








Probing the Electronic Structure of Dinuclear Carbon-Rich Complexes Containing an Octa-3,5-diene-1,7-diyndiyl Bridging Ligand



Abstract
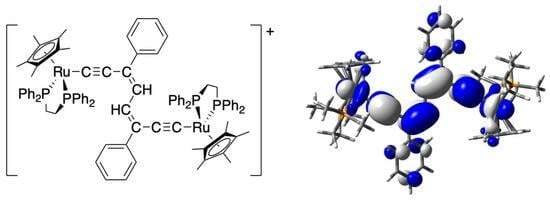
:
1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. Electronic Structure Calculations and Spectroscopy
3. Experimental Details
3.1. Synthesis of [{Ru(dppe)Cp*}2{μ-C=C=C(MeS-4-C6H4)–H2C–CH2–C(MeS-4-C6H4)=C=C}][PF6]2 ([3b][PF6]2)

3.2. Synthesis of [{RuCl(dppe)2}2{μ-C=C=C(MeS-4-C6H4)–H2C–CH2–C(MeS-4-C6H4)=C=C}][PF6]2 ([4b][PF6]2)

3.3. Synthesis of [{Ru(dppe)Cp*}2{μ-C≡CC(R)=HC–CH=C(R)C≡C}] (R = Ph (5a), MeS-4-C6H4 (5b)

3.4. Synthesis of [{RuCl(dppe)2}2{μ-C≡CC(MeS-4-C6H4)=HC–CH=C(MeS-4-C6H4)C≡C}] (6b)

3.5. Synthesis of [{RuCl(dppe)2}2{μ-C≡CC(Ph)=HC–CH=C(Ph)C≡C}]PF6 ([6a]PF6)

3.6. Preparation of [{RuCl(dppe)2}2{μ-C=C=C(Ph)–HC=CH–C(Ph)=C=C}][PF6]2 ([6a][PF6]2)

3.7. Preparation of trans-[Ru{C≡CC(=O)Ph}Cl(dppe)2] (2aox)

3.8. Preparation of trans-[Ru{C≡CC(=O)-4-MeS-C6H4}Cl(dppe)2] (2box)

4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aguirre-Etcheverry, P.; O’Hare, D. Electronic Communication through Unsaturated Hydrocarbon Bridges in Homobimetallic Organometallic Complexes. Chem. Rev. 2010, 110, 4839–4864. [Google Scholar] [CrossRef] [PubMed]
- Launay, J.P. Long-distance intervalence electron transfer. Chem. Soc. Rev. 2001, 30, 386–397. [Google Scholar] [CrossRef]
- Launay, J.P. Mixed-Valent Compounds and their Properties—Recent Developments. Eur. J. Inorg. Chem. 2020, 2020, 329–341. [Google Scholar] [CrossRef]
- Launay, J.P. An orbital approach of electron transfer in multisite systems. Implications for carbon-rich spacers. Polyhedron 2015, 86, 151–166. [Google Scholar] [CrossRef]
- Renz, M.; Theilacker, K.; Lambert, C.; Kaupp, M. A Reliable Quantum-Chemical Protocol for the Characterization of Organic Mixed-Valence Compounds. J. Am. Chem. Soc. 2009, 131, 16292–16302. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, E.C.; Brown, N.J.; Edge, R.; Helliwell, M.; Roberts, H.N.; Tuna, F.; Beeby, A.; Collison, D.; Low, P.J.; Whiteley, M.W. Orbital Symmetry Control of Electronic Coupling in a Symmetrical, All-Carbon-Bridged “Mixed Valence” Compound: Synthesis, Spectroscopy, and Electronic Structure of [{Mo(dppe)(η-C7H7)}2(μ-C4)]n+ (n = 0, 1, or 2). Organometallics 2012, 31, 157–169. [Google Scholar] [CrossRef]
- Pieslinger, G.E.; Aramburu-Troselj, B.M.; Cadranel, A.; Baraldo, L.M. Influence of the Electronic Configuration in the Properties of d6-d5 Mixed-Valence Complexes. Inorg. Chem. 2014, 53, 8221–8229. [Google Scholar] [CrossRef] [PubMed]
- Gückel, S.; Safari, P.; Ghazvini, S.M.B.H.; Hall, M.R.; Gluyas, J.B.G.; Kaupp, M.; Low, P.J. Iron Versus Ruthenium: Evidence for the Distinct Differences in the Electronic Structures of Hexa-1,3,5-triyn-1,6-diyl-bridged Complexes [Cp*(dppe)M}{μ-(C≡C)3}{M(dppe)Cp*}]+ (M = Fe, Ru). Organometallics 2021, 40, 346–357. [Google Scholar] [CrossRef]
- Launay, J.P. Electron transfer in molecular binuclear complexes and relation with electron transport through nanojunctions. Coord. Chem. Rev. 2013, 257, 1544–1554. [Google Scholar] [CrossRef]
- Zhang, J.; Ouyang, J.; Ye, Y.X.; Li, Z.; Lin, Q.; Chen, T.; Zhang, Z.; Xiang, S.C. Mixed-Valence Cobalt(II/III) Metal–Organic Framework for Ammonia Sensing with Naked-Eye Color Switching. ACS Appl. Mater. Interfaces 2018, 10, 27465–27471. [Google Scholar] [CrossRef]
- Corrente, G.A.; Cospito, S.; Capodilupo, A.L.; Beneduci, A. Mixed-Valence Compounds as a New Route for Electrochromic Devices with High Coloration Efficiency in the Whole Vis-NIR Region. Appl. Sci. 2020, 10, 8372. [Google Scholar] [CrossRef]
- Ma, X.; Pang, C.; Li, S.; Li, J.; Wang, M.; Xiong, Y.; Su, L.; Luo, J.; Xu, Z.; Lin, L. Biomimetic Synthesis of Ultrafine Mixed-Valence Metal–Organic Framework Nanowires and Their Application in Electrochemiluminescence Sensing. ACS Appl. Mater. Interfaces 2021, 13, 41987–41996. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Long, G.J.; Rebbouh, L.; Grandjean, F.; Beatty, A.M.; Fehlner, T.P. Properties of a Mixed-Valence (FeII)2(FeIII)2 Square Cell for Utilization in the Quantum Cellular Automata Paradigm for Molecular Electronics. J. Am. Chem. Soc. 2005, 127, 17819–17831. [Google Scholar] [CrossRef] [PubMed]
- Gluyas, J.B.G.; Gückel, S.; Kaupp, M.; Low, P.J. Rational Control of Conformational Distributions and Mixed-Valence Characteristics in Diruthenium Complexes. Chem. Eur. J. 2016, 22, 16138–16146. [Google Scholar] [CrossRef] [PubMed]
- Parthey, M.; Gluyas, J.B.G.; Fox, M.A.; Low, P.J.; Kaupp, M. Mixed-Valence Ruthenium Complexes Rotating through a Conformational Robin-Day Continuum. Chem. Eur. J. 2014, 20, 6895–6908. [Google Scholar] [CrossRef] [PubMed]
- Costuas, K.; Cador, O.; Justaud, F.; Le Stang, S.; Paul, F.; Monari, A.; Evangelisti, S.; Toupet, L.; Lapinte, C.; Halet, J.F. 3,5-Bis(ethynyl)pyridine and 2,6-Bis(ethynyl)pyridine Spanning Two Fe(Cp*)(dppe) Units: Role of the Nitrogen Atom on the Electronic and Magnetic Couplings. Inorg. Chem. 2011, 50, 12601–12622. [Google Scholar] [CrossRef]
- Fitzgerald, E.C.; Ladjarafi, A.; Brown, N.J.; Collison, D.; Costuas, K.; Edge, R.; Halet, J.F.; Justaud, F.; Low, P.J.; Meghezzi, H.; et al. Spectroscopic Evidence for Redox Isomerism in the 1,4-Diethynylbenzene-Bridged Heterobimetallic Cation [{Fe(dppe)Cp*}(μ-C≡CC6H4C≡C){Mo(dppe)(η-C7H7)}]PF6. Organometallics 2011, 30, 4180–4195. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, M.X.; Sun, C.F.; Xu, M.; Hartl, F.; Yin, J.; Yu, G.A.; Rao, L.; Liu, S.H. Diruthenium Complexes with Bridging Diethynyl Polyaromatic Ligands: Synthesis, Spectroelectrochemistry, and Theoretical Calculations. Organometallics 2015, 34, 3967–3978. [Google Scholar] [CrossRef]
- Bruce, M.; Low, P. Transition metal complexes containing all-carbon ligands. Adv. Organomet. Chem. 2004, 50, 179–444. [Google Scholar]
- Halet, J.F.; Lapinte, C. Charge delocalization vs localization in carbon-rich iron mixed-valence complexes: A subtle interplay between the carbon spacer and the (dppe)Cp*Fe organometallic electrophore. Coord. Chem. Rev. 2013, 257, 1584–1613. [Google Scholar] [CrossRef]
- Zheng, Q.L.; Schneider, J.F.; Amini, H.; Hampel, F.; Gladysz, J.A. Wire like diplatinum, triplatinum, and tetraplatinum complexes featuring X[PtC≡CC≡CC≡CC≡C]mPtX segments; iterative syntheses and functionalization for measurements of single molecule properties. Dalton Trans. 2019, 48, 5800–5816. [Google Scholar] [CrossRef] [PubMed]
- Ballmann, S.; Hieringer, W.; Secker, D.; Zheng, Q.L.; Gladysz, J.A.; Gorling, A.; Weber, H.B. Molecular Wires in Single-Molecule Junctions: Charge Transport and Vibrational Excitations. ChemPhysChem 2010, 11, 2256–2260. [Google Scholar] [CrossRef] [PubMed]
- Szafert, S.; Paul, F.; Meyer, W.E.; Gladysz, J.A.; Lapinte, C. Synthesis and reactivity of new heterodinuclear iron/rhenium Cx complexes of the formula (η5-C5Me5)Re(NO)(PPh3)(C≡C)n(η2-dppe)Fe(η5-C5Me5) (n = 3, 4): Redox properties and a dicobalt hexacarbonyl adduct. C. R. Chim. 2008, 11, 693–701. [Google Scholar] [CrossRef]
- Meyer, W.E.; Amoroso, A.J.; Horn, C.R.; Jaeger, M.; Gladysz, J.A. Synthesis and Oxidation of Dirhenium C4, C6, and C8 Complexes of the Formula (η5-C5Me5)Re(NO)(PR3)(C≡C)n(R3P)(ON)Re(η5-C5Me5) (R = 4-C6H4R’, c-C6H11): In Search of Dications and Radical Cations with Enhanced Stabilities. Organometallics 2001, 20, 1115–1127. [Google Scholar] [CrossRef]
- Gendron, F.; Burgun, A.; Skelton, B.W.; White, A.H.; Roisnel, T.; Bruce, M.I.; Halet, J.F.; Lapinte, C.; Costuas, K. Iron and Ruthenium sigma-Polyynyls of the General Formula [{M(dppe)Cp*}-(C≡C)n-R]0/+ (M = Fe, Ru): An Experimental and Theoretical Investigation. Organometallics 2012, 31, 6796–6811. [Google Scholar] [CrossRef]
- Bruce, M.I.; Costuas, K.; Davin, T.; Ellis, B.G.; Halet, J.F.; Lapinte, C.; Low, P.J.; Smith, M.E.; Skelton, B.W.; Toupet, L.; et al. Iron versus ruthenium: Dramatic changes in electronic structure result from replacement of one Fe by Ru in [{Cp*(dppe)Fe}-CC-CC-{Fe(dppe)Cp*}]n+) (n = 0, 1, 2). Organometallics 2005, 24, 3864–3881. [Google Scholar] [CrossRef]
- Gückel, S.; Gluyas, J.B.G.; El-Tarhuni, S.; Sobolev, A.N.; Whiteley, M.W.; Halet, J.-F.; Lapinte, C.; Kaupp, M.; Low, P.J. Iron versus Ruthenium: Clarifying the Electronic Differences between Prototypical Mixed-Valence Organometallic Butadiyndiyl Bridged Molecular Wires. Organometallics 2018, 37, 1432–1445. [Google Scholar] [CrossRef]
- Jiao, H.J.; Costuas, K.; Gladysz, J.A.; Halet, J.F.; Guillemot, M.; Toupet, L.; Paul, F.; Lapinte, C. Bonding and electronic structure in consanguineous and conjugal iron and rhenium sp carbon chain complexes [MC4M’]n+: Computational analyses of the effect of the metal. J. Am. Chem. Soc. 2003, 125, 9511–9522. [Google Scholar] [CrossRef]
- Paul, F.; Meyer, W.E.; Toupet, L.; Jiao, H.J.; Gladysz, J.A.; Lapinte, C. A “conjugal” consanguineous family of butadiynediyl-derived complexes: Synthesis and electronic ground states of neutral, radical cationic, and dicationic iron/rhenium C4 species. J. Am. Chem. Soc. 2000, 122, 9405–9414. [Google Scholar] [CrossRef]
- Roberts, H.N.; Brown, N.J.; Edge, R.; Fitzgerald, E.C.; Ta, Y.T.; Collison, D.; Low, P.J.; Whiteley, M.W. Synthesis, Redox Chemistry, and Electronic Structure of the Butadiynyl and Hexatriynyl Complexes [Mo{(C≡C)nC≡CR}(L2) (η-C7H7)]z+ (n = 1, 2; z = 0, 1; R = SiMe3, H.; L2 = 2,2′-bipyridine, Ph2PCH2CH2PPh2). Organometallics 2012, 31, 6322–6335. [Google Scholar] [CrossRef]
- Brown, N.J.; Collison, D.; Edge, R.; Fitzgerald, E.C.; Low, P.J.; Helliwell, M.; Ta, Y.T.; Whiteley, M.W. Metal-stabilised diynyl radicals: Structure and reactivity of [Mo(C≡C-C≡CSiMe3)L2(η-C7H7)]•+ (L2 = 2,2′-bipyridine or dppe). Chem. Commun. 2010, 46, 2253–2255. [Google Scholar] [CrossRef] [PubMed]
- Zhuravlev, F.; Gladysz, J.A. Electronic structure and chain-length effects in diplatimun polyynediyl complexes trans, trans-[(X)(R3P)2Pt(C≡C)nPt(PR3)2(X)]: A computational investigation. Chem. Eur. J. 2004, 10, 6510–6522. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.I.; Cole, M.L.; Ellis, B.G.; Gaudio, M.; Nicholson, B.K.; Parker, C.R.; Skelton, B.W.; White, A.H. The series of carbon-chain complexes {Ru(dppe)Cp*}2{μ-(C≡C)x} (x = 4–8, 11): Synthesis, structures, properties and some reactions. Polyhedron 2015, 86, 43–56. [Google Scholar] [CrossRef]
- Johnson, T.R.; Walton, D.R.M. Silylation as a Protective Method in Acetylene Chemistry—Polyyne Chain Extensions Using Reagents, Et3Si(C≡C)mH (m = 1, 2, 4) in Mixed Oxidative Couplings. Tetrahedron 1972, 28, 5221–5236. [Google Scholar] [CrossRef]
- Eastmond, R.; Walton, D.R.M.; Johnson, T.R. Silylation as a Protective Method for Terminal Alkynes in Oxidative Couplings—General Synthesis of Parent Polyynes H(C≡C)nH (n = 4–10, 12). Tetrahedron 1972, 28, 4601–4616. [Google Scholar] [CrossRef]
- Chalifoux, W.A.; Tykwinski, R.R. Synthesis of polyynes to model the sp-carbon allotrope carbyne. Nat. Chem. 2010, 2, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Burgun, A.; Gendron, F.; Schauer, P.A.; Skelton, B.W.; Low, P.J.; Costuas, K.; Halet, J.F.; Bruce, M.I.; Lapinte, C. Straightforward Access to Tetrametallic Complexes with a Square Array by Oxidative Dimerization of Organometallic Wires. Organometallics 2013, 32, 5015–5025. [Google Scholar] [CrossRef]
- Xia, H.P.; Jia, G.C. C5H5-bridged dimeric ruthenium complexes. Organometallics 1997, 16, 1–4. [Google Scholar] [CrossRef]
- Xia, H.P.; Yeung, R.C.Y.; Jia, G.C. Synthesis of symmetrical C5H5-bridged dimeric ruthenium complexes. Organometallics 1997, 16, 3557–3560. [Google Scholar] [CrossRef]
- Ribou, A.C.; Launay, J.P.; Sachtleben, M.L.; Li, H.; Spangler, C.W. Intervalence electron transfer in mixed valence diferrocenylpolyenes. Decay law of the metal-metal coupling with distance. Inorg. Chem. 1996, 35, 3735–3740. [Google Scholar] [CrossRef]
- Liu, S.H.; Chen, Y.H.; Wan, K.L.; Wen, T.B.; Zhou, Z.Y.; Lo, M.F.; Williams, I.D.; Jia, G.C. Synthesis and characterization of linear (CH)8-bridged bimetallic ruthenium complexes. Organometallics 2002, 21, 4984–4992. [Google Scholar] [CrossRef]
- Liu, S.H.; Xia, H.P.; Wen, T.B.; Zhou, Z.Y.; Jia, G.C. Synthesis and characterization of bimetallic ruthenium complexes with (CH)6 and related bridges. Organometallics 2003, 22, 737–743. [Google Scholar] [CrossRef]
- Yuan, P.; Liu, S.H.; Xiong, W.C.; Yin, J.; Yu, G.A.; Sung, H.Y.; Williams, I.D.; Jia, G.C. Synthesis and characterization of (CH=CH)3-bridged heterobimetallic ferrocene-ruthenium complexes. Organometallics 2005, 24, 1452–1457. [Google Scholar] [CrossRef]
- Sahnoune, H.; Mahias, V.; Halet, J.F.; Lapinte, C. 1,4-Dimethoxybutadienediyl-Bridged Diiron Compounds in Three Oxidation States: Evaluation of Delocalization Effects. Organometallics 2019, 38, 2724–2737. [Google Scholar] [CrossRef]
- Low, P.J. Twists and turns: Studies of the complexes and properties of bimetallic complexes featuring phenylene ethynylene and related bridging ligands. Coord. Chem. Rev. 2013, 257, 1507–1532. [Google Scholar] [CrossRef]
- Costuas, K.; Rigaut, S. Polynuclear carbon-rich organometallic complexes: Clarification of the role of the bridging ligand in the redox properties. Dalton Trans. 2011, 40, 5643–5658. [Google Scholar] [CrossRef] [PubMed]
- Haasler, M.; Maier, T.M.; Grotjahn, R.; Gückel, S.; Arbuznikov, A.V.; Kaupp, M. A Local Hybrid Functional with Wide Applicability and Good Balance between (De)Localization and Left–Right Correlation. J. Chem. Theory Comput. 2020, 16, 5645–5657. [Google Scholar] [CrossRef]
- Kaupp, M.; Karton, A.; Bischoff, F.A. [Al2O4]−, a Benchmark Gas-Phase Class II Mixed-Valence Radical Anion for the Evaluation of Quantum-Chemical Methods. J. Chem. Theory Comput. 2016, 12, 3796–3806. [Google Scholar] [CrossRef]
- Parthey, M.; Kaupp, M. Quantum-chemical insights into mixed-valence systems: Within and beyond the Robin-Day scheme. Chem. Soc. Rev. 2014, 43, 5067–5088. [Google Scholar] [CrossRef]
- Renz, M.; Kess, M.; Diedenhofen, M.; Klamt, A.; Kaupp, M. Reliable Quantum Chemical Prediction of the Localized/Delocalized Character of Organic Mixed-Valence Radical Anions. From Continuum Solvent Models to Direct-COSMO-RS. J. Chem. Theory Comput. 2012, 8, 4189–4203. [Google Scholar] [CrossRef]
- Renz, M.; Kaupp, M. Predicting the Localized/Delocalized Character of Mixed-Valence Diquinone Radical Anions. Toward the Right Answer for the Right Reason. J. Phys. Chem. A 2012, 116, 10629–10637. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Wierzbicki, I.; Cotic, A.; Cadranel, A. Photoinduced Intervalence Charge Transfers: Spectroscopic Tools to Study Fundamental Phenomena and Applications. ChemPhysChem 2022, 23, e202200384. [Google Scholar] [CrossRef] [PubMed]
- Pieslinger, G.E.; Ramirez-Wierzbicki, I.; Cadranel, A. The Excited-State Creutz-Taube Ion. Angew. Chem. Int. Ed. 2022, 61, e202211747. [Google Scholar] [CrossRef] [PubMed]
- Glover, S.D.; Goeltz, J.C.; Lear, B.J.; Kubiak, C.P. Inter- or intramolecular electron transfer between triruthenium clusters: We’ll cross that bridge when we come to it. Coord. Chem. Rev. 2010, 254, 331–345. [Google Scholar] [CrossRef]
- Safari, P.; Gückel, S.; Gluyas, J.B.G.; Moggach, S.A.; Kaupp, M.; Low, P.J. The Use of Bridging Ligand Substituents to Bias the Population of Localized and Delocalized Mixed-Valence Conformers in Solution. Chem. Eur. J. 2022, 28, e202200926. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.P.; Grotjahn, R.; Naher, M.; Ghazvini, S.; Mazzucato, D.M.; Korb, M.; Moggach, S.A.; Lambert, C.; Kaupp, M.; Low, P.J. Quantum Interference in Mixed-Valence Complexes: Tuning Electronic Coupling Through Substituent Effects. Angew. Chem. Int. Ed. 2022, 61, e202211000. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, M.J.; Frogley, B.J.; Hill, A.F.; Sharma, M.; Smith, M.K.; Ward, J.S. Hydrogenating an organometallic carbon chain: Buten-yn-diyl (CH=CHC≡C) as a missing link. Dalton Trans. 2019, 48, 16534–16554. [Google Scholar] [CrossRef]
- Wuttke, E.; Pevny, F.; Hervault, Y.M.; Norel, L.; Drescher, M.; Winter, R.F.; Rigaut, S. Fully Delocalized (Ethynyl)(vinyl)phenylene Bridged Triruthenium Complexes in up to Five Different Oxidation States. Inorg. Chem. 2012, 51, 1902–1915. [Google Scholar] [CrossRef]
- Olivier, C.; Costuas, K.; Choua, S.; Maurel, V.; Turek, P.; Saillard, J.Y.; Touchard, D.; Rigaut, S. “Chain-Like” Trimetallic Ruthenium Complexes with C7 Carbon-Rich Bridges: Experimental and Theoretical Investigations of Electronic Communication Tuning in Five Distinct Oxidation States. J. Am. Chem. Soc. 2010, 132, 5638–5651. [Google Scholar] [CrossRef]
- Vacher, A.; Benameur, A.; Ndiaye, C.M.; Touchard, D.; Rigaut, S. Linked C-7 Carbon-Rich Bridges: A New Dimension for Ruthenium Redox-Active Organometallics. Organometallics 2009, 28, 6096–6100. [Google Scholar] [CrossRef]
- Rigaut, S.; Olivier, C.; Costuas, K.; Choua, S.; Fadhel, O.; Massue, J.; Turek, P.; Saillard, J.Y.; Dixneuf, P.H.; Touchard, D. C7 and C9 carbon-rich bridges in diruthenium systems: Synthesis, spectroscopic, and theoretical investigations of different oxidation states. J. Am. Chem. Soc. 2006, 128, 5859–5876. [Google Scholar] [CrossRef] [PubMed]
- Rigaut, S.; Perruchon, J.; Guesmi, S.; Fave, C.; Touchard, D.; Dixneuf, P.H. Carbon-rich ruthenium complexes containing Bis(allenylidene) and mixed alkynyl-allenylidene bridges. Eur. J. Inorg. Chem. 2005, 2005, 447–460. [Google Scholar] [CrossRef]
- Hall, M.R.; Korb, M.; Moggach, S.A.; Low, P.J. Further Chemistry of Ruthenium Alkenyl Acetylide Complexes: Routes to Allenylidene Complexes via a Series of Electrophilic Addition Reactions. Organometallics 2020, 39, 2838–2853. [Google Scholar] [CrossRef]
- Hall, M.R.; Korb, M.; Moggach, S.A.; Low, P.J. Oxidative Coupling of Ruthenium Alkenyl Acetylide Complexes as a Route to Dinuclear Complexes Featuring Carbon-Rich Bridging Ligands. Organometallics 2022, 41, 2958–2973. [Google Scholar] [CrossRef]
- Li, Y.; Blacque, O.; Fox, T.; Luber, S.; Polit, W.; Winter, R.F.; Venkatesan, K.; Berke, H. Electronic communication in phosphine substituted bridged dirhenium complexes—Clarifying ambiguities raised by the redox non-innocence of the C4H2- and C4-bridges. Dalton Trans. 2016, 45, 5783–5799. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.I.; Ellis, B.G.; Low, P.J.; Skelton, B.W.; White, A.H. Syntheses, structures, and spectro-electrochemistry of {Cp*(PP)Ru}C≡CC≡C{Ru(PP)Cp*} (PP = dppm, dppe) and their mono- and dications. Organometallics 2003, 22, 3184–3198. [Google Scholar] [CrossRef]
- Hall, M.R.; Moggach, S.A.; Low, P.J. Syntheses and Structures of trans-bis(Alkenylacetylide) Ruthenium Complexes. Chem. Asian, J. 2021, 16, 3385–3403. [Google Scholar] [CrossRef]
- Kaupp, M.; Renz, M.; Parthey, M.; Stolte, M.; Wurthner, F.; Lambert, C. Computational and spectroscopic studies of organic mixed-valence compounds: Where is the charge? Phys. Chem. Chem. Phys. 2011, 13, 16973–16986. [Google Scholar] [CrossRef]
- Ladjarafi, A.; Costuas, K.; Meghezzi, H.; Halet, J.F. Electronic structure of modelized vs. real carbon-chain containing organometallic dinuclear complexes: Similarities and differences. J. Mol. Model. 2015, 21, 71. [Google Scholar] [CrossRef]
- Scott, A.P.; Radom, L. Harmonic vibrational frequencies: An evaluation of Hartree-Fock, Moller-Plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors. J. Phys. Chem. 1996, 100, 16502–16513. [Google Scholar] [CrossRef]
- Bruce, M.I.; Costuas, K.; Ellis, B.G.; Halet, J.F.; Low, P.J.; Moubaraki, B.; Murray, K.S.; Ouddai, N.; Perkins, G.J.; Skelton, B.W.; et al. Redox-active complexes containing group 8 metal centers linked by C2 bridges. Organometallics 2007, 26, 3735–3745. [Google Scholar] [CrossRef]
- Bruce, M.I.; Low, P.J.; Costuas, K.; Halet, J.F.; Best, S.P.; Heath, G.A. Oxidation chemistry of metal-bonded C4 chains: A combined chemical, spectroelectrochemical, and computational study. J. Am. Chem. Soc. 2000, 122, 1949–1962. [Google Scholar] [CrossRef]
- Parthey, M.; Gluyas, J.B.G.; Schauer, P.A.; Yufit, D.S.; Howard, J.A.K.; Kaupp, M.; Low, P.J. Refining the Interpretation of Near-Infrared Band Shapes in a Polyynediyl Molecular Wire. Chem. Eur. J. 2013, 19, 9780–9784. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.I.; Hall, B.C.; Low, P.J.; Smith, M.E.; Skelton, B.W.; White, A.H. Heterometallic complexes containing C4 chains. X-ray structures of {Cp(OC)3W}C≡CC≡C{Ir(CO)(PPh3)2(O2)} and cis-Pt{C≡C[W(CO)3Cp]}2(PEt3)2. Inorg Chim Acta 2000, 300, 633–644. [Google Scholar] [CrossRef]
- Brady, M.; Weng, W.Q.; Zhou, Y.L.; Seyler, J.W.; Amoroso, A.J.; Arif, A.M.; Bohme, M.; Frenking, G.; Gladysz, J.A. Consanguineous families of coordinated carbon: A ReC4Re assembly that is isolable in three oxidation states, including crystallographically characterized ReC≡CC≡CRe and Re+=C=C=C=C=Re+ adducts and a radical cation in which charge is delocalized between rhenium termini. J. Am. Chem. Soc. 1997, 119, 775–788. [Google Scholar]
- Weng, W.G.; Bartik, T.; Gladysz, J.A. Towards One-Dimensional Carbon Wires Connecting Single Metal Centers—A Cumulenic C5 Chain That Mediates Charge-Transfer between Rhenium and Manganese Termini. Angew. Chem. Int. Ed. 1994, 33, 2199–2202. [Google Scholar] [CrossRef]
- Seyler, J.W.; Weng, W.Q.; Zhou, Y.L.; Gladysz, J.A. An Isolable Organometallic Cation-Radical in Which a C4 Chain Conducts Charge between 2 Chiral and Configurationally Stable Rhenium Termini. Organometallics 1993, 12, 3802–3804. [Google Scholar] [CrossRef]
- Lear, B.J.; Chisholm, M.H. Oxalate Bridged MM (MM = Mo2, MoW, and W2) Quadruply Bonded Complexes as Test Beds for Current Mixed Valence Theory: Looking beyond the Intervalence Charge Transfer Transition. Inorg. Chem. 2009, 48, 10954–10971. [Google Scholar] [CrossRef]
- Szeghalmi, A.V.; Erdmann, M.; Engel, V.; Schmitt, M.; Amthor, S.; Kriegisch, V.; Noll, G.; Stahl, R.; Lambert, C.; Leusser, D.; et al. How delocalized is N,N,N′,N′-tetraphenylphianylenediamine radical cation? An experimental and theoretical study on the electronic and molecular structure. J. Am. Chem. Soc. 2004, 126, 7834–7845. [Google Scholar] [CrossRef]
- Badger, B.; Brocklehurst, B. Absorption spectra of dimer cations. Part 4—Theoretical considerations and dimer structure. Trans. Faraday Soc. 1970, 66, 2939–2947. [Google Scholar] [CrossRef]
- Connelly, N.G.; Geiger, W.E. Chemical redox agents for organometallic chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Puschmann, H.; Dolomanov, O. Olex(2): A Comprehensive Molecular Graphics Tool for Small-Molecule Structures. Acta Crystallogr. A 2006, 62, S246. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision, A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- O’Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Abinitio Effective Core Potentials for Molecular Calculations—Potentials for Main Group Elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Abinitio Effective Core Potentials for Molecular Calculations—Potentials for K to Au Including the Outermost Core Orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Abinitio Effective Core Potentials for Molecular Calculations—Potentials for the Transition-Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Petersson, G.A.; Allaham, M.A. A Complete Basis Set Model Chemistry. 2. Open-Shell Systems and the Total Energies of the 1st-Row Atoms. J. Chem. Phys. 1991, 94, 6081–6090. [Google Scholar] [CrossRef]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Allaham, M.A.; Shirley, W.A.; Mantzaris, J. A Complete Basis Set Model Chemistry.1. The Total Energies of Closed-Shell Atoms and Hydrides of the 1st-Row Elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Klamt, A. The COSMO and COSMO-RS solvation models. Wires Comput. Mol. Sci. 2011, 1, 699–709. [Google Scholar] [CrossRef]
- Klamt, A. Calculation of UV/Vis spectra in solution. J. Phys. Chem. 1996, 100, 3349–3353. [Google Scholar] [CrossRef]
- Martin, R.L. Natural transition orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]
- Krejcik, M.; Danek, M.; Hartl, F. Simple Construction of an Infrared Optically Transparent Thin-Layer Electrochemical-Cell—Applications to the Redox Reactions of Ferrocene, Mn2(CO)10 and Mn(CO)3(3,5-Di-Tert-Butyl-Catecholate)−. J. Electroanal. Chem. 1991, 317, 179–187. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| [6a]PF6 | |
|---|---|
| Ru1-P1 | 2.3729 (11) |
| Ru1-P2 | 2.3946 (11) |
| Ru1-P3 | 2.3803 (11) |
| Ru1-P4 | 2.3934 (11) |
| Ru1-Cl1 | 2.4591 (11) |
| Ru1-C1 | 1.958 (4) |
| C1-C2 | 1.217 (6) |
| C2-C3 | 1.400 (6) |
| C3-C4 | 1.401 (6) |
| C4-C4A | 1.403 (9) |
| C3-C5 | 1.490 (6) |
| Ru1-C1-C2 | 175.1 (4) |
| C1-C2-C3 | 175.4 (4) |
| C2-C3-C4 | 121.1 (4) |
| C3-C4-C4A | 124.3 (5) |
| C4-C3-C5 | 119.4 (4) |
| 2aox | 2box | |
|---|---|---|
| Ru1-P1 | 2.3884(5) | 2.3944(15) |
| Ru1-P2 | 2.3979(5) | 2.3529(14) |
| Ru1-P3 | 2.3407(5) | 2.3860(15) |
| Ru1-P4 | 2.3612(5) | 2.3856(15) |
| Ru1-Cl1 | 2.5067(5) | 2.5053(15) |
| Ru1-C1 | 1.986(2) | 1.972(6) |
| C1-C2 | 1.196(3) | 1.218(9) |
| C2-C3 | 1.428(3) | 1.424(9) |
| C3-C4 | 1.495(3) | 1.497(9) |
| O1-C3 | 1.245(3) | 1.240(8) |
| Ru1-C1-C2 | 175.58(17) | 178.2(5) |
| C1-C2-C3 | 167.6(2) | 166.0(7) |
| C2-C3-C4 | 119.45(18) | 119.3(6) |
| 1a† | [1a†]+ | 5a† | [5a†]+ | [5a†]2+ | |
|---|---|---|---|---|---|
| Ru1-P1 | 2.3137 | 2.3572 | 2.3226/2.3136 | 2.3260/2.3253 | 2.3446/2.3434 |
| Ru1-P2 | 2.3084 | 2.3393 | 2.3071/2.3075 | 2.3166/2.3176 | 2.3292/2.3287 |
| Ru1-C1 | 2.0293 | 1.9633 | 2.0278/2.0286 | 1.9661/1.9687 | 1.9114/1.9117 |
| C1-C2 | 1.2283 | 1.2372 | 1.2301/2.0286 | 1.2437/1.2433 | 1.2591/1.2587 |
| C2-C3 | 1.4307 | 1.4195 | 1.4250/1.4251 | 1.3884/1.3891 | 1.3565/1.3571 |
| C3-C4 | 1.3481 | 1.3546 | 1.3699/1.3702 | 1.4069/1.4065 | 1.4539/1.4538 |
| C4-C4′ | 1.4289 | 1.3895 | 1.3532 | ||
| Ru1-C1-C2 | 176.18 | 175.04 | 176.71/175.84 | 175.64/176.61 | 175.77/175.31 |
| C1-C2-C3 | 178.99 | 179.17 | 178.77/178.59 | 177.33/178.87 | 178.22/177.15 |
| C2-C3-C4 | 121.91 | 119.23 | 122.46/122.20 | 121.74/121.81 | 120.65/120.55 |
| C3-C4-C4′ | 125.01/122.20 | 124.83/124.90 | 124.41/124.23 |
| n-Hexane | Cyclohexane | THF | CH2Cl2 | PhCN | |
|---|---|---|---|---|---|
| 5a | 476 | 496 | 511 | 513 | 520 |
| 5b | 490 | 489 | 521 | 533 | 533 |
| 5c | 516 | 525 | 555 | 562 | 549 |
| 5d | 630 | 620 | 685 | 707 | 720 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hall, M.R.; Moggach, S.A.; Low, P.J. Probing the Electronic Structure of Dinuclear Carbon-Rich Complexes Containing an Octa-3,5-diene-1,7-diyndiyl Bridging Ligand. Inorganics 2024, 12, 20. https://doi.org/10.3390/inorganics12010020
Hall MR, Moggach SA, Low PJ. Probing the Electronic Structure of Dinuclear Carbon-Rich Complexes Containing an Octa-3,5-diene-1,7-diyndiyl Bridging Ligand. Inorganics. 2024; 12(1):20. https://doi.org/10.3390/inorganics12010020
Chicago/Turabian StyleHall, Michael R., Stephen A. Moggach, and Paul J. Low. 2024. "Probing the Electronic Structure of Dinuclear Carbon-Rich Complexes Containing an Octa-3,5-diene-1,7-diyndiyl Bridging Ligand" Inorganics 12, no. 1: 20. https://doi.org/10.3390/inorganics12010020
APA StyleHall, M. R., Moggach, S. A., & Low, P. J. (2024). Probing the Electronic Structure of Dinuclear Carbon-Rich Complexes Containing an Octa-3,5-diene-1,7-diyndiyl Bridging Ligand. Inorganics, 12(1), 20. https://doi.org/10.3390/inorganics12010020