
Tip of the Iceberg: A New Wave of Iron–Sulfur Cluster Proteins Found in Viruses



Abstract
:1. Iron–Sulfur Clusters as Essential Cofactors in Cells from Bacteria to Humans

2. Viral Genome Evolution: Harnessing the Host Machinery and FeS Clusters
3. FeS Clusters in Virally Encoded Proteins of Five Distinct Virus Families
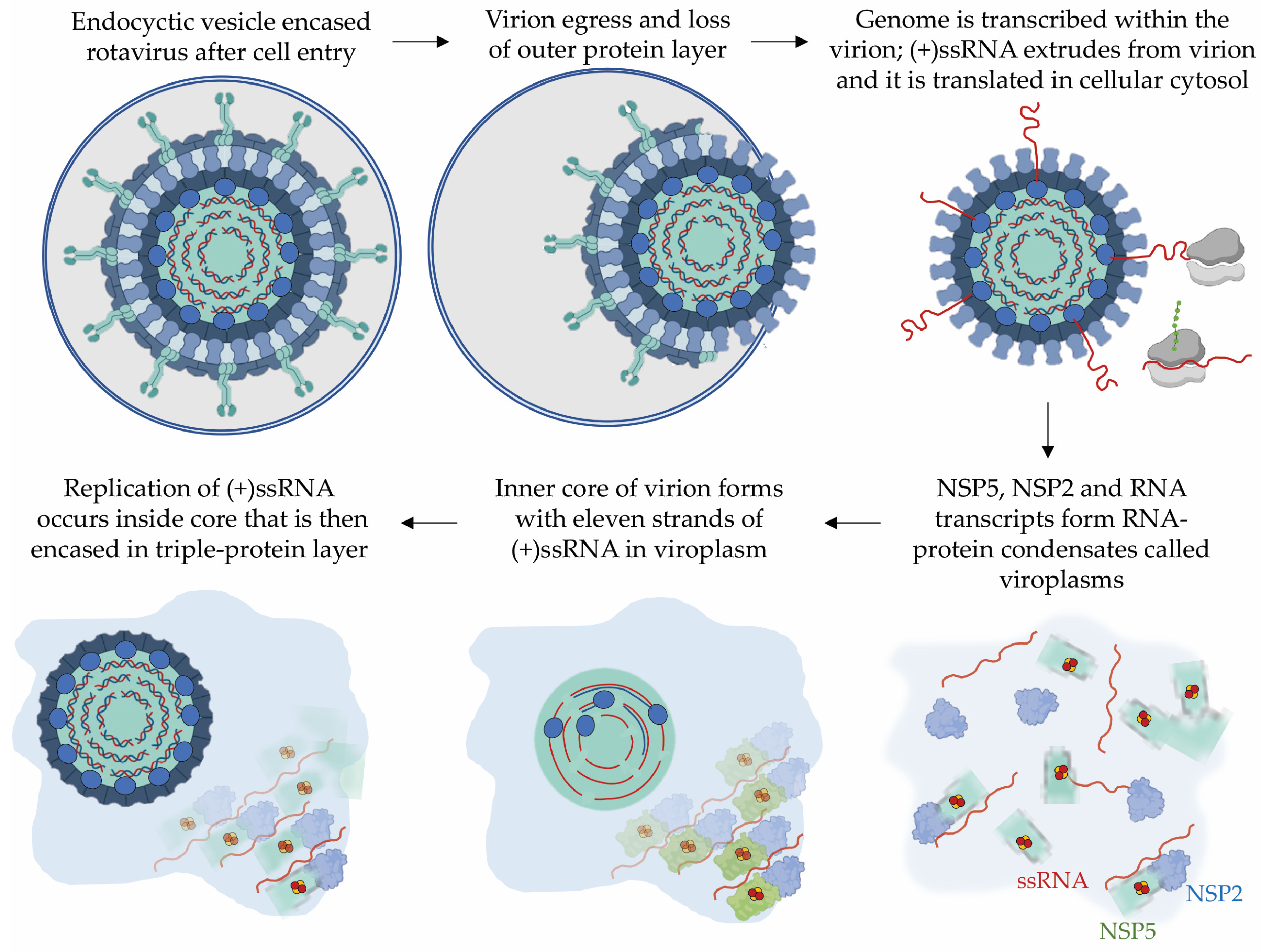
3.1. The Rotavirus Non-Structural Protein 5, NSP5
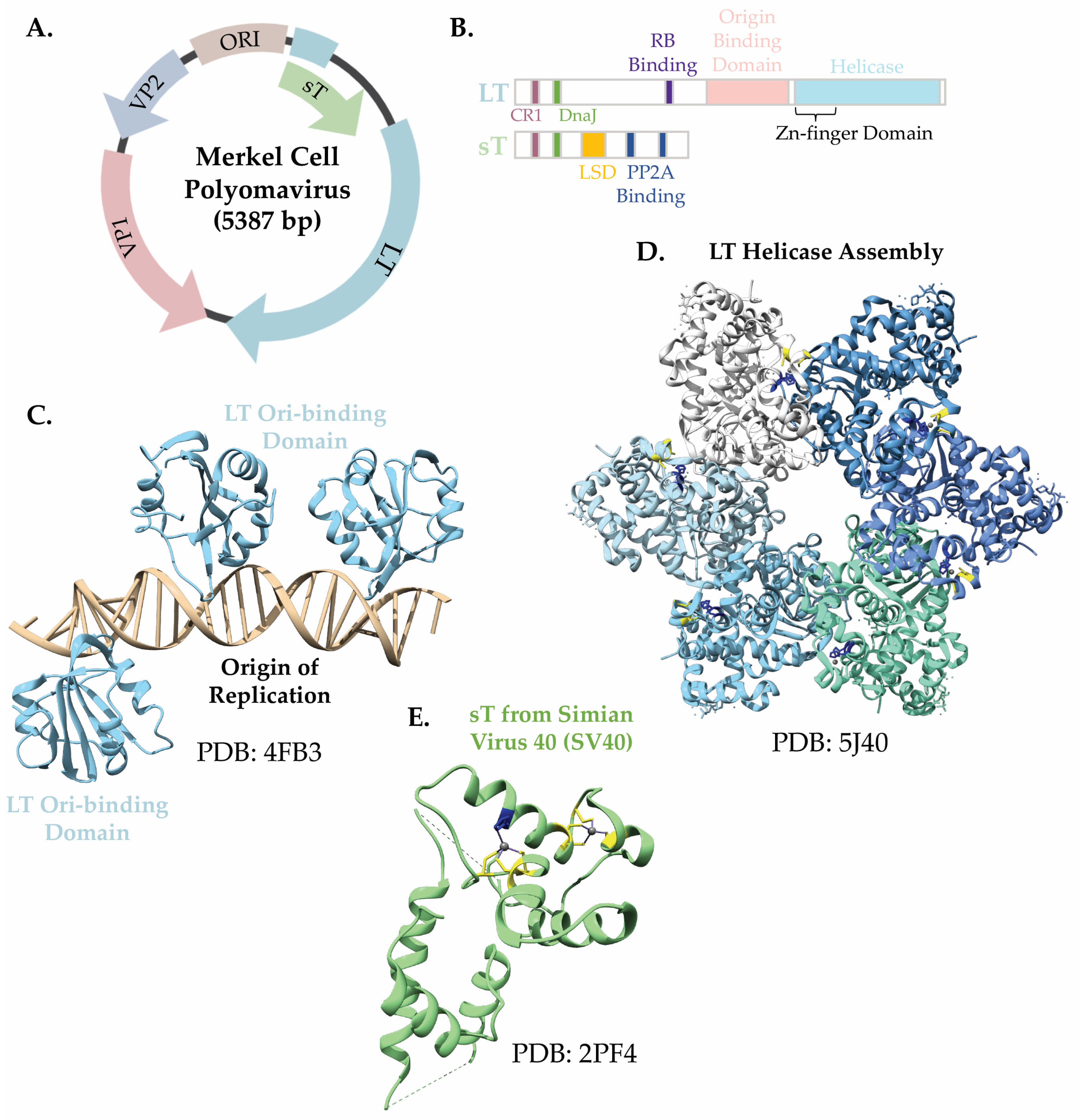
3.2. The Merkel Cell Polyomavirus Small T Antigen (sT)
3.3. The SARS-CoV-2 nsp12 and nsp13
3.4. The Hepatitis B HBx Protein
3.5. The Mimivirus GciS Protein
4. Conclusions and Perspectives
4.1. Drawing Similarities
4.2. So Why Do Viruses Utilize FeS Clusters?
4.3. A Note on Importance
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beinert, H.; Holm, R.H.; Munck, E. Iron-Sulfur Clusters: Nature’s Modular, Multipurpose Structures. Science 1997, 277, 653–659. [Google Scholar] [CrossRef]
- Sanchez, M.; Sabio, L.; Galvez, N.; Capdevila, M.; Dominguez-Vera, J.M. Iron chemistry at the service of life. IUBMB Life 2017, 69, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Letts, J.A.; Sazanov, L.A. Clarifying the supercomplex: The higher-order organization of the mitochondrial electron transport chain. Nat. Struct. Mol. Biol. 2017, 24, 800–808. [Google Scholar] [CrossRef]
- Rouault, T.A.; Maio, N. Biogenesis and functions of mammalian iron-sulfur proteins in the regulation of iron homeostasis and pivotal metabolic pathways. J. Biol. Chem. 2017, 292, 12744–12753. [Google Scholar] [CrossRef] [PubMed]
- Sickerman, N.S.; Ribbe, M.W.; Hu, Y. Nitrogenase Cofactor Assembly: An Elemental Inventory. ACC Chem. Res. 2017, 50, 2834–2841. [Google Scholar] [CrossRef]
- Bartels, P.L.; Stodola, J.L.; Burgers, P.M.J.; Barton, J.K. A Redox Role for the [4Fe4S] Cluster of Yeast DNA Polymerase delta. J. Am. Chem. Soc. 2017, 139, 18339–18348. [Google Scholar] [CrossRef] [PubMed]
- Barton, J.K.; Silva, R.M.B.; O’Brien, E. Redox Chemistry in the Genome: Emergence of the [4Fe4S] Cofactor in Repair and Replication. Annu. Rev. Biochem. 2019, 88, 163–190. [Google Scholar] [CrossRef]
- O’Brien, E.; Holt, M.E.; Thompson, M.K.; Salay, L.E.; Ehlinger, A.C.; Chazin, W.J.; Barton, J.K. The [4Fe4S] cluster of human DNA primase functions as a redox switch using DNA charge transport. Science 2017, 355, eaag1789. [Google Scholar] [CrossRef]
- Pinto, M.N.; Ter Beek, J.; Ekanger, L.A.; Johansson, E.; Barton, J.K. The [4Fe4S] Cluster of Yeast DNA Polymerase epsilon Is Redox Active and Can Undergo DNA-Mediated Signaling. J. Am. Chem. Soc. 2021, 143, 16147–16153. [Google Scholar] [CrossRef]
- Barthelme, D.; Dinkelaker, S.; Albers, S.V.; Londei, P.; Ermler, U.; Tampe, R. Ribosome recycling depends on a mechanistic link between the FeS cluster domain and a conformational switch of the twin-ATPase ABCE1. Proc. Natl. Acad. Sci. USA 2011, 108, 3228–3233. [Google Scholar] [CrossRef]
- Liu, G.; Sil, D.; Maio, N.; Tong, W.H.; Bollinger, J.M., Jr.; Krebs, C.; Rouault, T.A. Heme biosynthesis depends on previously unrecognized acquisition of iron-sulfur cofactors in human amino-levulinic acid dehydratase. Nat. Commun. 2020, 11, 6310. [Google Scholar] [CrossRef] [PubMed]
- Dailey, H.A.; Finnegan, M.G.; Johnson, M.K. Human ferrochelatase is an iron-sulfur protein. Biochemistry 1994, 33, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Booker, S.J.; Lloyd, C.T. Twenty Years of Radical SAM! The Genesis of the Superfamily. ACS Bio Med Chem Au 2022, 2, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shi, H.; Rajan, M.; Canarie, E.R.; Hong, S.; Simoneschi, D.; Pagano, M.; Bush, M.F.; Stoll, S.; Leibold, E.A.; et al. FBXL5 Regulates IRP2 Stability in Iron Homeostasis via an Oxygen-Responsive [2Fe2S] Cluster. Mol. Cell 2020, 78, 31–41.e5. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A.; Maio, N. How Oxidation of a Unique Iron-Sulfur Cluster in FBXL5 Regulates IRP2 Levels and Promotes Regulation of Iron Metabolism Proteins. Mol. Cell 2020, 78, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Holm, R.H.; Lo, W. Structural Conversions of Synthetic and Protein-Bound Iron-Sulfur Clusters. Chem. Rev. 2016, 116, 13685–13713. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, Y.; Aptekmann, A.A.; Mahlich, Y.; Cook, L.; Senn, S.; Miller, M.; Nanda, V.; Ferreiro, D.; Falkowski, P. Quantifying structural relationships of metal-binding sites suggests origins of biological electron transfer. Sci. Adv. 2022, 8, eabj3984. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.A. Iron-sulphur clusters and the problem with oxygen. Mol. Microbiol. 2006, 59, 1073–1082. [Google Scholar] [CrossRef]
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours-implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef]
- Thomas, K.A. Angiogenesis. In Encyclopedia of Cell Biology; Academic Press: Cambridge, MA, USA, 2016; pp. 102–116. ISBN 9780123947963. [Google Scholar]
- Pandelia, M.E.; Lanz, N.D.; Booker, S.J.; Krebs, C. Mossbauer spectroscopy of Fe/S proteins. Biochim. Biophys. Acta 2015, 1853, 1395–1405. [Google Scholar] [CrossRef]
- Cutsail, G.E., 3rd; Telser, J.; Hoffman, B.M. Advanced paramagnetic resonance spectroscopies of iron-sulfur proteins: Electron nuclear double resonance (ENDOR) and electron spin echo envelope modulation (ESEEM). Biochim. Biophys. Acta 2015, 1853, 1370–1394. [Google Scholar] [CrossRef] [PubMed]
- Crack, J.C.; Le Brun, N.E. Native Mass Spectrometry of Iron-Sulfur Proteins. In Fe-S Proteins: Methods and Protocols; Dos Santos, P.C., Ed.; Springer: New York, NY, USA, 2021; pp. 231–258. [Google Scholar] [CrossRef]
- Maio, N.; Rouault, T.A. Outlining the Complex Pathway of Mammalian Fe-S Cluster Biogenesis. Trends Biochem. Sci. 2020, 45, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Genova, M.L.; Lenaz, G. Functional role of mitochondrial respiratory supercomplexes. Biochim. Biophys. Acta 2014, 1837, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Carroll, B.L.; Zahn, K.E.; Hanley, J.P.; Wallace, S.S.; Dragon, J.A.; Doublie, S. Caught in motion: Human NTHL1 undergoes interdomain rearrangement necessary for catalysis. Nucleic Acids Res. 2021, 49, 13165–13178. [Google Scholar] [CrossRef] [PubMed]
- Maio, N.; Rouault, T.A. Mammalian iron sulfur cluster biogenesis: From assembly to delivery to recipient proteins with a focus on novel targets of the chaperone and co-chaperone proteins. IUBMB Life 2022, 74, 684–704. [Google Scholar] [CrossRef] [PubMed]
- Ciesielski, S.J.; Schilke, B.A.; Osipiuk, J.; Bigelow, L.; Mulligan, R.; Majewska, J.; Joachimiak, A.; Marszalek, J.; Craig, E.A.; Dutkiewicz, R. Interaction of J-protein co-chaperone Jac1 with Fe-S scaffold Isu is indispensable in vivo and conserved in evolution. J. Mol. Biol. 2012, 417, 1–12. [Google Scholar] [CrossRef]
- Chakrabbarti, M.; Lindahl, P.A. The utility of Mossbauer spectroscopy in eukaryotic cell biology and animal physiology. In Iron-Sulfur Clusters in Chemistry and Biology; Rouault, T.A., Ed.; Walter de Gruyter: Berlin, Germany; Boston, MA, USA, 2014; pp. 49–71. [Google Scholar]
- Maio, N.; Rouault, T.A. Iron-sulfur cluster biogenesis in mammalian cells: New insights into the molecular mechanisms of cluster delivery. Biochim. Biophys. Acta 2015, 1853, 1493–1512. [Google Scholar] [CrossRef]
- Li, K.; Tong, W.-H.; Hughes, R.M.; Rouault, T.A. Roles of the Mammalian Cytosolic Cysteine Desulfurase, ISCS, and Scaffold Protein, ISCU, in Iron-Sulfur Cluster Assembly. J. Biol. Chem. 2006, 281, 12344–12351. [Google Scholar] [CrossRef]
- Tong, W.H.; Rouault, T.A. Distinct iron-sulfur cluster assembly complexes exist in the cytosol and mitochondria of human cells. EMBO 2000, 19, 5692–5700. [Google Scholar] [CrossRef]
- Tong, W.H.; Rouault, T.A. Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron-sulfur cluster biogenesis and iron homeostasis. Cell Metab. 2006, 3, 199–210. [Google Scholar] [CrossRef]
- Srour, B.; Gervason, S.; Hoock, M.H.; Monfort, B.; Want, K.; Larkem, D.; Trabelsi, N.; Landrot, G.; Zitolo, A.; Fonda, E.; et al. Iron Insertion at the Assembly Site of the ISCU Scaffold Protein Is a Conserved Process Initiating Fe-S Cluster Biosynthesis. J. Am. Chem. Soc. 2022, 144, 17496–17515. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Proteau, A.; Villarroya, M.; Moukadiri, I.; Zhang, L.; Trempe, J.F.; Matte, A.; Armengod, M.E.; Cygler, M. Structural basis for Fe-S cluster assembly and tRNA thiolation mediated by IscS protein-protein interactions. PLoS Biol. 2010, 8, e1000354. [Google Scholar] [CrossRef] [PubMed]
- Adam, A.C.; Bornhovd, C.; Prokisch, H.; Neupert, W.; Hell, K. The Nfs1 interacting protein Isd11 has an essential role in Fe/S cluster biogenesis in mitochondria. EMBO J. 2006, 25, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ghosh, M.C.; Tong, W.H.; Rouault, T.A. Human ISD11 is essential for both iron-sulfur cluster assembly and maintenance of normal cellular iron homeostasis. Hum. Mol. Genet. 2009, 18, 3014–3025. [Google Scholar] [CrossRef]
- Terali, K.; Beavil, R.L.; Pickersgill, R.W.; van der Giezen, M. The effect of the adaptor protein Isd11 on the quaternary structure of the eukaryotic cysteine desulphurase Nfs1. Biochem. Biophys. Res. Commun. 2013, 440, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.J.; Frey, A.G.; Palenchar, D.J.; Achar, S.; Bullough, K.Z.; Vashisht, A.; Wohlschlegel, J.A.; Philpott, C.C. A PCBP1-BolA2 chaperone complex delivers iron for cytosolic [2Fe-2S] cluster assembly. Nat. Chem. Biol. 2019, 15, 872–881. [Google Scholar] [CrossRef]
- Shi, H.; Bencze, K.Z.; Stemmler, T.L.; Philpott, C.C. A cytosolic iron chaperone that delivers iron to ferritin. Science 2008, 320, 1207–1210. [Google Scholar]
- Nandal, A.; Ruiz, J.C.; Subramanian, P.; Ghimire-Rijal, S.; Sinnamon, R.A.; Stemmler, T.L.; Bruick, R.K.; Philpott, C.C. Activation of the HIF prolyl hydroxylase by the iron chaperones PCBP1 and PCBP2. Cell Metab. 2011, 14, 647–657. [Google Scholar] [CrossRef]
- Leidgens, S.; Bullough, K.Z.; Shi, H.; Li, F.; Shakoury-Elizeh, M.; Yabe, T.; Subramanian, P.; Hsu, E.; Natarajan, N.; Nandal, A.; et al. Each member of the poly-r(C)-binding protein 1 (PCBP) family exhibits iron chaperone activity toward ferritin. J. Biol. Chem. 2013, 288, 17791–17802. [Google Scholar] [CrossRef]
- Frey, A.G.; Nandal, A.; Park, J.H.; Smith, P.M.; Yabe, T.; Ryu, M.S.; Ghosh, M.C.; Lee, J.; Rouault, T.A.; Park, M.H.; et al. Iron chaperones PCBP1 and PCBP2 mediate the metallation of the dinuclear iron enzyme deoxyhypusine hydroxylase. Proc. Natl. Acad. Sci. USA 2014, 111, 8031–8036. [Google Scholar] [CrossRef]
- Boniecki, M.T.; Freibert, S.A.; Muhlenhoff, U.; Lill, R.; Cygler, M. Structure and functional dynamics of the mitochondrial Fe/S cluster synthesis complex. Nat. Commun. 2017, 8, 1287. [Google Scholar] [CrossRef] [PubMed]
- Cai, K.; Tonelli, M.; Frederick, R.O.; Markley, J.L. Human Mitochondrial Ferredoxin 1 (FDX1) and Ferredoxin 2 (FDX2) Both Bind Cysteine Desulfurase and Donate Electrons for Iron-Sulfur Cluster Biosynthesis. Biochemistry 2017, 56, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Chandramouli, K.; Unciuleac, M.C.; Naik, S.; Dean, D.R.; Huynh, B.H.; Johnson, M.K. Formation and properties of [4Fe-4S] clusters on the IscU scaffold protein. Biochemistry 2007, 46, 6804–6811. [Google Scholar] [CrossRef] [PubMed]
- Tse, E.C.M.; Zwang, T.J.; Barton, J.K. The Oxidation State of [4Fe4S] Clusters Modulates the DNA-Binding Affinity of DNA Repair Proteins. J. Am. Chem. Soc. 2017, 139, 12784–12792. [Google Scholar] [CrossRef] [PubMed]
- Voisine, C.; Cheng, Y.C.; Ohlson, M.; Schilke, B.; Hoff, K.; Beinert, H.; Marszalek, J.; Craig, E.A. Jac1, a mitochondrial J-type chaperone, is involved in the biogenesis of Fe/S clusters in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2001, 98, 1483–1488. [Google Scholar] [CrossRef] [PubMed]
- Cupp-Vickery, J.R.; Peterson, J.C.; Ta, D.T.; Vickery, L.E. Crystal structure of the molecular chaperone HscA substrate binding domain complexed with the IscU recognition peptide ELPPVKIHC. J. Mol. Biol. 2004, 342, 1265–1278. [Google Scholar] [CrossRef]
- Maio, N.; Singh, A.; Uhrigshardt, H.; Saxena, N.; Tong, W.H.; Rouault, T.A. Cochaperone binding to LYR motifs confers specificity of iron sulfur cluster delivery. Cell Metab. 2014, 19, 445–457. [Google Scholar] [CrossRef]
- Marszalek, J.; Craig, E.A. Interaction of client-the scaffold on which FeS clusters are build-with J-domain protein Hsc20 and its evolving Hsp70 partners. Front. Mol. Biosci. 2022, 9, 1034453. [Google Scholar] [CrossRef]
- Maio, N.; Kim, K.S.; Singh, A.; Rouault, T.A. A Single Adaptable Cochaperone-Scaffold Complex Delivers Nascent Iron-Sulfur Clusters to Mammalian Respiratory Chain Complexes I–III. Cell Metab. 2017, 25, 945–953.e6. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Craig, E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010, 11, 579–592. [Google Scholar] [CrossRef]
- Sahi, C.; Craig, E.A. Network of general and specialty J protein chaperones of the yeast cytosol. Proc. Natl. Acad. Sci. USA 2007, 104, 7163–7168. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, F.; Iametti, S.; Morleo, A.; Ta, D.; Vickery, L.E. Facilitated transfer of IscU-[2Fe2S] clusters by chaperone-mediated ligand exchange. Biochemistry 2011, 50, 9641–9650. [Google Scholar] [CrossRef] [PubMed]
- Dutkiewicz, R.; Schilke, B.; Cheng, S.; Knieszner, H.; Craig, E.A.; Marszalek, J. Sequence-specific interaction between mitochondrial Fe-S scaffold protein Isu and Hsp70 Ssq1 is essential for their in vivo function. J. Biol. Chem. 2004, 279, 29167–29174. [Google Scholar] [CrossRef]
- Hoff, K.G.; Cupp-Vickery, J.R.; Vickery, L.E. Contributions of the LPPVK motif of the iron-sulfur template protein IscU to interactions with the Hsc66-Hsc20 chaperone system. J. Biol. Chem. 2003, 278, 37582–37589. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A. Mammalian iron-sulphur proteins: Novel insights into biogenesis and function. Nat. Rev. Mol. Cell Biol. 2015, 16, 45–55. [Google Scholar] [CrossRef]
- Gari, K.; Ortiz, A.M.L.; Borel, V.; Flynn, H.; Skehel, J.M.; Boulton, S.J. MMS19 links cytoplasmic iron-sulfur cluster assembly to DNA metabolism. Science 2012, 337, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Stehling, O.; Vashisht, A.A.; Mascarenhas, J.; Jonsson, Z.O.; Sharma, T.; Netz, D.J.; Pierik, A.J.; Wohlschlegel, J.A.; Lill, R. MMS19 assembles iron-sulfur proteins required for DNA metabolism and genomic integrity. Science 2012, 337, 195–199. [Google Scholar] [CrossRef]
- Kim, K.S.; Maio, N.; Singh, A.; Rouault, T.A. Cytosolic HSC20 integrates de novo iron-sulfur cluster biogenesis with the CIAO1-mediated transfer to recipients. Hum. Mol. Genet. 2018, 27, 837–852. [Google Scholar] [CrossRef]
- Lill, R.; Freibert, S.A. Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis. Annu. Rev. Biochem. 2020, 89, 471–499. [Google Scholar] [CrossRef]
- Kassube, S.A.; Thoma, N.H. Structural insights into Fe-S protein biogenesis by the CIA targeting complex. Nat. Struct. Mol. Biol. 2020, 27, 735–742. [Google Scholar] [CrossRef]
- Ye, H.; Rouault, T.A. Human iron-sulfur cluster assembly, cellular iron homeostasis, and disease. Biochemistry 2010, 49, 4945–4956. [Google Scholar] [CrossRef] [PubMed]
- Maio, N.; Orbach, R.; Zaharieva, I.; Töpf, A.; Donkervoort, S.; Munot, P.; Mueller, J.; Willis, T.; Verma, S.; Peric, S.; et al. Loss of Function of the Cytoplasmic Fe-S Assembly Protein CIAO1 Causes a Neuromuscular Disorder with Compromise of Nucleocytoplasmic Fe-S Enzymes. medRxiv 2023. [Google Scholar] [CrossRef]
- Crick, F.H.C.; Watson, J.D. Structure of Small Viruses. Nature 1956, 177, 473–475. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.I.; Kazlauskas, D.; Iranzo, J.; Lucia-Sanz, A.; Kuhn, J.H.; Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origins and Evolution of the Global RNA Virome. mBio 2018, 9, e02329-18. [Google Scholar] [CrossRef]
- Twarock, R.; Luque, A. Structural puzzles in virology solved with an overarching icosahedral design principle. Nat. Commun. 2019, 10, 4414. [Google Scholar] [CrossRef]
- Colson, P.; De Lamballerie, X.; Yutin, N.; Asgari, S.; Bigot, Y.; Bideshi, D.K.; Cheng, X.W.; Federici, B.A.; Van Etten, J.L.; Koonin, E.V.; et al. “Megavirales”, a proposed new order for eukaryotic nucleocytoplasmic large DNA viruses. Arch. Virol. 2013, 158, 2517–2521. [Google Scholar] [CrossRef]
- Raoult, D.; Audic, S.; Robert, C.; Abergel, C.; Renesto, P.; Ogata, H.; Scola, B.; Suzan, M.; Claverie, J. The 1.2-Mb Genome Sequence of Mimivirus. Science 2004, 306, 1344–1350. [Google Scholar] [CrossRef]
- Martin, D.; Charpilienne, A.; Parent, A.; Boussac, A.; D’Autreaux, B.; Poupon, J.; Poncet, D. The rotavirus nonstructural protein NSP5 coordinates a [2Fe-2S] iron-sulfur cluster that modulates interaction to RNA. FASEB J. 2013, 27, 1074–1083. [Google Scholar] [CrossRef]
- Ozsari, T.; Bora, G.; Kaya, B.; Yakut, K. The Prevalence of Rotavirus and Adenovirus in the Childhood Gastroenteritis. Jundishapur J. Microbiol. 2016, 9, e34867. [Google Scholar] [CrossRef] [PubMed]
- Papa, G.; Borodavka, A.; Desselberger, U. Viroplasms: Assembly and Functions of Rotavirus Replication Factories. Viruses 2021, 13, 1349. [Google Scholar] [CrossRef]
- Martin, D.; Ouldali, M.; Menetrey, J.; Poncet, D. Structural organisation of the rotavirus nonstructural protein NSP5. J. Mol. Biol. 2011, 413, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Fabbretti, E.; Afrikanova, I.; Vascotto, F.; Burrone, O.R. Two non-structural rotavirus proteins, NSP2 and NSP5, form viroplasm-like structures in vivo. J. Gen. Virol. 1999, 80, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Papa, G.; Venditti, L.; Arnoldi, F.; Schraner, E.M.; Potgieter, C.; Borodavka, A.; Eichwald, C.; Burrone, O.R. Recombinant Rotaviruses Rescued by Reverse Genetics Reveal the Role of NSP5 Hyperphosphorylation in the Assembly of Viral Factories. J. Virol. 2019, 94, e01110-19. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.H.; Wang, R.; Nakamaru-Ogiso, E.; Knight, S.A.; Buck, C.B.; You, J. The Oncogenic Small Tumor Antigen of Merkel Cell Polyomavirus Is an Iron-Sulfur Cluster Protein That Enhances Viral DNA Replication. J. Virol. 2016, 90, 1544–1556. [Google Scholar] [CrossRef]
- Wendzicki, J.A.; Moore, P.S.; Chang, Y. Large T and small T antigens of Merkel cell polyomavirus. Curr. Opin. Virol. 2015, 11, 38–43. [Google Scholar] [CrossRef]
- Kwun, H.J.; Guastafierro, A.; Shuda, M.; Meinke, G.; Bohm, A.; Moore, P.S.; Chang, Y. The minimum replication origin of merkel cell polyomavirus has a unique large T-antigen loading architecture and requires small T-antigen expression for optimal replication. J. Virol. 2009, 83, 12118–12128. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.C.; Stang, A.; DeCaprio, J.A.; Cerroni, L.; Lebbe, C.; Veness, M.; Nghiem, P. Merkel cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17077. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal Integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef]
- Rapchak, K.; Yagobian, S.D.; Moore, J.; Khattri, M.; Shuda, M. Merkel cell polyomavirus small T antigen is a viral transcription activator that is essential for viral genome maintenance. PLoS Pathog. 2022, 18, e1011039. [Google Scholar] [CrossRef]
- Kwun, H.J.; Shuda, M.; Feng, H.; Camacho, C.J.; Moore, P.S.; Chang, Y. Merkel cell polyomavirus small T antigen controls viral replication and oncoprotein expression by targeting the cellular ubiquitin ligase SCFFbw7. Cell Host Microbe 2013, 14, 125–135. [Google Scholar] [CrossRef]
- Maio, N.; Lafont, B.A.P.; Sil, D.; Li, Y.; Bollinger, J.M., Jr.; Krebs, C.; Pierson, T.C.; Linehan, W.M.; Rouault, T.A. Fe-S cofactors in the SARS-CoV-2 RNA-dependent RNA polymerase are potential antiviral targets. Science 2021, 373, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Maio, N.; Raza, M.K.; Li, Y.; Zhang, D.; Bollinger, J.M., Jr.; Krebs, C.; Rouault, T.A. An iron-sulfur cluster in zinc-binding domain of the SARS-CoV-2 helicase modulates its RNA binding and unwinding activities. Proc. Natl. Acad. Sci. USA 2023, 120, e2303860120. [Google Scholar] [CrossRef] [PubMed]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020, 584, 154–156. [Google Scholar] [CrossRef]
- Yan, L.; Ge, J.; Zheng, L.; Zhang, Y.; Gao, Y.; Wang, T.; Huang, Y.; Yang, Y.; Gao, S.; Li, M.; et al. Cryo-EM Structure of an Extended SARS-CoV-2 Replication and Transcription Complex Reveals an Intermediate State in Cap Synthesis. Cell 2021, 184, 184–193.e10. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, Q.; Malone, B.; Llewellyn, E.; Pechersky, Y.; Maruthi, K.; Eng, E.T.; Perry, J.K.; Campbell, E.A.; Shaw, D.E.; et al. Ensemble cryo-EM reveals conformational states of the nsp13 helicase in the SARS-CoV-2 helicase replication-transcription complex. Nat. Struct. Mol. Biol. 2022, 29, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Brant, A.C.; Tian, W.; Majerciak, V.; Yang, W.; Zheng, Z.M. SARS-CoV-2: From its discovery to genome structure, transcription, and replication. Cell Biosci. 2021, 11, 136. [Google Scholar] [CrossRef]
- Long, S. SARS-CoV-2 Subgenomic RNAs: Characterization, Utility, and Perspectives. Viruses 2021, 13, 1923. [Google Scholar] [CrossRef]
- Malone, B.; Campbell, E.A.; Darst, S.A. CoV-er all the bases: Structural perspectives of SARS-CoV-2 RNA synthesis. Enzymes 2021, 49, 1–37. [Google Scholar] [CrossRef]
- Newman, J.A.; Douangamath, A.; Yadzani, S.; Yosaatmadja, Y.; Aimon, A.; Brandao-Neto, J.; Dunnett, L.; Gorrie-Stone, T.; Skyner, R.; Fearon, D.; et al. Structure, mechanism and crystallographic fragment screening of the SARS-CoV-2 NSP13 helicase. Nat. Commun. 2021, 12, 4848. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Huang, Y.; Yuen, K.Y. Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. 2009, 234, 1117–1127. [Google Scholar] [CrossRef]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol. Cell 2020, 79, 710–727. [Google Scholar] [CrossRef] [PubMed]
- Belshaw, R.; Pybus, O.G.; Rambaut, A. The evolution of genome compression and genomic novelty in RNA viruses. Genome Res. 2007, 17, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Gribble, J.; Stevens, L.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Pruijssers, A.J.; Routh, A.L.; Denison, M.R. The coronavirus proofreading exoribonuclease mediates extensive viral recombination. PLoS Pathog. 2021, 17, e1009226. [Google Scholar] [CrossRef]
- Yan, L.; Yang, Y.; Li, M.; Zhang, Y.; Zheng, L.; Ge, J.; Huang, Y.C.; Liu, Z.; Wang, T.; Gao, S.; et al. Coupling of N7-methyltransferase and 3′-5′ exoribonuclease with SARS-CoV-2 polymerase reveals mechanisms for capping and proofreading. Cell 2021, 184, 3474–3485.e11. [Google Scholar] [CrossRef]
- Dos Ramos, F.; Carrasco, M.; Doyle, T.; Brierley, I. Programmed-1 ribosomal frameshifting in the SARS coronavirus. Biochem. Soc. Trans. 2004, 32, 1081–1083. [Google Scholar] [CrossRef] [PubMed]
- Thiel, V.; Ivanov, K.A.; Putics, A.; Hertzig, T.; Schelle, B.; Bayer, S.; Weissbrich, B.; Snijder, E.J.; Rabenau, H.; Doerr, H.W.; et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003, 84, 2305–2315. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Lokugamage, K.G.; Makino, S. Viral and Cellular mRNA Translation in Coronavirus-Infected Cells. Adv. Virus Res. 2016, 96, 165–192. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019, 10, 2342. [Google Scholar] [CrossRef]
- Saikrishnan, K.; Powell, B.; Cook, N.J.; Webb, M.R.; Wigley, D.B. Mechanistic basis of 5′-3′ translocation in SF1B helicases. Cell 2009, 137, 849–859. [Google Scholar] [CrossRef]
- Singleton, M.R.; Dillingham, M.S.; Wigley, D.B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007, 76, 23–50. [Google Scholar] [CrossRef]
- Mickolajczyk, K.J.; Shelton, P.M.M.; Grasso, M.; Cao, X.; Warrington, S.E.; Aher, A.; Liu, S.; Kapoor, T.M. Force-dependent stimulation of RNA unwinding by SARS-CoV-2 nsp13 helicase. Biophys. J. 2021, 120, 1020–1030. [Google Scholar] [CrossRef]
- Jang, K.J.; Jeong, S.; Kang, D.Y.; Sp, N.; Yang, Y.M.; Kim, D.E. A high ATP concentration enhances the cooperative translocation of the SARS coronavirus helicase nsP13 in the unwinding of duplex RNA. Sci. Rep. 2020, 10, 4481. [Google Scholar] [CrossRef] [PubMed]
- Sommers, J.A.; Loftus, L.N.; Jones, M.P., 3rd; Lee, R.A.; Haren, C.E.; Dumm, A.J.; Brosh, R.M., Jr. Biochemical analysis of SARS-CoV-2 Nsp13 helicase implicated in COVID-19 and factors that regulate its catalytic functions. J. Biol. Chem. 2023, 299, 102980. [Google Scholar] [CrossRef]
- Shimberg, G.D.; Michalek, J.L.; Oluyadi, A.A.; Rodrigues, A.V.; Zucconi, B.E.; Neu, H.M.; Ghosh, S.; Sureschandra, K.; Wilson, G.M.; Stemmler, T.L.; et al. Cleavage and polyadenylation specificity factor 30: An RNA-binding zinc-finger protein with an unexpected 2Fe-2S cluster. Proc. Natl. Acad. Sci. USA 2016, 113, 4700–4705. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.; Murdoch, C.C.; Edmonds, K.A.; Jordan, M.R.; Monteith, A.J.; Perera, Y.R.; Rodriguez Nassif, A.M.; Petoletti, A.M.; Beavers, W.N.; Munneke, M.J.; et al. Zn-regulated GTPase metalloprotein activator 1 modulates vertebrate zinc homeostasis. Cell 2022, 185, 2148–2163.e27. [Google Scholar] [CrossRef]
- Maio, N.; Cherry, S.; Schultz, D.C.; Hurst, B.L.; Linehan, W.M.; Rouault, T.A. TEMPOL inhibits SARS-CoV-2 replication and development of lung disease in the Syrian hamster model. iScience 2022, 25, 105074. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.C.; Tong, W.H.; Zhang, D.; Ollivierre-Wilson, H.; Singh, A.; Krishna, M.C.; Mitchell, J.B.; Rouault, T.A. Tempol-mediated activation of latent iron regulatory protein activity prevents symptoms of neurodegenerative disease in IRP2 knockout mice. Proc. Natl. Acad. Sci. USA 2008, 105, 12028–12033. [Google Scholar] [PubMed]
- Ghosh, M.C.; Zhang, D.L.; Ollivierre, H.; Eckhaus, M.A.; Rouault, T.A. Translational repression of HIF2alpha expression in mice with Chuvash polycythemia reverses polycythemia. J. Clin. Investig. 2018, 128, 1317–1325. [Google Scholar] [CrossRef]
- Burns, G.S.; Thompson, A.J. Viral hepatitis B: Clinical and epidemiological characteristics. Cold Spring Harb. Perspect. Med. 2014, 4, a024935. [Google Scholar] [CrossRef]
- Schollmeier, A.; Glitscher, M.; Hildt, E. Relevance of HBx for Hepatitis B Virus-Associated Pathogenesis. Int. J. Mol. Sci. 2023, 24, 4964. [Google Scholar] [CrossRef]
- Brechot, C. Hepatitis B virus (HBV) and hepatocellular carcinoma. J. Hepatol. 1987, 4, 269–279. [Google Scholar] [CrossRef]
- Wu, H.Y.; Guy, J.S.; Yoo, D.; Vlasak, R.; Urbach, E.; Brian, D.A. Common RNA replication signals exist among group 2 coronaviruses: Evidence for in vivo recombination between animal and human coronavirus molecules. Virology 2003, 315, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, S.G.; Milich, D.R.; McLachlan, A. Hepatitis B virus core antigen has two nuclear localization sequences in the Arginine-rich carboxyl terminus. J. Virol. 1991, 65, 575–582. [Google Scholar] [PubMed]
- Tsukuda, S.; Watashi, K. Hepatitis B virus biology and life cycle. Antivir. Res. 2020, 182, 104925. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Ploss, A. Hepatitis B virus cccDNA is formed through distinct repair processes of each strand. Nat. Commun. 2021, 12, 1591. [Google Scholar] [CrossRef]
- Dane, D.S.; Cameron, C.H. Virus-like particles in serum of patients with Australia-antigen associated hepatitis. Lancet 1970, 295, 649–698. [Google Scholar] [CrossRef]
- van Hemert, F.J.; van de Klundert, M.A.; Lukashov, V.V.; Kootstra, N.A.; Berkhout, B.; Zaaijer, H.L. Protein X of hepatitis B virus: Origin and structure similarity with the central domain of DNA glycosylase. PLoS ONE 2011, 6, e23392. [Google Scholar] [CrossRef]
- Ueda, C.; Langton, M.; Chen, J.; Pandelia, M.E. The HBx protein from Hepatitis B Virus coordinates a redox-active Fe-S cluster. J. Biol. Chem. 2022, 298, 101698. [Google Scholar] [CrossRef]
- Gu, J.M.; Lim, S.O.; Oh, S.J.; Yoon, S.M.; Seong, J.K.; Jung, G. HBx modulates iron regulatory protein 1-mediated iron metabolism via reactive oxygen species. Virus Res. 2008, 133, 167–177. [Google Scholar] [CrossRef]
- Bettany, A.J.; Eisenstein, R.S.; Munro, H.N. Mutagenesis of the iron-regulatory element further defines a role for RNA secondary structure in the regulation of ferritin and transferrin receptor expression. J. Biol. Chem. 1992, 267, 16531–16537. [Google Scholar] [CrossRef]
- Aziz, N.; Munro, H.N. Iron regulates ferritin mRNA translation through a segment of its 5′ untranslated region. Proc. Natl. Acad. Sci. USA 1987, 84, 8478–8482. [Google Scholar] [CrossRef]
- Rouault, T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, D.; Xing, W.; Beran, R.K.; Chemuru, S.; Rohrs, H.; Niedziela-Majka, A.; Marchand, B.; Mehra, U.; Zabransky, A.; Dolezal, M.; et al. Hepatitis B Virus X Protein Function Requires Zinc Binding. J. Virol. 2019, 93, e00250-19. [Google Scholar] [CrossRef] [PubMed]
- Pritts, J.D.; Michel, S.L.J. Fe-S clusters masquerading as zinc finger proteins. J. Inorg. Biochem. 2022, 230, 111756. [Google Scholar] [CrossRef]
- Beilschmidt, L.K.; Ollagnier de Choudens, S.; Fournier, M.; Sanakis, I.; Hograindleur, M.A.; Clemancey, M.; Blondin, G.; Schmucker, S.; Eisenmann, A.; Weiss, A.; et al. ISCA1 is essential for mitochondrial Fe(4)S(4) biogenesis in vivo. Nat. Commun. 2017, 8, 15124. [Google Scholar] [CrossRef] [PubMed]
- Monttinen, H.A.M.; Bicep, C.; Williams, T.A.; Hirt, R.P. The genomes of nucleocytoplasmic large DNA viruses: Viral evolution writ large. Microb. Genom. 2021, 7, 649. [Google Scholar] [CrossRef]
- Villalta, A.; Srour, B.; Lartigue, A.; Clemancey, M.; Byrne, D.; Chaspoul, F.; Loquet, A.; Guigliarelli, B.; Blondin, G.; Abergel, C.; et al. Evidence for [2Fe-2S](2+) and Linear [3Fe-4S](1+) Clusters in a Unique Family of Glycine/Cysteine-Rich Fe-S Proteins from Megavirinae Giant Viruses. J. Am. Chem. Soc. 2023, 145, 2733–2738. [Google Scholar] [CrossRef]
- Zhang, B.; Bandyopadhyay, S.; Shakamuri, P.; Naik, S.G.; Huynh, B.H.; Couturier, J.; Rouhier, N.; Johnson, M.K. Monothiol glutaredoxins can bind linear [Fe3S4]+ and [Fe4S4]2+ clusters in addition to [Fe2S2]2+ clusters: Spectroscopic characterization and functional implications. J. Am. Chem. Soc. 2013, 135, 15153–15164. [Google Scholar] [CrossRef]
- Koonin, E.V.; Krupovic, M.; Agol, V.I. The Baltimore Classification of Viruses 50 Years Later: How Does It Stand in the Light of Virus Evolution? Microbiol. Mol. Biol. Rev. 2021, 85, e0005321. [Google Scholar] [CrossRef]
- Peersen, O.B. A Comprehensive Superposition of Viral Polymerase Structures. Viruses 2019, 11, 745. [Google Scholar] [CrossRef]
- de Farias, S.T.; Dos Santos Junior, A.P.; Rego, T.G.; Jose, M.V. Origin and Evolution of RNA-Dependent RNA Polymerase. Front. Genet. 2017, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Arias, L.; Sebastian-Martin, A.; Alvarez, M. Viral reverse transcriptases. Virus Res. 2017, 234, 153–176. [Google Scholar] [CrossRef] [PubMed]
- Sessions, A.L.; Doughty, D.M.; Welander, P.V.; Summons, R.E.; Newman, D.K. The continuing puzzle of the great oxidation event. Curr. Biol. 2009, 19, R567–R574. [Google Scholar] [CrossRef]
- Mahmoudabadi, G.; Milo, R.; Phillips, R. Energetic cost of building a virus. Proc. Natl. Acad. Sci. USA 2017, 114, E4324–E4333. [Google Scholar] [CrossRef]
- Şimşek, B.; Özilgen, M.; Utku, F.Ş. How much energy is stored in SARS-CoV-2 and its structural elements? Energy Storage 2022, 4, e298. [Google Scholar] [CrossRef]
- Sazanov, L.A. A giant molecular proton pump: Structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 2015, 16, 375–388. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Genome | Family | Host and Tropism | FeS Protein(s) | Stoichiometry and Oxidation States | Role of Protein(s) | Proposed Role of FeS Cluster | Reference | Date |
|---|---|---|---|---|---|---|---|---|---|
| Rotavirus | Segmented dsRNA | Reoviridae | Human, mature enterocytes of the villi of the small intestine | NSP5 | [2Fe-2S] | Viral RNA Replication and Packaging, Viroplasm Formation | Either packaging and/or replication of viral genome by modulating RNA interaction | [71] | 2013 |
| Merkel Cell Polyomavirus | dsDNA | Polyomaviridae | Human, Merkel Cells | sT | [2Fe-2S], [4Fe-4S] | Promotes LT-mediated viral DNA replication by the host cell polymerases | FeS coordination is important for sT promotion of viral replication | [77] | 2015 |
| SARS-CoV-2 | (+)ssRNA | Coronaviridae | Human, ACE2 and TMPRSS2 expressing cells | nsp12 | [4Fe-4S]2+, [4Fe-4S]2+ | Polymerase, Viral RNA replication and transcription | Allows coordination between replication proteins in the complex. Enhances processivity of polymerase. | [84] | 2021 |
| nsp13 | [4Fe-4S]2+, Zn2+, Zn2+ | dsRNA Helicase (5’ -> 3’) | Enhances binding specificity for RNA substrate and increases unwinding ability | [85] | 2023 | ||||
| Hepatitis B | Partially dsDNA | Hepadnaviridae | Human, Hepatocytes | HbX | [2Fe-2S]2+ -> [4Fe-4S]2+ | Viral replication, Plays role in carcinogenesis | Potential Scaffold-like protein for directing cellular FeS cluster production. Regulation of Fe homeostasis proteins. Cluster conversion leads to accumulation of ROS | [121] | 2022 |
| Mimivirus | dsDNA | Mimivirinae | Acanthamoeba, Eukaryotic Unicellular Organism | GciS | [2Fe-2S]2+, [3Fe-4S]1+ | Unknown | Iron-sulfur Cluster Intermediate Scaffold | [130] | 2023 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heffner, A.L.; Maio, N. Tip of the Iceberg: A New Wave of Iron–Sulfur Cluster Proteins Found in Viruses. Inorganics 2024, 12, 34. https://doi.org/10.3390/inorganics12010034
Heffner AL, Maio N. Tip of the Iceberg: A New Wave of Iron–Sulfur Cluster Proteins Found in Viruses. Inorganics. 2024; 12(1):34. https://doi.org/10.3390/inorganics12010034
Chicago/Turabian StyleHeffner, Audrey L., and Nunziata Maio. 2024. "Tip of the Iceberg: A New Wave of Iron–Sulfur Cluster Proteins Found in Viruses" Inorganics 12, no. 1: 34. https://doi.org/10.3390/inorganics12010034
APA StyleHeffner, A. L., & Maio, N. (2024). Tip of the Iceberg: A New Wave of Iron–Sulfur Cluster Proteins Found in Viruses. Inorganics, 12(1), 34. https://doi.org/10.3390/inorganics12010034