




Hydrogen Incorporation in RuxTi1−xO2 Mixed Oxides Promotes Total Oxidation of Propane

 ,
,  and
and
Abstract
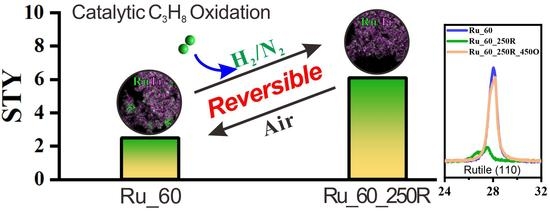
:
1. Introduction
2. Experimental Results
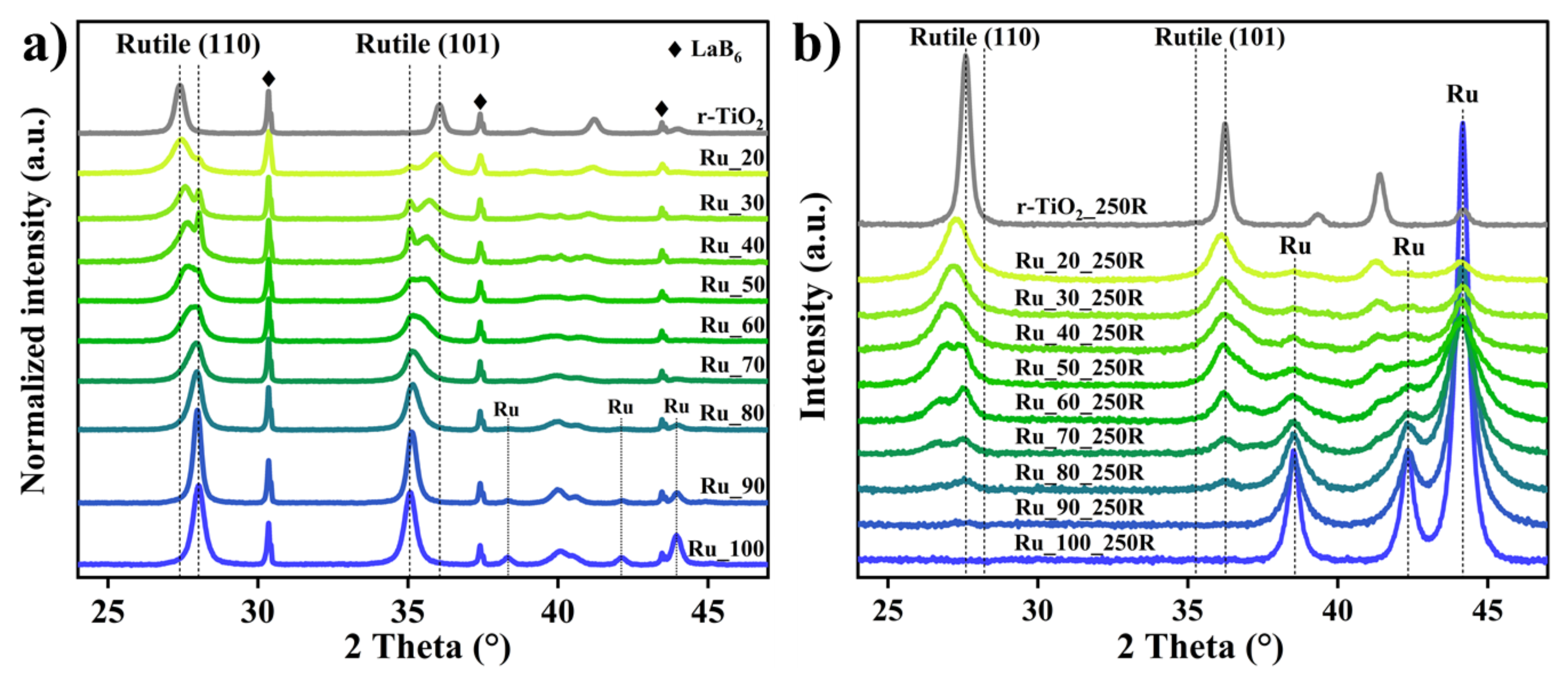
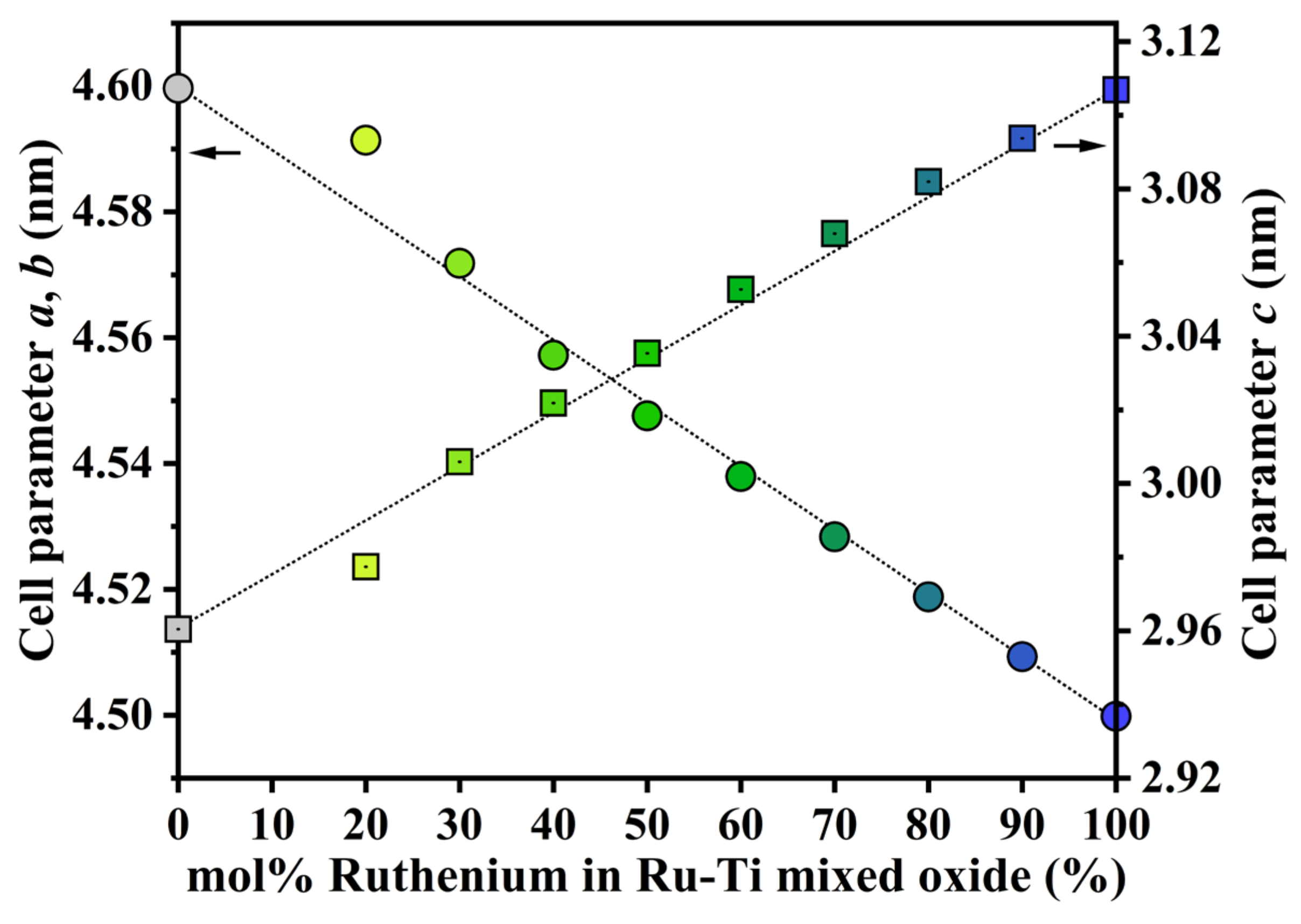
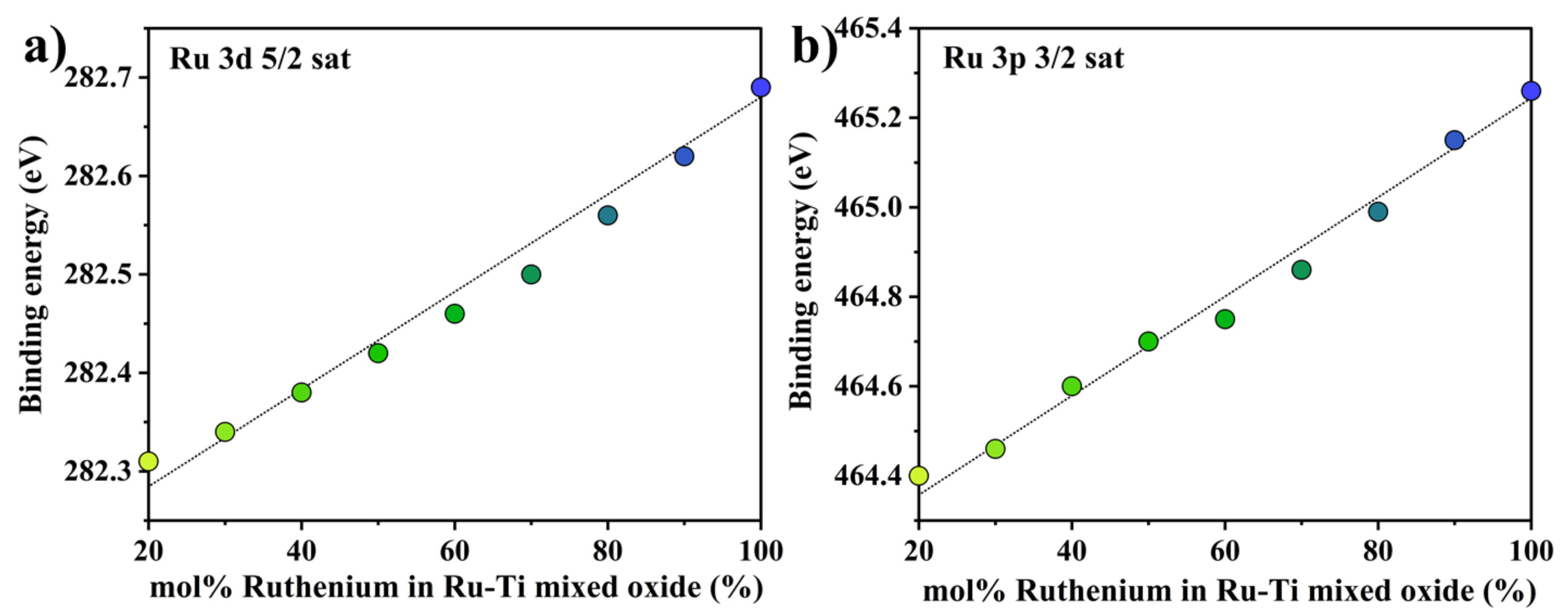
2.1. Characterization of the Fresh Ruthenium−Titanium Mixed Oxide Samples
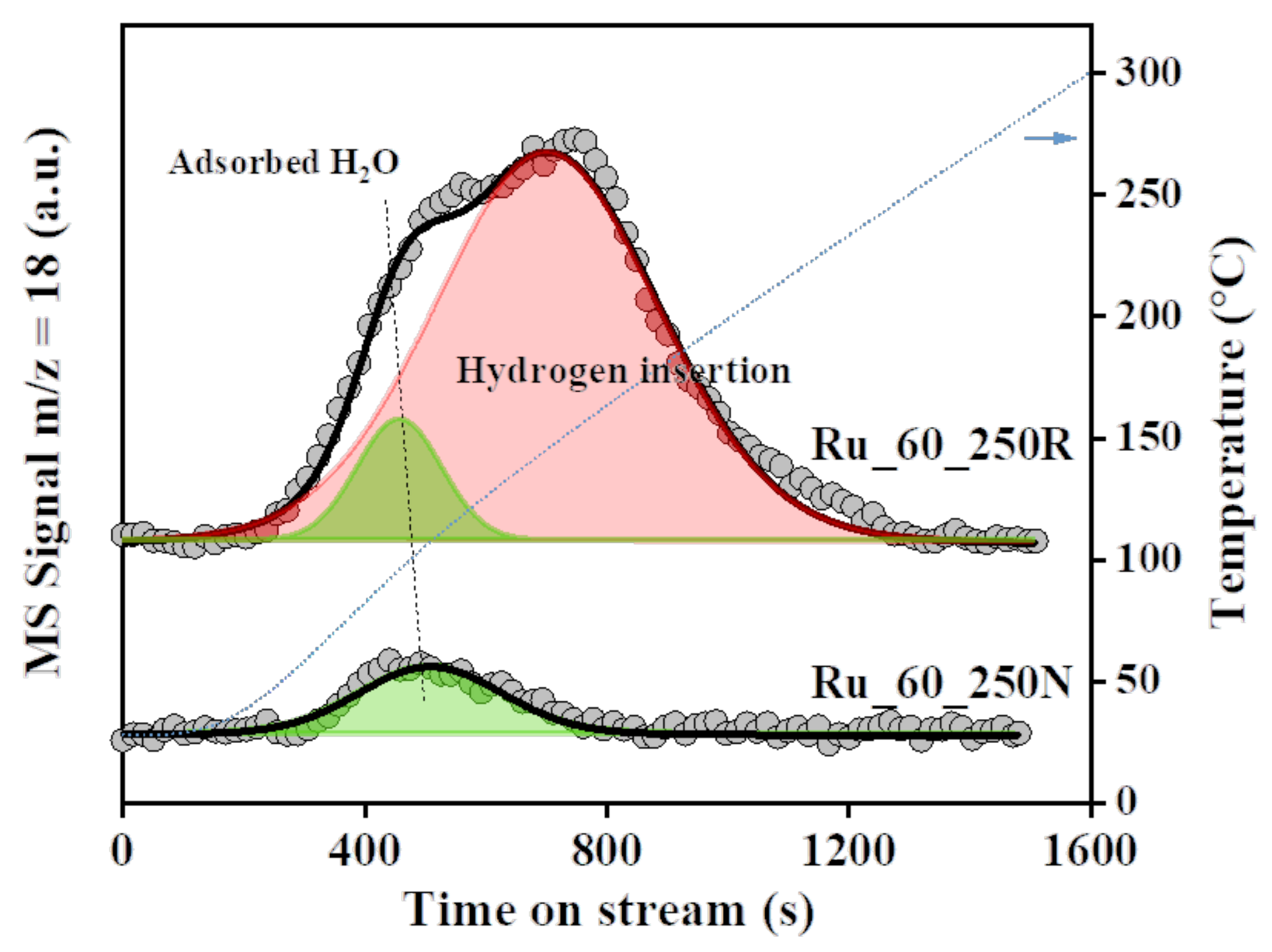
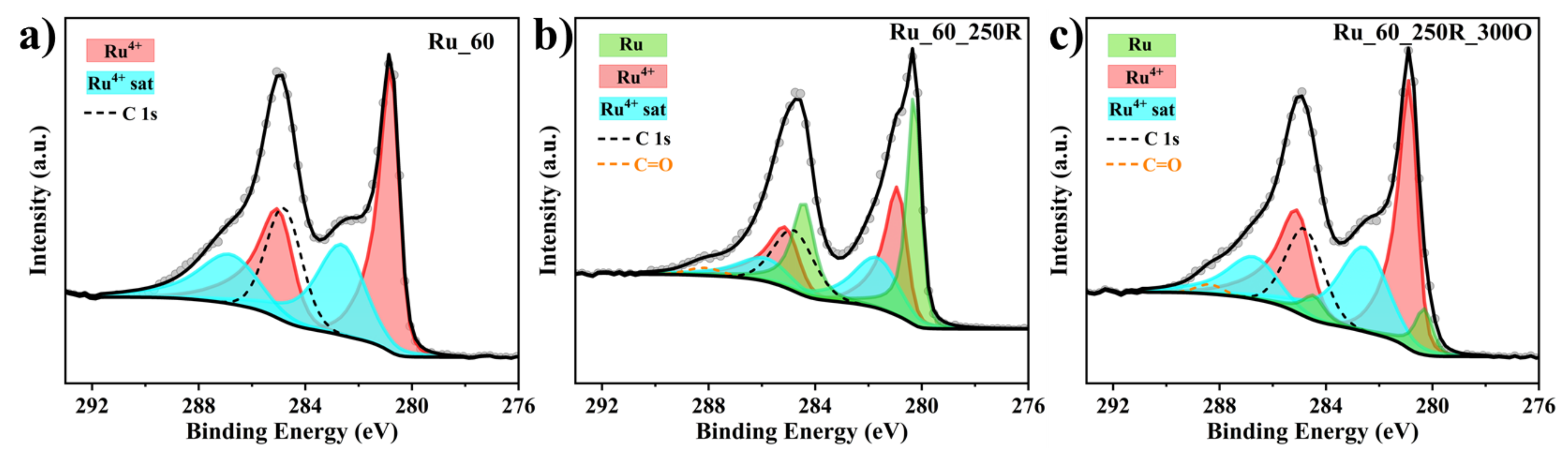
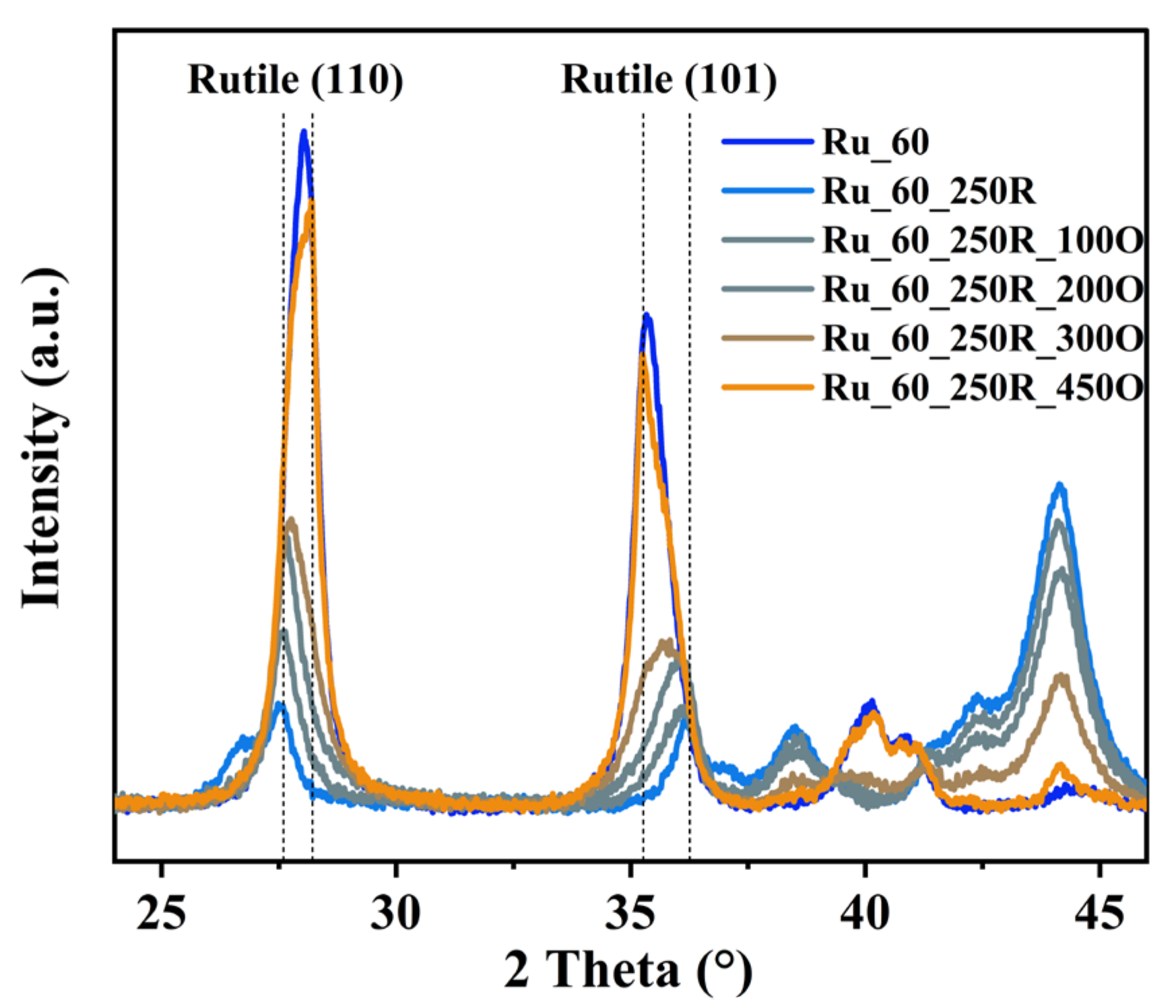
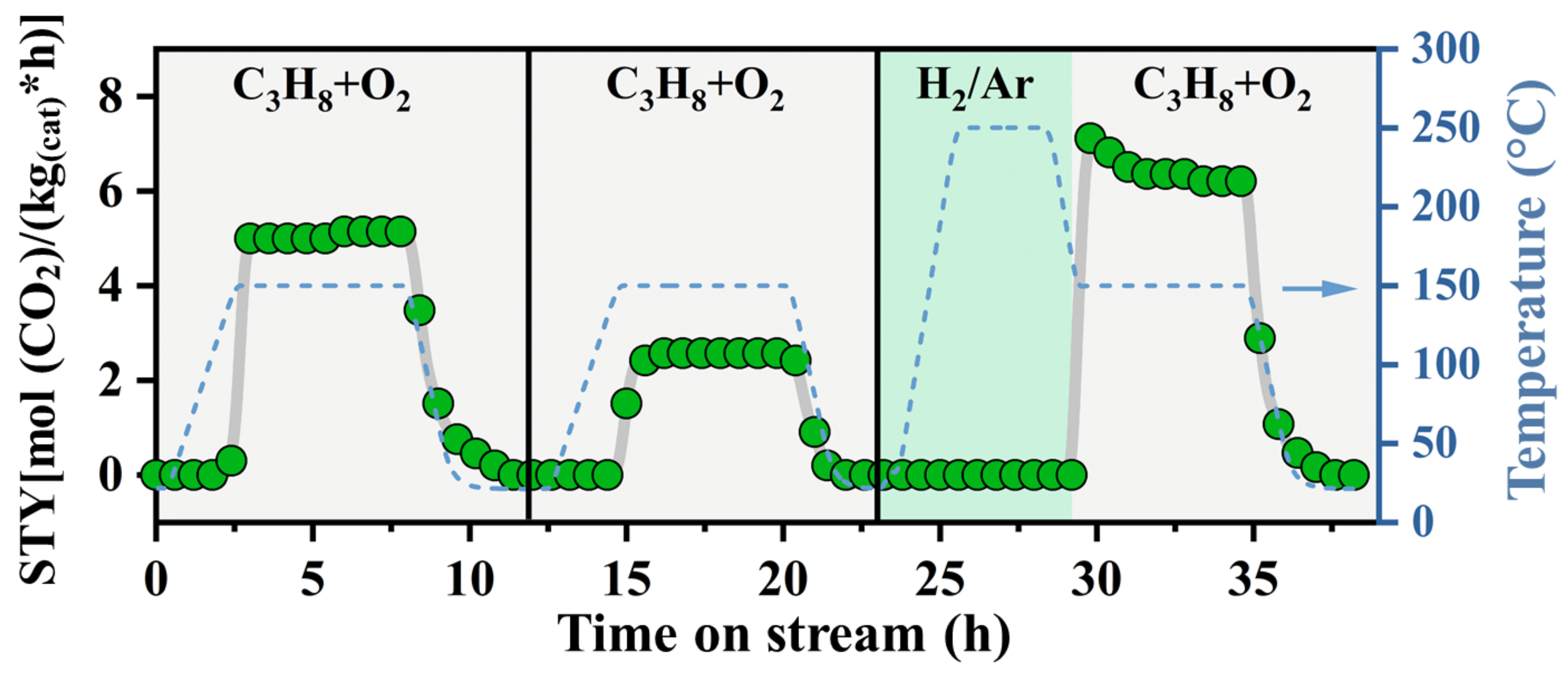
2.2. Hydrogen-Induced Changes of Ruthenium−Titanium Mixed Oxides
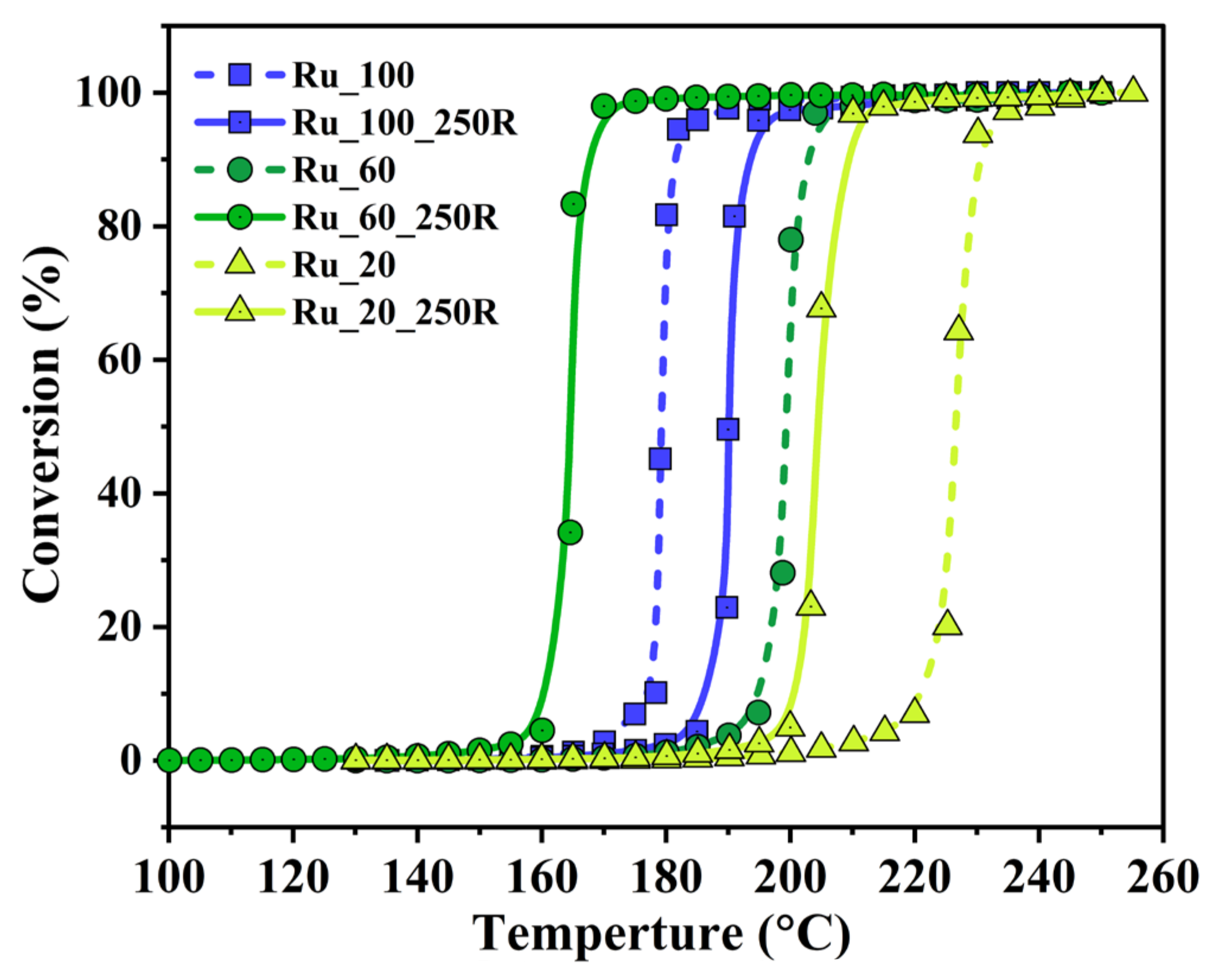
2.3. Catalytic Tests: Propane Combustion
3. Discussion
3.1. Formation of Ru_x and Ru_x_250R
3.2. Improved Oxidation Catalysis of Ru_x_250R in Comparison to Ru_x
4. Materials and Methods
4.1. Materials
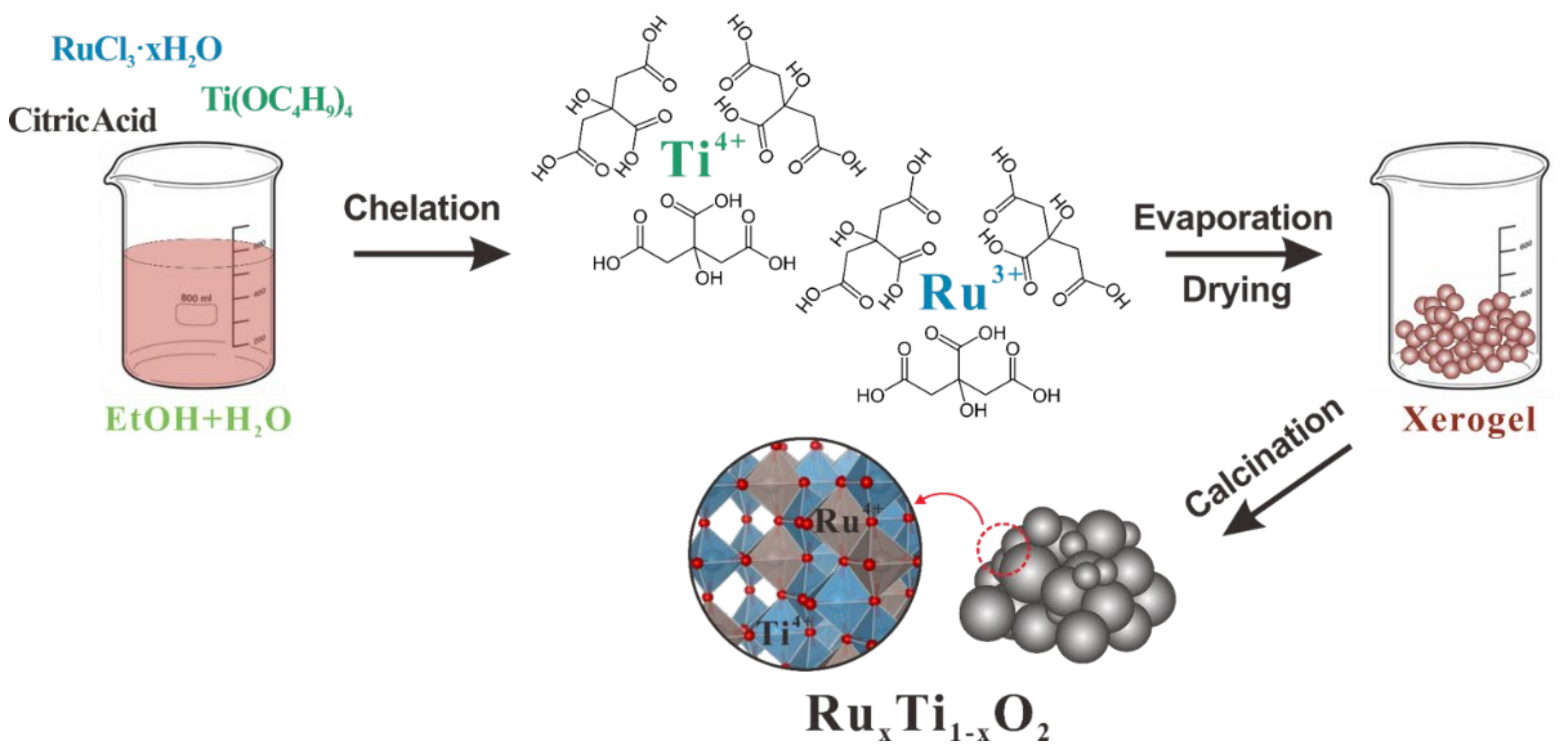
4.2. Catalysts Preparation
4.3. Catalysts Characterization
4.4. Catalytic Tests
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mavrikakis, M.; Hammer, B.; Nørskov, J.K. Effect of Strain on the Reactivity of Metal Surfaces. Phys. Rev. Lett. 1998, 81, 2819–2822. [Google Scholar] [CrossRef] [Green Version]
- Hammer, B.; Nørskov, J.K. Chemisorption and Reactivity on Supported Clusters and Thin Films; Kluwer Academic: Dordrecht, The Netherlands, 1997; pp. 285–351. [Google Scholar]
- Buvat, G.; Eslamibidgoli, M.J.; Youssef, A.H.; Garbarino, S.; Ruediger, A.; Eikerling, M.; Guay, D. Effect of IrO6 Octahedron Distortion on the OER Activity at (100) IrO2 Thin Film. ACS Catal. 2020, 10, 806–817. [Google Scholar] [CrossRef]
- Wang, H.; Xu, S.; Tsai, C.; Li, Y.; Liu, C.; Zhao, J.; Liu, Y.; Yuan, H.; Abild-Pedersen, F.; Prinz, F.B.; et al. Direct and Continuous Strain Control of Catalysts with Tunable Battery Electrode Materials. Science 2016, 354, 1031–1036. [Google Scholar] [CrossRef]
- Strasser, P.; Kühl, S. Dealloyed Pt-based Core-shell Oxygen Reduction Electrocatalysts. Nano Energy 2016, 29, 166–177. [Google Scholar] [CrossRef] [Green Version]
- Gawande, M.B.; Goswami, A.; Asefa, T.; Guo, H.; Biradar, A.V.; Peng, D.-L.; Zboril, R.; Varma, R.S. Core-shell Nanoparticles: Synthesis and Applications in Catalysis and Electrocatalysis. Chem. Soc. Rev. 2015, 44, 7540–7590. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, X.; Jiang, G.; Zhu, H.; Guo, S.; Su, D.; Lu, G.; Sun, S. Tuning Nanoparticle Structure and Surface Strain for Catalysis Optimization. J. Am. Chem. Soc. 2014, 136, 7734–7739. [Google Scholar] [CrossRef] [Green Version]
- Kibler, L.A.; El-Aziz, A.M.; Hoyer, R.; Kolb, D.M. Tuning Reaction Rates by Lateral Strain in a Palladium Monolayer. Angew. Chem. Int. Ed. 2005, 44, 2080–2084. [Google Scholar] [CrossRef] [PubMed]
- Strasser, P.; Koh, S.; Anniyev, T.; Greeley, J.; More, K.; Yu, C.; Liu, Z.; Kaya, S.; Nordlund, D.; Ogasawara, H.; et al. Lattice-strain Control of the Activity in Dealloyed Core-shell Fuel Cell Catalysts. Nat. Chem. 2010, 2, 454–460. [Google Scholar] [CrossRef]
- Xia, Z.; Guo, S. Strain Engineering of Metal-based Nanomaterials for Energy Electrocatalysis. Chem. Soc. Rev. 2019, 48, 3265–3278. [Google Scholar] [CrossRef]
- Wang, L.; Zeng, Z.; Gao, W.; Maxson, T.; Raciti, D.; Giroux, M.; Pan, X.; Wang, C.; Greeley, J. Tunable Intrinsic Strain in Two-dimensional Transition Metal Electrocatalysts. Science 2019, 363, 870–874. [Google Scholar] [CrossRef]
- You, B.; Tang, M.T.; Tsai, C.; Abild-Pedersen, F.; Zheng, X.; Li, H. Enhancing Electrocatalytic Water Splitting by Strain Engineering. Adv. Mater. 2019, 31, 1807001. [Google Scholar] [CrossRef] [PubMed]
- Alayoglu, S.; Nilekar, A.U.; Mavrikakis, M.; Eichhorn, B. Ru-Pt Core-shell Nanoparticles for Preferential Oxidation of Carbon Monoxide in Hydrogen. Nat. Mater. 2008, 7, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Schlapka, A.; Lischka, M.; Groß, A.; Käsberger, U.; Jakob, P. Surface Strain versus Substrate Interaction in Heteroepitaxial Metal Layers: Pt on Ru(0001). Phys. Rev. Lett. 2003, 91, 016101. [Google Scholar] [CrossRef] [Green Version]
- Teschner, D.; Borsodi, J.; Wootsch, A.; Révay, Z.; Hävecker, M.; Knop-Gericke, A.; Jackson, S.D.; Schlögl, R. The Roles of Subsurface Carbon and Hydrogen in Palladium-Catalyzed Alkyne Hydrogenation. Science 2008, 320, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Wilde, M.; Fukutani, K.; Ludwig, W.; Brandt, B.; Fischer, J.-H.; Schauermann, S.; Freund, H.-J. Influence of Carbon Deposition on the Hydrogen Distribution in Pd Nanoparticles and Their Reactivity in Olefin Hydrogenation. Angew. Chem. Int. Ed. 2008, 47, 9289–9293. [Google Scholar] [CrossRef] [PubMed]
- Copéret, C.; Estes, D.P.; Larmier, K.; Searles, K. Isolated Surface Hydrides: Formation, Structure, and Reactivity. Chem. Rev. 2016, 116, 8463–8505. [Google Scholar] [CrossRef]
- Wu, Z.; Cheng, Y.; Tao, F.; Daemen, L.; Foo, G.S.; Nguyen, L.; Zhang, X.; Beste, A.; Ramirez-Cuesta, A.J. Direct Neutron Spectroscopy Observation of Cerium Hydride Species on a Cerium Oxide Catalyst. J. Am. Chem. Soc. 2017, 139, 9721–9727. [Google Scholar] [CrossRef]
- Werner, K.; Weng, X.; Calaza, F.; Sterrer, M.; Kropp, T.; Paier, J.; Sauer, J.; Wilde, M.; Fukutani, K.; Shaikhutdinov, S.; et al. Toward an Understanding of Selective Alkyne Hydrogenation on Ceria: On the Impact of O Vacancies on H2 Interaction with CeO2(111). J. Am. Chem. Soc. 2017, 139, 17608–17616. [Google Scholar] [CrossRef]
- Cao, T.; You, R.; Zhang, X.; Chen, S.; Li, D.; Zhang, Z.; Huang, W. An in situ DRIFTS Mechanistic Study of CeO2-catalyzed Acetylene Semihydrogenation Reaction. Phys. Chem. Chem. Phys. 2018, 20, 9659–9670. [Google Scholar] [CrossRef]
- Cheng, H.; Wen, M.; Ma, X.; Kuwahara, Y.; Mori, K.; Dai, Y.; Huang, B.; Yamashita, H. Hydrogen Doped Metal Oxide Semiconductors with Exceptional and Tunable Localized Surface Plasmon Resonances. J. Am. Chem. Soc. 2016, 138, 9316–9324. [Google Scholar] [CrossRef]
- Li, Z.; Werner, K.; Qian, K.; You, R.; Płucienik, A.; Jia, A.; Wu, L.; Zhang, L.; Pan, H.; Kuhlenbeck, H.; et al. Oxidation of Reduced Ceria by Incorporation of Hydrogen. Angew. Chem. Int. Ed. 2019, 58, 14686–14693. [Google Scholar] [CrossRef] [Green Version]
- Vilé, G.; Bridier, B.; Wichert, J.; Pérez-Ramírez, J. Ceria in Hydrogenation Catalysis: High Selectivity in the Conversion of Alkynes to Olefins. Angew. Chem. Int. Ed. 2012, 51, 8620–8623. [Google Scholar] [CrossRef]
- Vilé, G.; Colussi, S.; Krumeich, F.; Trovarelli, A.; Pérez-Ramírez, J. Opposite Face Sensitivity of CeO2 in Hydrogenation and Oxidation Catalysis. Angew. Chem. Int. Ed. 2014, 53, 12069–12072. [Google Scholar] [CrossRef]
- Carrasco, J.; Vilé, G.; Fernández-Torre, D.; Pérez, J.; Pérez-Ramírez, R.; Ganduglia-Pirovano, M.V. Molecular-Level Understanding of CeO2 as a Catalyst for Partial Alkyne Hydrogenation. J. Phys. Chem. C 2014, 118, 5352–5360. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Timmer, P.; Luciano, A.S.; Wang, Y.; Weber, T.; Glatthaar, L.; Guo, Y.; Smarsly, B.M.; Over, H. Inserted Hydrogen Promotes Oxidation Catalysis of Mixed Ru0.3Ti0.7O2 as Exemplified with Total Propane Oxidation and the HCl Oxidation Reaction. Catal. Sci. Technol. 2023, 13, 1395–1408. [Google Scholar] [CrossRef]
- Colomer, M.T.; Jurado, J.R. Structural, Microstructural, and Electrical Transport Properties of TiO2-RuO2 Ceramic Materials Obtained by Polymeric Sol-Gel Route. Chem. Mater. 2000, 12, 923–930. [Google Scholar] [CrossRef]
- Wang, X.; Shao, Y.; Liu, X.; Tang, D.; Wu, B.; Tang, Z.; Wang, X.; Lin, W. Phase Stability and Phase Structure of Ru–Ti–O Complex Oxide Electrocatalyst. J. Am. Ceram. Soc. 2015, 98, 1915–1924. [Google Scholar] [CrossRef]
- Ashcroft, N.; Denton, A. Vegard’s Law. Phys. Rev. A 1991, 43, 3161–3164. [Google Scholar]
- Özkan, E.; Cop, P.; Benfer, F.; Hofmann, A.; Votsmeier, M.; Guerra, J.M.; Giar, M.; Heiliger, C.; Over, H.; Smarsly, B.M. Rational Synthesis Concept for Cerium Oxide Nanoparticles: On the Impact of Particle Size on the Oxygen Storage Capacity. J. Phys. Chem. C 2020, 124, 8736–8748. [Google Scholar] [CrossRef]
- Sivakami, R.; Dhanuskodi, S.; Karvembu, R. Estimation of Lattice Strain in Nanocrystalline RuO2 by Williamson-Hall and Size-strain Plot Methods. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2016, 152, 43–50. [Google Scholar] [CrossRef]
- Over, H.; Muhler, M. Catalytic CO Oxidation over Ruthenium—Bridging the Pressure Gap. Prog. Surf. Sci. 2003, 72, 3–17. [Google Scholar] [CrossRef]
- Over, H.; Seitsonen, A.P.; Lundgren, E.; Smedh, M.; Andersen, J.N. On the Origin of the Ru-3d5/2 Satellite Feature from RuO2(110). Surf. Sci. 2002, 504, L196–L200. [Google Scholar] [CrossRef]
- Khalid, O.; Weber, T.; Drazic, G.; Djerdj, I.; Over, H. Mixed RuxIr1-xO2 Oxide Catalyst with Well-Defined and Varying Composition Applied to CO Oxidation. J. Phys. Chem. C 2020, 124, 18670–18683. [Google Scholar] [CrossRef]
- Assmann, J.; Narkhede, V.; Khodeir, L.; Löffler, E.; Hinrichsen, O.; Birkner, A.; Over, H.; Muhler, M. On the Nature of the Active State of Supported Ruthenium Catalysts Used for the Oxidation of Carbon Monoxide: Steady-state and Transient Kinetics Combined with in Situ Infrared Spectroscopy. J. Phys. Chem. B 2004, 108, 14634–14642. [Google Scholar] [CrossRef]
- Wang, Z.; Khalid, O.; Wang, W.; Wang, Y.; Weber, T.; Luciano, A.S.; Zhan, W.; Smarsly, B.M.; Over, H. Comparison Study of the Effect of CeO2-based Carrier Materials on the Total Oxidation of CO, Methane, and Propane over RuO2. Catal. Sci. Technol. 2021, 11, 6839–6853. [Google Scholar] [CrossRef]
- Walsh, F.; Wills, R. The Continuing Development of Magnéli Phase Titanium Sub-Oxides and Ebonex® Electrodes. Electrochim. Acta 2010, 55, 6342–6351. [Google Scholar] [CrossRef]
- Malik, H.; Sarkar, S.; Mohanty, S.; Carlson, K. Modeling and Synthesis of Magneli Phases in Ordered Titanium Oxide Nanotubes with Preserved Morphology. Sci. Rep. 2020, 10, 8050. [Google Scholar] [CrossRef]
- Wang, Z.; Huang, Z.; Brosnahan, J.T.; Zhang, S.; Guo, Y.; Guo, Y.; Wang, L.; Wang, Y.; Zhan, W. Ru/CeO2 Catalyst with Optimized CeO2 Support Morphology and Surface Facets for Propane Combustion. Environ. Sci. Technol. 2019, 53, 5349–5358. [Google Scholar] [CrossRef]
- Wu, J.; Chen, B.; Yan, J.; Zheng, X.; Wang, X.; Deng, W.; Dai, Q. Ultra-active Ru Supported on CeO2 Nanosheets for Catalytic Combustion of Propane: Experimental Insights into Interfacial Active Sites. J. Chem. Eng. 2022, 438, 135501. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Cell Parameter a/b (nm) a | Cell Parameter c (nm) b | Grain Size (RuO2) (nm) c | Grain Size (Ru–Ti) (nm) c | Ru–Ti/(RuO2 + Ru–Ti) (%) d |
|---|---|---|---|---|---|
| Ru_100 | 4.500 | 3.107 | 18 ± 0.5 | - | 0 |
| Ru_90 | 4.509 | 3.094 | 27 ± 1 | 25 ± 2 | 81.2 |
| Ru_80 | 4.519 | 3.082 | 32 ± 0.5 | 15 ± 0.3 | 89.9 |
| Ru_70 | 4.528 | 3.068 | 33 ± 1 | 12 ± 0.2 | 90.9 |
| Ru_60 | 4.538 | 3.053 | 36± 1 | 10 ± 0.5 | 93.7 |
| Ru_50 | 4.548 | 3.035 | 46 ± 8 | 9 ± 0.5 | 96.4 |
| Ru_40 | 4.558 | 3.022 | 45 ± 6 | 10 ± 0.5 | 88.8 |
| Ru_30 | 4.572 | 3.006 | 43 ± 5 | 12 ± 2 | 81.9 |
| Ru_20 | 4.591 | 2.977 | 28 ± 6 | 16 ± 1 | 92.5 |
| Catalysts | SBET (m2/g) | Ru/(Ti + Ru)(mol%) a | Ru/(Ti + Ru)(mol%) b | (OH + Oxygenated Carbon)/O 1s Total b |
|---|---|---|---|---|
| Ru_100 | 9 | 100 | 100 | 39.90 |
| Ru_90 | 6 | 89.3 | 70.65 | 14.67 |
| Ru_80 | 10 | 78.8 | 59.81 | 5.42 |
| Ru_70 | 14 | 68.1 | 60.53 | 4.85 |
| Ru_60 | 19 | 56.7 | 54.4 | 4.42 |
| Ru_50 | 26 | 46.8 | 47.74 | 2.50 |
| Ru_40 | 32 | 40.9 | 37.96 | 4.69 |
| Ru_30 | 31 | 30.0 | 29.08 | 3.38 |
| Ru_20 | 34 | 17.4 | 17.61 | 4.11 |
| Ru_60_250R | 19 | 56.2 | 52.12 | 4.43 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Wang, Y.; Timmer, P.; Spriewald-Luciano, A.; Weber, T.; Glatthaar, L.; Guo, Y.; Smarsly, B.M.; Over, H. Hydrogen Incorporation in RuxTi1−xO2 Mixed Oxides Promotes Total Oxidation of Propane. Inorganics 2023, 11, 330. https://doi.org/10.3390/inorganics11080330
Wang W, Wang Y, Timmer P, Spriewald-Luciano A, Weber T, Glatthaar L, Guo Y, Smarsly BM, Over H. Hydrogen Incorporation in RuxTi1−xO2 Mixed Oxides Promotes Total Oxidation of Propane. Inorganics. 2023; 11(8):330. https://doi.org/10.3390/inorganics11080330
Chicago/Turabian StyleWang, Wei, Yu Wang, Phillip Timmer, Alexander Spriewald-Luciano, Tim Weber, Lorena Glatthaar, Yun Guo, Bernd M. Smarsly, and Herbert Over. 2023. "Hydrogen Incorporation in RuxTi1−xO2 Mixed Oxides Promotes Total Oxidation of Propane" Inorganics 11, no. 8: 330. https://doi.org/10.3390/inorganics11080330
APA StyleWang, W., Wang, Y., Timmer, P., Spriewald-Luciano, A., Weber, T., Glatthaar, L., Guo, Y., Smarsly, B. M., & Over, H. (2023). Hydrogen Incorporation in RuxTi1−xO2 Mixed Oxides Promotes Total Oxidation of Propane. Inorganics, 11(8), 330. https://doi.org/10.3390/inorganics11080330