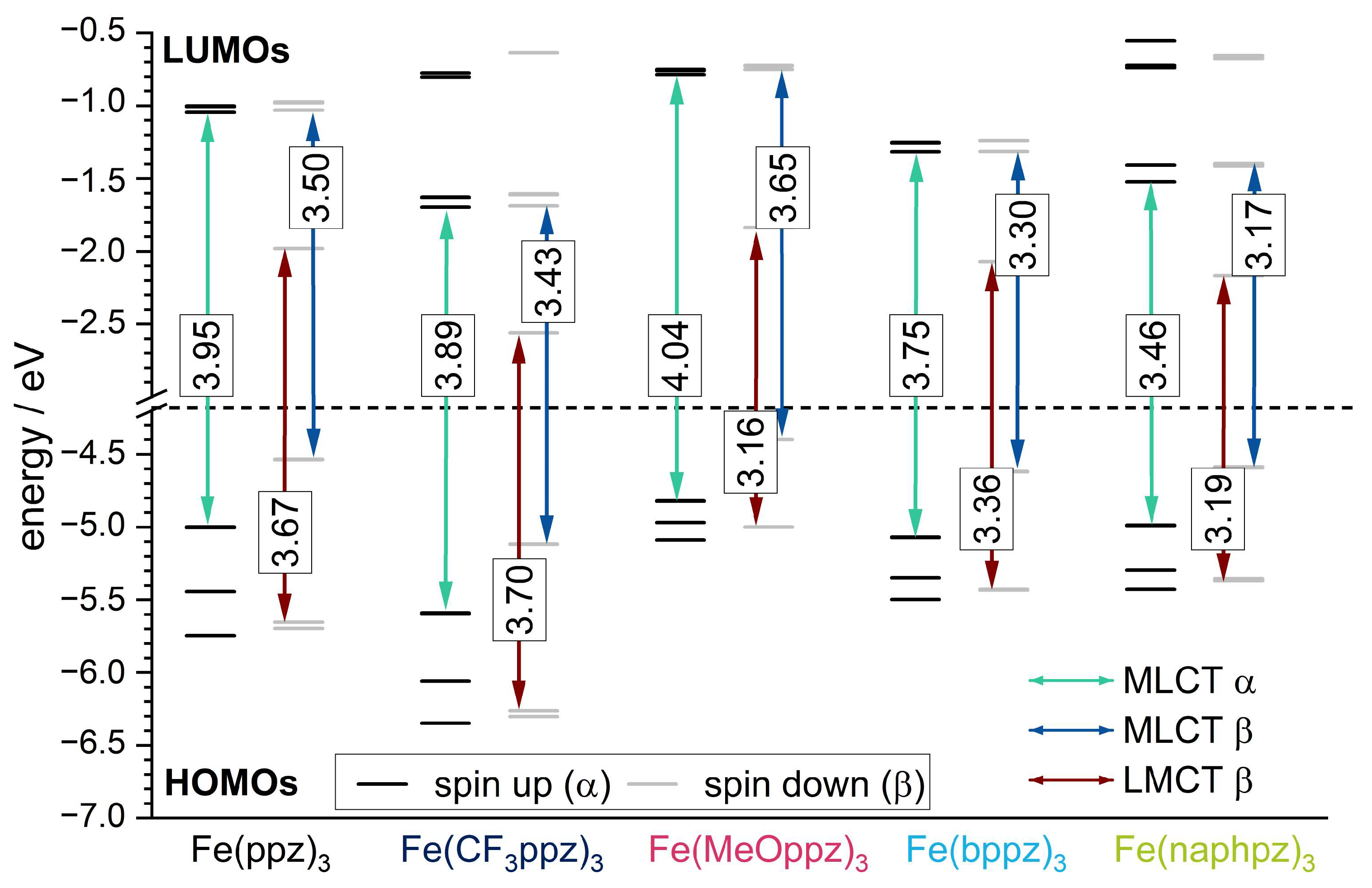
3. Computational Calculations
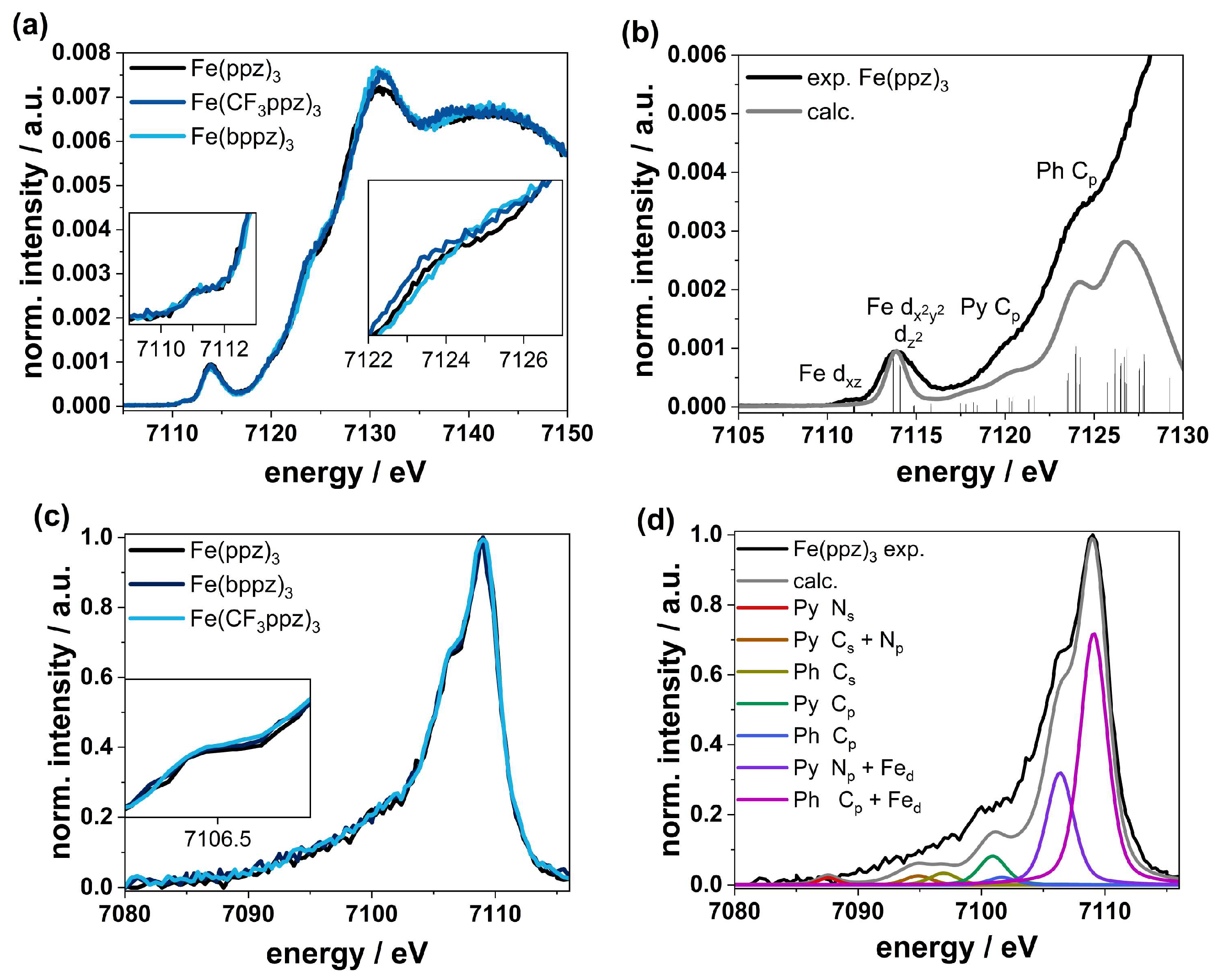
4. Hard X-ray Spectroscopy
5. Behavior under Irradiation
6. Conclusions
7. General Procedures
Complex Synthesis





8. Materials and Methods
8.1. NMR Spectroscopy
8.2. Quantum Chemical Calculations
8.3. X-ray Absorption and Emission Spectroscopy
8.4. Single-Crystal X-ray Diffraction
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balzani, V.; Campagna, S. Photochemistry and Photophysics of Coordination Compounds I; Springer: Berlin/Heidelberg, Germany, 2007; Volume 280. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed]
- Wenger, O.S. Long-range electron transfer in artificial systems with d6 and d8 metal photosensitizers. Coord. Chem. Rev. 2009, 253, 1439–1457. [Google Scholar] [CrossRef]
- Goldsmith, J.I.; Hudson, W.R.; Lowry, M.S.; Anderson, T.H.; Bernhard, S. Discovery and high-throughput screening of heteroleptic iridium complexes for photoinduced hydrogen production. J. Am. Chem. Soc. 2005, 127, 7502–7510. [Google Scholar] [CrossRef]
- Day, J.I.; Teegardin, K.; Weaver, J.; Chan, J. Advances in Photocatalysis: A Microreview of Visible Light Mediated Ruthenium and Iridium Catalyzed Organic Transformations. Org. Process Res. Dev. 2016, 20, 1156–1163. [Google Scholar] [CrossRef]
- Corredor, J.; Rivero, M.J.; Rangel, C.M.; Gloaguen, F.; Ortiz, I. Comprehensive review and future perspectives on the photocatalytic hydrogen production. J. Chem. Technol. Biot. 2019, 94, 3049–3063. [Google Scholar] [CrossRef]
- Wenger, O.S. Is Iron the New Ruthenium? Chem. Eur. J. 2019, 25, 6043–6052. [Google Scholar] [CrossRef] [PubMed]
- Dierks, P.; Vukadinovic, Y.; Bauer, M. Photoactive iron complexes: More sustainable, but still a challenge. Inorg. Chem. Front. 2022, 9, 206–220. [Google Scholar] [CrossRef]
- Cannizzo, A.; Milne, C.J.; Consani, C.; Gawelda, W.; Bressler, C.; van Mourik, F.; Chergui, M. Light-induced spin crossover in Fe(II)-based complexes: The full photocycle unraveled by ultrafast optical and X-ray spectroscopies. Coord. Chem. Rev. 2010, 254, 2677–2686. [Google Scholar] [CrossRef]
- Sauvage, J.-P.; Collin, J.-P.; Chambron, J.-C.; Gullirez, S.; Coudret, C.; Balzani, V.; Barigelletti, F.; de Cola, L.; Flamigni, L. ChemInform Abstract: Ruthenium(II) and Osmium(II) Bis(terpyridine) Complexes in Covalently- Linked Multicomponent Systems: Synthesis, Electrochemical Behavior, Absorption Spectra, and Photochemical and Photophysical Properties. Chem. Rev. 1994, 25, 993–1019. [Google Scholar] [CrossRef]
- Damrauer, N.H.; Cerullo, G.; Yeh, A.; Boussie, T.R.; Shank, C.V.; McCusker, J.K. Femtosecond Dynamics of Excited-State Evolution in. Science 1997, 275, 54–57. [Google Scholar] [CrossRef]
- Jakubikova, E.; Chen, W.; Dattelbaum, D.M.; Rein, F.N.; Rocha, R.C.; Martin, R.L.; Batista, E.R. Electronic Structure and Spectroscopy of [Ru(tpy)2]2+, [Ru(tpy)(bpy)(H2O)]2+, and [Ru(tpy)(bpy)(Cl)]+. Inorg. Chem. 2009, 48, 10720–10725. [Google Scholar] [CrossRef] [PubMed]
- McCusker, J.K. Electronic structure in the transition metal block and its implications for light harvesting. Science 2019, 363, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Persson, P.; Sundström, V.; Wärnmark, K. Fe N-Heterocyclic Carbene Complexes as Promising Photosensitizers. Acc. Chem. Res. 2016, 49, 1477–1485. [Google Scholar] [CrossRef]
- Kaufhold, S.; Wärnmark, K. Design and Synthesis of Photoactive Iron N-Heterocyclic Carbene Complexes. Catalysts 2020, 10, 132. [Google Scholar] [CrossRef]
- Liu, Y.; Harlang, T.; Canton, S.E.; Chábera, P.; Suárez-Alcántara, K.; Fleckhaus, A.; Vithanage, D.A.; Göransson, E.; Corani, A.; Lomoth, R.; et al. Towards longer-lived metal-to-ligand charge transfer states of iron(II) complexes: An N-heterocyclic carbene approach. Chem. Comm. 2013, 49, 6412–6414. [Google Scholar] [CrossRef] [PubMed]
- Lindh, L.; Chábera, P.; Rosemann, N.W.; Uhlig, J.; Wärnmark, K.; Yartsev, A.; Sundström, V.; Persson, P. Photophysics and Photochemistry of Iron Carbene Complexes for Solar Energy Conversion and Photocatalysis. Catalysts 2020, 10, 315. [Google Scholar] [CrossRef]
- Steube, J.; Kruse, A.; Bokareva, O.S.; Reuter, T.; Demeshko, S.; Schoch, R.; Argüello Cordero, M.A.; Krishna, A.; Hohloch, S.; Meyer, F.; et al. Janus-type emission from a cyclometalated iron(III) complex. Nat. Chem. 2023, 15, 468–474. [Google Scholar] [CrossRef]
- Chábera, P.; Liu, Y.; Prakash, O.; Thyrhaug, E.; Nahhas, A.E.; Honarfar, A.; Essén, S.; Fredin, L.A.; Harlang, T.C.B.; Kjær, K.S.; et al. A low-spin Fe(III) complex with 100-ps ligand-to-metal charge transfer photoluminescence. Nature 2017, 543, 695–699. [Google Scholar] [CrossRef]
- Kjær, K.S.; Kaul, N.; Prakash, O.; Chábera, P.; Rosemann, N.W.; Honarfar, A.; Gordivska, O.; Fredin, L.A.; Bergquist, K.-E.; Häggström, L.; et al. Luminescence and reactivity of a charge-transfer excited iron complex with nanosecond lifetime. Science 2019, 363, 249–253. [Google Scholar] [CrossRef]
- Koeberl, C.; Young, E.R.; Oldacre, A. Iron hits the mark. Science 2019, 363, 224–225. [Google Scholar] [CrossRef]
- Chábera, P.; Lindh, L.; Rosemann, N.W.; Prakash, O.; Uhlig, J.; Yartsev, A.; Wärnmark, K.; Sundström, V.; Persson, P. Photofunctionality of iron(III) N-heterocyclic carbenes and related d transition metal complexes. Coord. Chem. Rev. 2021, 426, 213517. [Google Scholar] [CrossRef]
- Liu, Y.; Kjaer, K.S.; Fredin, L.A.; Chábera, P.; Harlang, T.; Canton, S.E.; Lidin, S.; Zhang, J.; Lomoth, R.; Bergquist, K.-E.; et al. A heteroleptic ferrous complex with mesoionic bis(1,2,3-triazol-5-ylidene) ligands: Taming the MLCT excited state of iron(II). Chem. Eur. J. 2015, 21, 3628–3639. [Google Scholar] [CrossRef] [PubMed]
- Medlycott, E.A.; Hanan, G.S. Designing tridentate ligands for ruthenium(II) complexes with prolonged room temperature luminescence lifetimes. Chem. Soc. Rev. 2005, 34, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Chábera, P.; Kjaer, K.S.; Prakash, O.; Honarfar, A.; Liu, Y.; Fredin, L.A.; Harlang, T.C.B.; Lidin, S.; Uhlig, J.; Sundström, V.; et al. FeII Hexa N-Heterocyclic Carbene Complex with a 528 ps Metal-to-Ligand Charge-Transfer Excited-State Lifetime. J. Phys. Chem. Lett. 2018, 9, 459–463. [Google Scholar] [CrossRef]
- Nam, E.J.; Kim, J.H.; Kim, B.-O.; Kim, S.M.; Park, N.G.; Kim, Y.S.; Kim, Y.K.; Ha, Y. A Synthesis and Luminescence Study of Ir(ppz)3 for Organic Light-Emitting Devices. Bull. Chem. Soc. Jpn. 2004, 77, 751–755. [Google Scholar] [CrossRef]
- Ren, X.; Alleyne, B.D.; Djurovich, P.I.; Adachi, C.; Tsyba, I.; Bau, R.; Thompson, M.E. Organometallic complexes as hole-transporting materials in organic light-emitting diodes. Inorg. Chem. 2004, 43, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Drevs, H. Elektronenvariable σ-Organochelatkomplexe: Pyrazolylphenylkomplexe des Eisen(II), Eisen(III) und Eisen(IV). Z. Chem. 1979, 19, 31–32. [Google Scholar] [CrossRef]
- Cristau, H.-J.; Cellier, P.P.; Spindler, J.-F.; Taillefer, M. Mild Conditions for Copper-Catalysed N-Arylation of Pyrazoles. Eur. J. Org. Chem. 2004, 2004, 695–709. [Google Scholar] [CrossRef]
- Cristau, H.-J.; Cellier, P.P.; Spindler, J.-F.; Taillefer, M. Highly efficient and mild copper-catalyzed N- and C-arylations with aryl bromides and iodides. Chem. Eur. J. 2004, 10, 5607–5622. [Google Scholar] [CrossRef]
- Marxer, A.; Siegrist, M. Über die Umsetzung von 1-Phenylpyrazol mit Äthylmagnesiumbromid. 8. Mitteilung über Grignard-Reaktionen [1]//Über die Umsetzung von 1-Phenylpyrazol mit Äthylmagnesiumbromid. Helv. Chim. Acta 1974, 57, 1988–2000. [Google Scholar] [CrossRef]
- Cotton, F.; Luck, R.L.; Son, K.-A. New polynuclear compounds of iron(II) chloride with oxygen donor ligands Part I. Fe4Cl8(THF)6: Synthesis and a single crystal X-ray structure determination. Inorg. Chim. Acta 1991, 179, 11–15. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Ito, E.; Yagai, S.; Kitamura, A.; Karatsu, T. Chirality in the Photochemical mer→fac Geometrical Isomerization of Tris(1-phenylpyrazolato,N,C2′)iridium(III). Eur. J. Inorg. Chem. 2009, 2009, 2104–2109. [Google Scholar] [CrossRef]
- Francés-Monerris, A.; Gros, P.C.; Assfeld, X.; Monari, A.; Pastore, M. Toward luminescent iron complexes: Unraveling the photophysics by computing potential energy surfaces. ChemPhotoChem 2019, 3, 666–683. [Google Scholar] [CrossRef]
- Deorukhkar, N.; Besnard, C.; Guénée, L.; Piguet, C. Tuning spin-crossover transition temperatures in non-symmetrical homoleptic meridional/facial [Fe(didentate)3]2+ complexes: What for and who cares about it? Dalton Trans. 2021, 50, 1206–1223. [Google Scholar] [CrossRef]
- Tamayo, A.B.; Alleyne, B.D.; Djurovich, P.I.; Lamansky, S.; Tsyba, I.; Ho, N.N.; Bau, R.; Thompson, M.E. Synthesis and characterization of facial and meridional tris-cyclometalated iridium(III) complexes. J. Am. Chem. Soc. 2003, 125, 7377–7387. [Google Scholar] [CrossRef]
- Lehr, M.; Paschelke, T.; Trumpf, E.; Vogt, A.-M.; Näther, C.; Sönnichsen, F.D.; McConnell, A.J. A Paramagnetic NMR Spectroscopy Toolbox for the Characterisation of Paramagnetic/Spin-Crossover Coordination Complexes and Metal-Organic Cages. Chem. Angew. Chem. Int. Ed. 2020, 59, 19344–19351. [Google Scholar] [CrossRef]
- Bomben, P.G.; Robson, K.C.D.; Sedach, P.A.; Berlinguette, C.P. On the viability of cyclometalated Ru(II) complexes for light-harvesting applications. Inorg. Chem. 2009, 48, 9631–9643. [Google Scholar] [CrossRef]
- Kaneko, R.; Wu, G.; Sugawa, K.; Otsuki, J.; Islam, A.; Han, L.; Bedja, I.; Gupta, R.K. Cyclometalated ruthenium complexes with 6-(ortho-methoxyphenyl)-2,2′-bipyridine as panchromatic dyes for dye-sensitized solar cells. J. Organomet. Chem. 2017, 833, 61–70. [Google Scholar] [CrossRef]
- Kisserwan, H.; Kamar, A.; Shoker, T.; Ghaddar, T.H. Photophysical properties of new cyclometalated ruthenium complexes and their use in dye sensitized solar cells. Dalton Trans. 2012, 41, 10643–10651. [Google Scholar] [CrossRef]
- Motley, T.C.; Troian-Gautier, L.; Brennaman, M.K.; Meyer, G.J. Excited-State Decay Pathways of Tris(bidentate) Cyclometalated Ruthenium(II) Compounds. Inorg. Chem. 2017, 56, 13579–13592. [Google Scholar] [CrossRef]
- Steube, J.; Burkhardt, L.; Päpcke, A.; Moll, J.; Zimmer, P.; Schoch, R.; Wölper, C.; Heinze, K.; Lochbrunner, S.; Bauer, M. Excited-State Kinetics of an Air-Stable Cyclometalated Iron(II) Complex. Chem. Eur. J. 2019, 25, 11826–11830. [Google Scholar] [CrossRef] [PubMed]
- Prakash, O.; Chábera, P.; Rosemann, N.W.; Huang, P.; Häggström, L.; Ericsson, T.; Strand, D.; Persson, P.; Bendix, J.; Lomoth, R.; et al. A Stable Homoleptic Organometallic Iron(IV) Complex. Chemistry 2020, 26, 12728–12732. [Google Scholar] [CrossRef] [PubMed]
- Ashley, D.C.; Jakubikova, E. Ray-Dutt and Bailar Twists in Fe(II)-Tris(2,2’-bipyridine): Spin States, Sterics, and Fe-N Bond Strengths. Inorg. Chem. 2018, 57, 5585–5596. [Google Scholar] [CrossRef] [PubMed]
- Paulus, B.C.; Adelman, S.L.; Jamula, L.L.; McCusker, J.K. Leveraging excited-state coherence for synthetic control of ultrafast dynamics. Nature 2020, 582, 214–218. [Google Scholar] [CrossRef]
- Grimme, S.; Brandenburg, J.G.; Bannwarth, C.; Hansen, A. Consistent structures and interactions by density functional theory with small atomic orbital basis sets. Chem. Phys. 2015, 143, 54107. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Shoaf, A.L.; Bayse, C.A. TD-DFT and structural investigation of natural photosensitive phenanthroperylene quinone derivatives. New J. Chem. 2016, 40, 413–422. [Google Scholar] [CrossRef]
- Atkins, A.J.; Bauer, M.; Jacob, C.R. The chemical sensitivity of X-ray spectroscopy: High energy resolution XANES versus X-ray emission spectroscopy of substituted ferrocenes. Phys. Chem. Chem. Phys. 2013, 15, 8095–8105. [Google Scholar] [CrossRef]
- Burkhardt, L.; Holzwarth, M.; Plietker, B.; Bauer, M. Detection and Characterization of Hydride Ligands in Iron Complexes by High-Resolution Hard X-ray Spectroscopy and Implications for Catalytic Processes. Inorg. Chem. 2017, 56, 13300–13310. [Google Scholar] [CrossRef]
- Atkins, A.J.; Jacob, C.R.; Bauer, M. Probing the electronic structure of substituted ferrocenes with high-resolution XANES spectroscopy. Chem. Eur. J. 2012, 18, 7021–7025. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M. HERFD-XAS and valence-to-core-XES: New tools to push the limits in research with hard X-rays? Phys. Chem. Chem. Phys. 2014, 16, 13827–13837. [Google Scholar] [CrossRef] [PubMed]
- Glatzel, P.; Bergmann, U. High resolution 1s core hole X-ray spectroscopy in 3d transition metal complexes—Electronic and structural information. Coord. Chem. Rev. 2005, 249, 65–95. [Google Scholar] [CrossRef]
- Bergmann, U.; Glatzel, P. X-ray emission spectroscopy. Photosyn. Res. 2009, 102, 255–266. [Google Scholar] [CrossRef]
- Delgado-Jaime, M.U.; DeBeer, S.; Bauer, M. Valence-to-core X-ray emission spectroscopy of iron-carbonyl complexes: Implications for the examination of catalytic intermediates. Chem. Eur. J. 2013, 19, 15888–15897. [Google Scholar] [CrossRef]
- Illanes, A. (Ed.) Enzyme Biocatalysis: Principles and Applications; Springer: Dordrecht, The Netherlands, 2008; ISBN 978-1-4020-8360-0. [Google Scholar]
- Eastham, K.; Scattergood, P.A.; Chu, D.; Boota, R.Z.; Soupart, A.; Alary, F.; Dixon, I.M.; Rice, C.R.; Hardman, S.J.O.; Elliott, P.I.P. Not All 3MC States Are the Same: The Role of 3MCcis States in the Photochemical N^N Ligand Release from Ru(bpy)2(N^N)2+ Complexes. Inorg. Chem. 2022, 61, 19907–19924. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.; Perdew, J.P. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. Chem. Phys. 2003, 119, 12129–12137. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree–Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree–Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Prediction and interpretation of the 57Fe isomer shift in Mössbauer spectra by density functional theory. Inorg. Chim. Acta 2002, 337, 181–192. [Google Scholar] [CrossRef]
- Delgado-Jaime, M.U.; DeBeer, S. Expedited analysis of DFT outputs: Introducing MOAnalyzer. J. Comput. Chem. 2012, 33, 2180–2185. [Google Scholar] [CrossRef]
- Plasser, F. TheoDORE: A toolbox for a detailed and automated analysis of electronic excited state computations. Chem. Phys. 2020, 152, 84–108. [Google Scholar] [CrossRef] [PubMed]
- Knizia, G.; Klein, J.E.M.N. Electron flow in reaction mechanisms--revealed from first principles. Angew. Chem. Int. Ed. Engl. 2015, 54, 5518–5522. [Google Scholar] [CrossRef]
- Gauthier, C.; Solé, V.A.; Signorato, R.; Goulon, J.; Moguiline, E. The ESRF beamline ID26: X-ray absorption on ultra dilute sample. J. Synchrotron. Rad. 1999, 6, 164–166. [Google Scholar] [CrossRef]
- Johann, H.H. Die Erzeugung lichtstarker Röntgenspektren mit Hilfe von Konkavkristallen. Z. Physik 1931, 69, 185–206. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | dØ (Fe-N) (Å) | dØ (Fe-C) (Å) | ∢Chelate Bite Angle (°) | ∢(C-Fe-N)axial (°) |
|---|---|---|---|---|
| Fe(ppz)3 | 2.0030(15) | 1.9508(13) | 87.07(7) | 171.54(7) |
| Fe(CF3ppz)3 | 2.0075(15) | 1.9520(16) | 85.88(6) | 170.46(6) |
| Fe(MeOppz)3 | 2.0129(15) | 1.9512(13) | 94.52(5) | 170.89(5) |
| Fe(bppz)3 | 2.0122(7) | 1.9536(7) | 91.68(1) | 172.90(1) |
| Fe(naphpz)3 | 2.0134(12) | 1.9530(17) | 93.59(7) | 169.99(7) |
| Complex | E1/2 FeII/III (V) | E1/2 FeIII/IV (V) | E1/2 (Ligand) (V) | ΔELMCT e (V) | λabs-max (nm) (ε (cm−1 M−1)) |
|---|---|---|---|---|---|
| Fe(ppz)3 | −1.80 (rev) | −0.26 (rev) | 1.17 (irrev) | 2.97 (418 nm) | 109 (6.18) |
| 346 (1.63) | |||||
| 450 (0.64) (λmax = 522) c | |||||
| Fe(CF3ppz)3 | −1.50 (rev) | 0.07 (rev) | 1.54 d (irrev) | 3.04 (408 nm) | 293 (2.82) |
| 350 (0.61) | |||||
| 417 (0.38) (λmax = 530) c | |||||
| Fe(MeOppz)3 | −1.78 (rev) | −0.23 (rev) | 1.11 (irrev) | 2.89 (429 nm) | 290 (6.45) |
| 356 (0.62) | |||||
| 453 (0.39) (λmax = 580) c | |||||
| Fe(bppz)3 | −1.73 (rev) | −0.21 (rev) | 1.17 (irrev) | 2.90 (428 nm) | 277 (2.99) |
| 343 (0.68) | |||||
| 440 (0.45) (λmax = 540) c | |||||
| Fe(naphpz)3 | −1.68 (rev) | −0.22 (rev) | 1.08 (irrev) | 2.76 (449 m) | 284 (3.35) |
| 362 (0.93) | |||||
| 442 (0.68) (λmax = 590) c |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirschhausen, T.; Fritsch, L.; Lux, F.; Steube, J.; Schoch, R.; Neuba, A.; Egold, H.; Bauer, M. Iron(III)-Complexes with N-Phenylpyrazole-Based Ligands. Inorganics 2023, 11, 282. https://doi.org/10.3390/inorganics11070282
Hirschhausen T, Fritsch L, Lux F, Steube J, Schoch R, Neuba A, Egold H, Bauer M. Iron(III)-Complexes with N-Phenylpyrazole-Based Ligands. Inorganics. 2023; 11(7):282. https://doi.org/10.3390/inorganics11070282
Chicago/Turabian StyleHirschhausen, Tanja, Lorena Fritsch, Franziska Lux, Jakob Steube, Roland Schoch, Adam Neuba, Hans Egold, and Matthias Bauer. 2023. "Iron(III)-Complexes with N-Phenylpyrazole-Based Ligands" Inorganics 11, no. 7: 282. https://doi.org/10.3390/inorganics11070282
APA StyleHirschhausen, T., Fritsch, L., Lux, F., Steube, J., Schoch, R., Neuba, A., Egold, H., & Bauer, M. (2023). Iron(III)-Complexes with N-Phenylpyrazole-Based Ligands. Inorganics, 11(7), 282. https://doi.org/10.3390/inorganics11070282