
Symmetrical and Unsymmetrical Dicopper Complexes Based on Bis-Oxazoline Units: Synthesis, Spectroscopic Properties and Reactivity

 , , and
, , and
Abstract
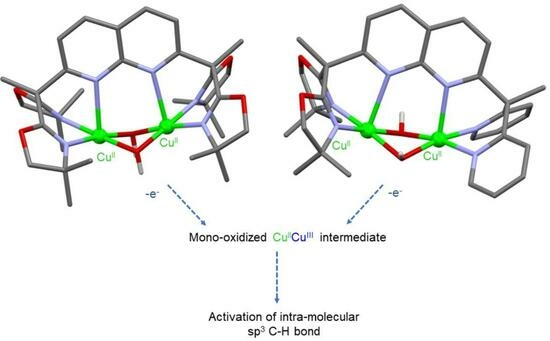
:
1. Introduction
2. Results and Discussion
2.1. Ligands Synthesis
2.2. Dicopper(I) Complexes
2.2.1. Synthesis and Characterizations
2.2.2. Reactivity
2.3. Dicopper(II) Complexes
2.3.1. Synthesis and Characterizations
2.3.2. Electrochemical Oxidation of Complexes 1 and 2
3. Materials and Methods
3.1. General
3.2. Ligands’ Syntheses
3.3. Complexes Syntheses
3.4. Crystallographic Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 |
|---|---|---|
| Chemical Formula | [C32H44Cu2N6O6](CF3O3S)2 | 2[C32H36Cu2N6O4)(CFO3S)2]∙CH3CN |
| Formula mass | 1033.95 | 2028.83 |
| Morphology | plate | plate |
| Color | blue | blue |
| Crystal size (mm) | 0.48 × 0.3 × 0.1 | 0.45 × 0.2 × 0.1 |
| Crystal system | monoclinic | triclinic |
| Space group | P1 21/n 1 | P-1 |
| a [Å] | 10.332 (2) | 12.377 (3) |
| b [Å] | 30.427 (6) | 14.361 (3) |
| c [Å] | 13.415 (3) | 23.657 (5) |
| α [°] | 90 | 84.30 (3) |
| β [°] | 92.80 (3) | 82.98 (3) |
| γ [°] | 90 | 83.00 (3) |
| Unit-cell volume [Å3] | 4212.2 (15) | 4127.1 (15) |
| Dx (g·cm−3) | 1.63 | 1.633 |
| T [K] | 200 | 200 |
| Z | 4 | 2 |
| μ [mm−1] | 1.202 | 1.222 |
| Total reflections | 68,415 | 77,009 |
| Unique reflections | 12,173 | 18,827 |
| Obsd. reflections | 9889 (F > 2σ) | 11,658 (F > 2σ) |
| Rint. | 0.0501 | 0.0961 |
| Ra | 0.0400 | 0.0868 |
| R(w)a | 0.0873 | 0.2176 |
| Goodness of fit S | 1.085 | 1.061 |
| ∆ρmin/∆ρmax (e·Å−3) | −0.641/0.574 | −1.310/1.754 |
| CCDC Number | 2,266,888 | 2,266,887 |
3.5. Spectroelectrochemistry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elwell, C.E.; Gagnon, N.L.; Neisen, B.D.; Dhar, D.; Spaeth, A.D.; Yee, G.M.; Tolman, W.B. Copper−Oxygen Complexes Revisited: Structures, Spectroscopy, and Reactivity. Chem. Rev. 2017, 117, 2059–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quist, D.A.; Diaz, D.E.; Liu, J.J.; Karlin, K.D. Activation of Dioxygen by Copper Metalloproteins and Insights from Model Complexes. J. Biol. Inorg. Chem. 2017, 22, 253–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keown, W.; Gary, J.B.; Stack, T.D.P. High-Valent Copper in Biomimetic and Biological Oxidations. J. Biol. Inorg. Chem. 2017, 22, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Trammell, R.; Rajabimoghadam, K.; Garcia-Bosch, I. Copper-Promoted Functionalization of Organic Molecules: From Biologically Relevant Cu/O2 Model Systems to Organometallic Transformations. Chem. Rev. 2019, 119, 2954–3031. [Google Scholar] [CrossRef]
- Garcia-Bosch, I.; Cowley, R.E.; Díaz, D.E.; Peterson, R.L.; Solomon, E.I.; Karlin, K.D. Substrate and Lewis Acid Coordination Promote O–O Bond Cleavage of an Unreactive L2CuII2(O22–) Species to Form L2CuIII2(O)2 Cores with Enhanced Oxidative Reactivity. J. Am. Chem. Soc. 2017, 139, 3186–3195. [Google Scholar] [CrossRef] [Green Version]
- Magallón, C.; Serrano-Plana, J.; Roldán-Gómez, S.; Ribas, X.; Costas, M.; Company, A. Preparation of a Coordinatively Saturated μ-H2:H2-Peroxodicopper(II) Compound. Inorg.Chim. Acta 2018, 481, 166–170. [Google Scholar] [CrossRef]
- Paul, M.; Teubner, M.; Grimm-Lebsanft, B.; Buchenau, S.; Hoffmann, A.; Rübhausen, M.; Herres-Pawlis, S. Influence of the Amine Donor on Hybrid Guanidine-Stabilized Bis(μ-Oxido) Dicopper(III) Complexes and Their Tyrosinase-like Oxygenation Activity towards Polycyclic Aromatic Alcohols. J. Inorg. Biochem. 2021, 224, 111541. [Google Scholar] [CrossRef]
- Tahsini, L.; Kotani, H.; Lee, Y.-M.; Cho, J.; Nam, W.; Karlin, K.D.; Fukuzumi, S. Electron-Transfer Reduction of Dinuclear Copper Peroxo and Bis-μ-Oxo Complexes Leading to the Catalytic Four-Electron Reduction of Dioxygen to Water. Chem.-Eur. J. 2012, 18, 1084–1093. [Google Scholar] [CrossRef]
- Li, S.T.; Braun-Cula, B.; Hoof, S.; Limberg, C. Copper(i) Complexes Based on Ligand Systems with Two Different Binding Sites: Synthesis, Structures and Reaction with O2. Dalton Trans. 2018, 47, 544–560. [Google Scholar] [CrossRef]
- Kodera, M.; Kano, K. Reversible O2-Binding and Activation with Dicopper and Diiron Complexes Stabilized by Various Hexapyridine Ligands. Stability, Modulation, and Flexibility of the Dinuclear Structure as Key Aspects for the Dimetal/O2 Chemistry. Bull. Chem. Soc. Jpn. 2007, 80, 662–676. [Google Scholar] [CrossRef] [Green Version]
- Dalle, K.E.; Gruene, T.; Dechert, S.; Demeshko, S.; Meyer, F. Weakly Coupled Biologically Relevant CuII2 (μ-η1:η1-O2) Cis-Peroxo Adduct That Binds Side-On to Additional Metal Ions. J. Am. Chem. Soc. 2014, 136, 7428–7434. [Google Scholar] [CrossRef] [PubMed]
- Karlin, K.D.; Lee, D.-H.; Kaderli, S.; Zuberbühler, A.D. Copper Dioxygen Complexes Stable at Ambient Temperature: Optimization of Ligand Design and Solvent. Chem. Commun. 1997, 5, 475–476. [Google Scholar] [CrossRef]
- Lohmiller, T.; Spyra, C.-J.; Dechert, S.; Demeshko, S.; Bill, E.; Schnegg, A.; Meyer, F. Antisymmetric Spin Exchange in a μ-1,2-Peroxodicopper(II) Complex with an Orthogonal Cu–O–O–Cu Arrangement and S = 1 Spin Ground State Characterized by THz-EPR. JACS Au 2022, 2, 1134–1143. [Google Scholar] [CrossRef] [PubMed]
- Börzel, H.; Comba, P.; Hagen, K.S.; Kerscher, M.; Pritzkow, H.; Schatz, M.; Schindler, S.; Walter, O. Copper−Bispidine Coordination Chemistry: Syntheses, Structures, Solution Properties, and Oxygenation Reactivity. Inorg. Chem. 2002, 41, 5440–5452. [Google Scholar] [CrossRef] [PubMed]
- Brückmann, T.; Becker, J.; Würtele, C.; Seuffert, M.T.; Heuler, D.; Müller-Buschbaum, K.; Weiß, M.; Schindler, S. Characterization of Copper Complexes with Derivatives of the Ligand (2-Aminoethyl)Bis(2-Pyridylmethyl)Amine (Uns-Penp) and Their Reactivity towards Oxygen. J. Inorg. Biochem. 2021, 223, 111544. [Google Scholar] [CrossRef]
- Jacobson, R.R.; Tyeklar, Z.; Farooq, A.; Karlin, K.D.; Liu, S.; Zubieta, J. A Copper-Oxygen (Cu2-O2) Complex. Crystal Structure and Characterization of a Reversible Dioxygen Binding System. J. Am. Chem. Soc. 1988, 110, 3690–3692. [Google Scholar] [CrossRef]
- Isaac, J.A.; Gennarini, F.; Lopez, I.; Thibon-Pourret, A.; David, R.; Gellon, G.; Gennaro, B.; Philouze, C.; Meyer, F.; Demeshko, S.; et al. Room-Temperature Characterization of a Mixed-Valent µ-Hydroxodicopper(II,III) Complex. Inorg. Chem. 2016, 55, 8263–8266. [Google Scholar] [CrossRef]
- Isaac, J.A.; Thibon-Pourret, A.; Durand, A.; Philouze, C.; Le Poul, N.; Belle, C. High-Valence CuIICuIII Species in Action: Demonstration of Aliphatic C–H Bond Activation at Room Temperature. Chem. Commun. 2019, 55, 12711–12714. [Google Scholar] [CrossRef]
- Isaac, J.A. Conception et Synthèse de Catalyseurs de Cuivre Bio-Inspirés Pour l’activation de Liaisons C-H. Ph.D. Thesis, Université Grenoble-Alpes, Grenoble, France, 2018. [Google Scholar]
- Desimoni, G.; Faita, G.; Jørgensen, K.A. C2-Symmetric Chiral Bis(Oxazoline) Ligands in Asymmetric Catalysis. Chem. Rev. 2006, 106, 3561–3651. [Google Scholar] [CrossRef]
- Walli, A.; Dechert, S.; Bauer, M.; Demeshko, S.; Meyer, F. BOX Ligands in Biomimetic Copper-Mediated Dioxygen Activation: A Hemocyanin Model: BOX Ligands in Copper-Mediated Dioxygen Activation. Eur. J. Inorg. Chem. 2014, 2014, 4660–4676. [Google Scholar] [CrossRef]
- Dagorne, S.; Bellemin-Laponnaz, S.; Welter, R. Synthesis and Structure of Neutral and Cationic Aluminum Complexes Incorporating Bis(Oxazolinato) Ligands. Organometallics 2004, 23, 3053–3061. [Google Scholar] [CrossRef]
- Bechlars, B.; D’Alessandro, D.M.; Jenkins, D.M.; Iavarone, A.T.; Glover, S.D.; Kubiak, C.P.; Long, J.R. High-Spin Ground States via Electron Delocalization in Mixed-Valence Imidazolate-Bridged Divanadium Complexes. Nat. Chem. 2010, 2, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Newkome, G.R.; Garbis, S.J.; Majestic, V.K.; Fronczek, F.R.; Chiari, G. Chemistry of Heterocyclic Compounds. 61. Synthesis and Conformational Studies of Macrocycles Possessing 1,8- or 1,5-Naphthyridino Subunits Connected by Carbon-Oxygen Bridges. J. Org. Chem. 1981, 46, 833–839. [Google Scholar] [CrossRef]
- Boelrijk, A.E.M.; Neenan, T.X.; Reedijk, J. Ruthenium Complexes with Naphthyridine Ligands. Synthesis, Characterization and Catalytic Activity in Oxidation Reactions. J. Chem. Soc. Dalton Trans. 1997, 23, 4561–4570. [Google Scholar] [CrossRef]
- Davenport, T.C.; Tilley, T.D. Dinucleating Naphthyridine-Based Ligand for Assembly of Bridged Dicopper(I) Centers: Three-Center Two-Electron Bonding Involving an Acetonitrile Donor. Angew. Chem. Int. Ed. 2011, 50, 12205–12208. [Google Scholar] [CrossRef] [PubMed]
- Davenport, T.C.; Tilley, T.D. Dinuclear First-Row Transition Metal Complexes with a Naphthyridine-Based Dinucleating Ligand. Dalton Trans. 2015, 44, 12244–12255. [Google Scholar] [CrossRef]
- Mirica, L.M.; Ottenwaelder, X.; Stack, T.D. Structure and Spectroscopy of Copper Dioxygen Complexes. Chem. Rev. 2004, 104, 1013–1045. [Google Scholar] [CrossRef]
- Hatcher, L.Q.; Karlin, K.D. Oxidant Types in Copper–Dioxygen Chemistry: The Ligand Coordination Defines the Cun-O2 Structure and Subsequent Reactivity. J. Biol. Inorg. Chem. 2004, 9, 669–683. [Google Scholar] [CrossRef]
- Lucas, H.R.; Li, L.; Sarjeant, A.A.N.; Vance, M.A.; Solomon, E.I.; Karlin, K.D. Toluene and Ethylbenzene Aliphatic C−H Bond Oxidations Initiated by a Dicopper(II)-μ-1,2-Peroxo Complex. J. Am. Chem. Soc. 2009, 131, 3230–3245. [Google Scholar] [CrossRef] [Green Version]
- Solomon, E.I.; Heppner, D.E.; Johnston, E.M.; Ginsbach, J.W.; Cirera, J.; Quyyum, M.; Kieber-Emmons, M.T.; Kjaergaard, C.H.; Hadt, R.G.; Tian, L. Copper Active Sites in Biology. Chem. Rev. 2014, 114, 3659–3853. [Google Scholar] [CrossRef] [Green Version]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J.; Verschoor, G.C. Synthesis, Structure, and Spectroscopic Properties of Copper(II) Compounds Containing Nitrogen–Sulphur Donor Ligands; the Crystal and Molecular Structure of Aqua [1,7-Bis(N-Methylbenzimidazol-2′-Yl)-2,6-Dithiaheptane]Copper(II) Perchlorate. J. Chem. Soc. Dalton Trans. 1984, 7, 1349–1356. [Google Scholar] [CrossRef]
- Halvagar, M.R.; Solntsev, P.V.; Lim, H.; Hedman, B.; Hodgson, K.O.; Solomon, E.I.; Cramer, C.J.; Tolman, W.B. Hydroxo-Bridged Dicopper(II,III) and -(III,III) Complexes: Models for Putative Intermediates in Oxidation Catalysis. J. Am. Chem. Soc. 2014, 136, 7269–7272. [Google Scholar] [CrossRef]
- Kochem, A.; Gennarini, F.; Yemloul, M.; Orio, M.; Le Poul, N.; Rivière, E.; Giorgi, M.; Faure, B.; Le Mest, Y.; Réglier, M.; et al. Characterization of a Dinuclear Copper(II) Complex and Its Fleeting Mixed-Valent Copper(II)/Copper(III) Counterpart. ChemPlusChem 2017, 82, 615–624. [Google Scholar] [CrossRef]
- Thibon-Pourret, A.; Gennarini, F.; David, R.; Isaac, J.A.; Lopez, I.; Gellon, G.; Molton, F.; Wojcik, L.; Philouze, C.; Flot, D.; et al. Effect of Monoelectronic Oxidation of an Unsymmetrical Phenoxido-Hydroxido Bridged Dicopper(II) Complex. Inorg. Chem. 2018, 57, 12364–12375. [Google Scholar] [CrossRef] [PubMed]
- Mabbott, G.A. An Introduction to Cyclic Voltammetry. J. Chem. Educ. 1983, 60, 697. [Google Scholar] [CrossRef]
- Elgrishi, N.; Rountree, K.J.; McCarthy, B.D.; Rountree, E.S.; Eisenhart, T.T.; Dempsey, J.L. A Practical Beginner’s Guide to Cyclic Voltammetry. J. Chem. Educ. 2018, 95, 197–206. [Google Scholar] [CrossRef]
- Robin, M.B.; Day, P. Mixed Valence Chemistry-A Survey and Classification. In Adv Inorg Chem Radiochem; Elsevier: Amsterdam, The Netherlands, 1968; Volume 10, pp. 247–422. ISBN 978-0-12-023610-7. [Google Scholar]
- Stoll, S.; Schweiger, A. EasySpin, a Comprehensive Software Package for Spectral Simulation and Analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Brunschwig, B.S.; Creutz, C.; Sutin, N. Optical Transitions of Symmetrical Mixed-Valence Systems in the Class II–III Transition Regime. Chem. Soc. Rev. 2002, 31, 168–184. [Google Scholar] [CrossRef]
- Winter, R.F. Half-Wave Potential Splittings ΔE1/2 as a Measure of Electronic Coupling in Mixed-Valent Systems: Triumphs and Defeats. Organometallics 2014, 33, 4517–4536. [Google Scholar] [CrossRef]
- Warren, J.J.; Tronic, T.A.; Mayer, J.M. Thermochemistry of Proton-Coupled Electron Transfer Reagents and Its Implications. Chem. Rev. 2010, 110, 6961–7001. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A Short History of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isaac, J.A.; Gellon, G.; Molton, F.; Philouze, C.; Le Poul, N.; Belle, C.; Thibon-Pourret, A. Symmetrical and Unsymmetrical Dicopper Complexes Based on Bis-Oxazoline Units: Synthesis, Spectroscopic Properties and Reactivity. Inorganics 2023, 11, 332. https://doi.org/10.3390/inorganics11080332
Isaac JA, Gellon G, Molton F, Philouze C, Le Poul N, Belle C, Thibon-Pourret A. Symmetrical and Unsymmetrical Dicopper Complexes Based on Bis-Oxazoline Units: Synthesis, Spectroscopic Properties and Reactivity. Inorganics. 2023; 11(8):332. https://doi.org/10.3390/inorganics11080332
Chicago/Turabian StyleIsaac, James A., Gisèle Gellon, Florian Molton, Christian Philouze, Nicolas Le Poul, Catherine Belle, and Aurore Thibon-Pourret. 2023. "Symmetrical and Unsymmetrical Dicopper Complexes Based on Bis-Oxazoline Units: Synthesis, Spectroscopic Properties and Reactivity" Inorganics 11, no. 8: 332. https://doi.org/10.3390/inorganics11080332
APA StyleIsaac, J. A., Gellon, G., Molton, F., Philouze, C., Le Poul, N., Belle, C., & Thibon-Pourret, A. (2023). Symmetrical and Unsymmetrical Dicopper Complexes Based on Bis-Oxazoline Units: Synthesis, Spectroscopic Properties and Reactivity. Inorganics, 11(8), 332. https://doi.org/10.3390/inorganics11080332