1. Introduction
The wobble nucleoside at position 34 of tRNA ubiquitously carries a thiomodification that enhances codon binding in vivo and is found frequently in all kingdoms of life [
1]. The most conserved example is the wobble uridine of tRNA
Lys, Gln, Glu in bacteria that encompasses a sulfur-based modification at position 2 (s
2) and a side chain modification, e.g., 5-methoxyaminomethyl at position 5 (mnm
5). The genes involved in these modifications are conserved in eukaryotes from yeast to mammals, however a different set of genes, with a similar outcome is present in bacteria [
2]. The modified uridine 34 is generally found in tRNAs that decode split codon-box family codons where the A- and G-ending codons encode a different amino acid than the U- and C-ending codons [
3,
4].
These modifications help to ensure the correct tRNA folding by stabilizing the conformation of the tRNA of the anticodon loop and facilitating their interactions with the aminoacyl-tRNA synthetase, thereby modulating mRNA decoding and ribosome processivity [
5]. In yeast, it was shown that single tRNA mutants lacking either the mcm
5 or the s
2U34 modification have numerous phenotypes, including higher stress sensitivity and changes in temperature [
1,
6]. Higher temperatures and stress exposure were shown to lower the levels of s
2 thiomodifications. In humans and other eukaryotes, hypomodification is linked to neurological and mitochondrial disorders like type 2 diabetes and cancer [
7,
8]. These phenotypes arise from changes in the composition of the proteome caused by a reduced level of the translation of certain proteins. The lack of s
2 modification was shown to result in a higher density of ribosomes on mRNAs with repeats of AAA, CAA, and GAA codons [
9,
10]. Thereby, mcm
5s
2U34 at the first anticodon position is important for the exact base pairing interactions with purines with the third codon position harboring an A and G [
11]. On the basis of the mnm
5s
2U34-modified crystal structure of tRNA
Lys from
E. coli, that reads both Lys codons AAA and AAG, the s
2 group was revealed to be specifically involved in stacking with U35 and anticodon loop stabilization [
12,
13].
In
E. coli, for the (c)mnm
5s
2U modification of tRNA
Lys, Gln, Glu, a specific sulfur-relay system has been identified, requiring the
l-cysteine desulfurase IscS and the proteins TusA, TusBCD, TusE and MnmA [
14]. Overall, the seven
E. coli genes, namely
tusA,
tusB,
tusC,
tusD,
tusE,
mnmA, and
iscS, were identified to be involved in the formation of s
2U34 in specific tRNAs for Lys, Gln, and Glu. Among them, the
tusBCD operon and their gene products form the TusBCD complex, composed of a (αβγ)
2 dimer. In vitro, the most efficient 2-thiouridine formation is achieved using purified tRNA and the purified proteins IscS, TusA, and the TusBCD complex, in addition to TusE and MnmA [
14]. TusA forms a complex with IscS and enhances its
L-cysteine desulfurase activity two-fold. After the transfer of the persulfide group to Cys19 of TusA, TusA transfers sulfur further onto the Cys278 of TusD present in the TusBCD complex. Subsequently, TusE accepts the sulfur from TusD and transfers it to the Cys199 of MnmA. In vitro studies have shown that IscS can also directly transfer sulfur to MnmA but with a lowered efficiency relative to the TusBCD complex [
14,
15]. MnmA is a member of the adenosine 5′-triphosphate (ATP)-pyrophosphatase family and bears a PP-loop signature motif [
15]. MnmA binds tRNA and ATP, and after binding, the tRNA is activated by an acyl-adenylated intermediate on U34. Subsequently, the persulfide sulfur of MnmA-Cys199 performs a nucleophilic attack on the acyl-adenylate intermediate, resulting in a formed tRNA thiocarbonyl group and a released adenosine 5′-monophosphate (AMP) molecule [
15,
16,
17]. Finally, the persulfide of Cys199 in MnmA is reduced by Cys102, and s
2U34-thiolated tRNA dissociates from MnmA. The newly formed disulfide bridge within MnmA has to be reduced before the next catalytic cycle. For the formation of the (c)mnm
5 sidechain modification, the MnmG–MnmE complex and MnmC are essential (
Figure 1). Here, both the mnm
5 modification and the s
2U modification in bacterial tRNAs were shown to be independently modified.
Two different routes of modification at the C5 position of uridine have been described, which are dependent on the substrate of MnmEG [
18]. Either uridine on U34 is converted into 5-methylaminomethyluridine (mnm
5) using ammonium as a substrate or 5-carboxymethylaminomethyluridine (cmnm
5) using the substrate glycine [
18] (
Figure 1). In turn, MnmC can convert mnm
5U into cmnm
5 using flavin adenine dinucleotide (FAD) as a cofactor, which is bound to the domain at the C-terminus. The N-terminal part of MnmC involved in donating a methyl group in a
S-adenosylmethionine (SAM)-dependent reaction to nm
5U finally forms the mnm
5U modification.
The pathway for the thiomodification of mnm
5s
2U34 tRNAs present in bacteria resembles the human τm
5s
2U34 tRNA modification pathway present in the mitochondria [
19]. In particular, the human MTU1 protein shares significant amino acid sequence homologies with bacterial MnmA [
5]. In contrast, for the mcm
5s
2U34 modifications that are located in the cytosol, the human proteins MOCS3, URM1, CTU1, and CTU2 are essential [
2,
20]. This tRNA thiolation pathway is different from the mnm
5s
2U34 thiolation pathway in bacteria described above, using the proteins MOCS3, URM1, CTU1, and CTU2 instead. However, similar proteins are present in archaea and thermophilic bacteria [
5,
21,
22]. While yeast mutant strains that contain an unmodified base at position U34 in tRNAs are not viable, the phenotype in yeast can be rescued by the overexpression of the respective hypomodified tRNAs [
9]. This observation has led to the suggestion that the main function of the mcm
5s
2U34 modification is to improve the decoding efficiency of cognate codons and not to prevent missense errors. In bacteria, the role of (c) mnm
5s
2U modifications is more complex than in eukaryotes. The overexpression of hypomodified tRNAs in bacteria does not improve the slow growth phenotype of
mnmA or
mnmG mutants but instead further reduces the slow growth of mutant strains [
13]. It has been suggested that increased translational errors are consequentially due to the missing tRNA modifications [
23]. This difference in translation correctness between
E. coli and yeast has been noted earlier since missense errors are about 10-fold lower in yeast compared to
E. coli [
13]. Therefore, yeast might have evolved mechanisms to reduce missense errors that are not currently present in
E. coli [
24]. While the pathway of mnm
5s
2U34 formation was believed to be iron–sulfur Fe-S cluster-independent in
E. coli [
25,
26], the MnmA homologous proteins NCS2 (yeast) and CTU1 (humans) are Fe-S-cluster-dependent proteins [
25]. Here, analysis of the MnmA sequences from bacteria and eukaryotes revealed that the MnmA homologs can be divided into two groups: one that contains a conserved C-DXXC-C motif (D-type) and one that contains a conserved CC-CXXC-C motif (C-type) [
5]. In addition, the D-type enzymes are believed to be Fe-S cluster-independent, while the C-type ones are classified to be Fe-S-cluster-dependent [
5].
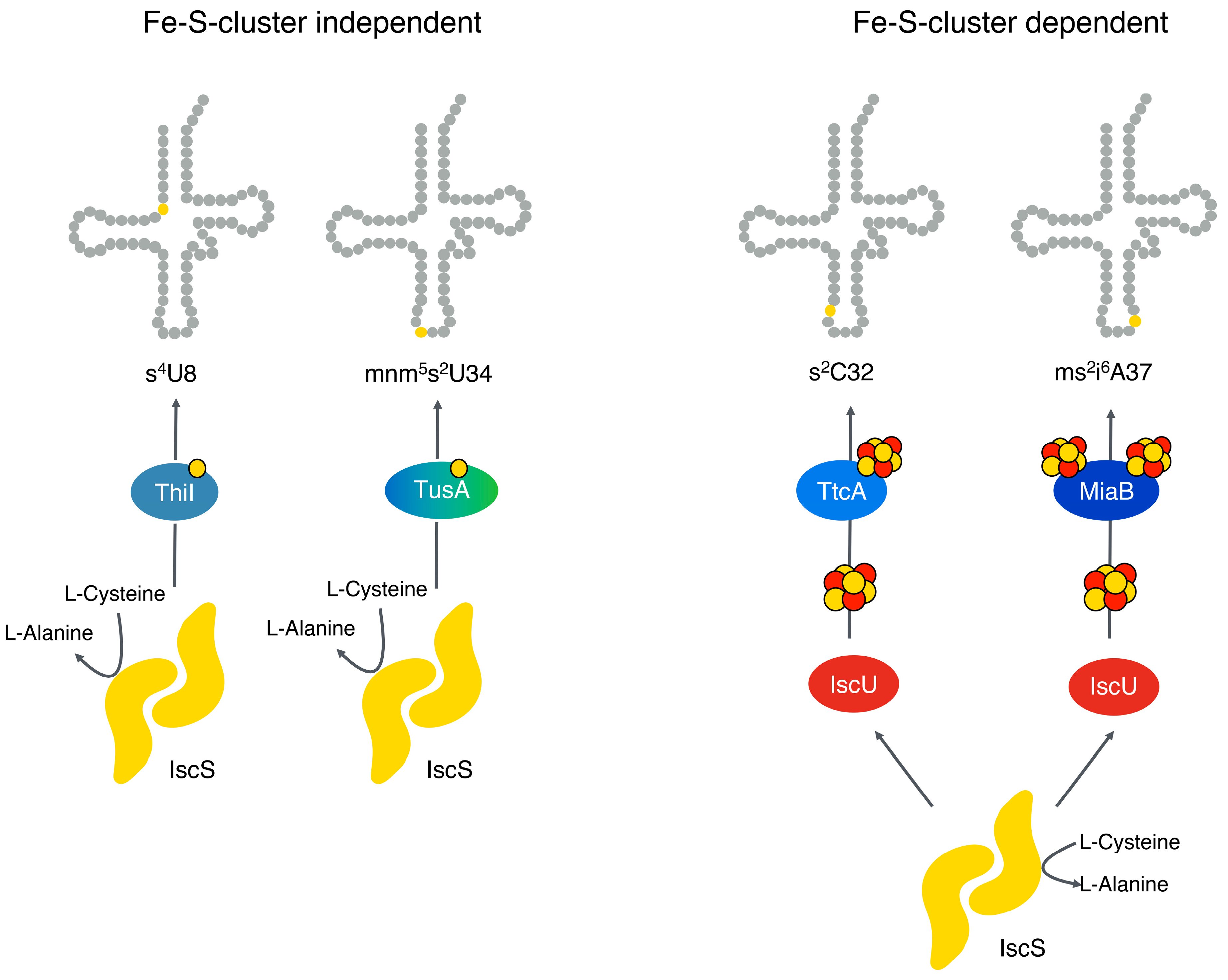
In contrast, the tRNA thiomodifications of 2-thiocytidine at position 32 (s
2C32) and of 2-methylthio-N6 isopentenyladenosine at position (ms
2i
6A37) for certain tRNAs are Fe-S cluster-dependent (
Figure 2) [
25]. In
E. coli, these tRNA modifications involve the proteins TtcA for the s
2C32 modification and MiaB for the ms
2i
6A37 modification (
Figure 2). TtcA binds one [4Fe-4S] cluster, while MiaB binds two [4Fe-4S] clusters and is a member of the radical SAM superfamily of proteins [
27,
28]. In contrast, the s
4U8 and (c)mnm
5s
2 U34 thiomodifications are so far described to be Fe-S cluster-independent in
E. coli [
2,
25,
26] (
Figure 2).
E. coli MnmA was believed to be an Fe-S cluster-independent protein based on the fact that no Fe-S cluster was identified in the crystal structure of the protein [
16,
17]. Recently, a report describing the purification of a [4Fe-4S] cluster containing the MnmA protein isolated under anaerobic conditions from
E. coli reported that this [4Fe-4S] cluster is essential for its activity [
29]. However, this report contradicts our previous published data, where we showed that by using an
E. coli iscS mutant strain complemented with a
sufS-expressing plasmid, the Fe-S cluster biosynthesis was rescued, and this strain was able to produce s
2C32 or ms
2i
6A37-modified tRNAs, but not s
4U8 and (c)mnm
5s
2U34 tRNAs [
26]. Our previous results showed clearly the differentiation of two tRNA thiolation pathways in
E. coli as being either Fe-S cluster-dependent or independent, as initially described by Suzuki [
25]. To clarify the contradiction with the recent publication by Zhou et al. [
29], we aimed to investigate the activity of the Fe-S cluster containing MnmA under our conditions.
3. Discussion
Here, we show that MnmA, the thiouridylase in
E. coli responsible for s
2U34 tRNA modifications, does not require a [4Fe-4S] cluster for activity. In contrast, when reconstituted with a [4Fe-4S] cluster, the protein was rather inactive and unable to transfer sulfur to tRNA. We show that the interaction of MnmA-rec with IscS decreased when the [4Fe-4S] cluster was present, while tRNA and ATP were still able to bind to MnmA-rec. The undetectable s
2U formation originating from an Fe-S cluster reconstituted on MnmA-rec could be based on its low activity and the decreased interaction of MnmA-rec with IscS, which consequently decreased the sulfur transfer reaction (
Table 1). However, this does not counter the fact that in the absence of Fe-S clusters, s
2U formation was indeed detected, which further clearly showed the Fe-S cluster’s independence on MnmA-catalyzed s
2U formation. The presence of a [4Fe-4S] cluster in the MnmA-rec protein was verified by EPR spectroscopy, with spin relaxation properties largely reflective of a reduced [4Fe-4S]
+ cluster. The bound Fe-S cluster exhibited largely similar
g-tensor parameters to that reported preliminarily elsewhere [
31,
32]. Reduced [4Fe-4S]
+ Fe-S clusters typically exhibited very fast spin relaxation properties and showed saturation only at very high microwave power and low temperatures. Both characteristics are observed in the spin relaxation of MnmA, which is largely consistent with a reduced [4Fe-4S]
+ cluster [
29]. Despite the relatively poor reduction in the bound [4Fe-4S] cluster with Na
2S
2O
4 (0.13 spins/MnmA monomer) observed, the Fe number of MnmA-rec (2.5 to 2.8 Fe/MnmA monomer) is nevertheless consistent with a majority of [4Fe-4S] clusters bound since no s
2U formation was observed. Therefore, the non-Fe-S-loaded protein does not seem to perform the sulfur transfer reaction or is inactive. Our results are consistent with the grouping of
E. coli MnmA to D-type thiouridylases, which does not bind a Fe-S cluster in the cell, while C-type MnmAs from thermophilic Archeae do [
5,
21]. Our data are also consistent with previous reports investigating Fe-S cluster mutants in
E. coli, in which the s
2U34 tRNA modification was not impaired, but the s
2C and ms
2I
6A modifications were impaired [
26]. Surprisingly, in the report by Zhou et al. [
29], the investigated Δ
iscUA/Δ
sufABCDSE strain coincided with a complete defect in Fe-S cluster assembly in which normal mnm
5s
2U34 modifications were identified. This result is consistent with previous reports, showing that the mnm
5s
2U34 modifications are Fe-S independent [
25]. However, in contrast, the authors rather interpreted this result as revealing that a yet unknown Fe-S cluster assembly pathway is responsible for providing the Fe-S clusters to MnmA in this strain [
29]. We rather consider this hypothesis as unlikely since a third Fe-S cluster assembly system is not known in
E. coli, and our data show that by reintroducing the
sufABCDSE operon to this strain, the Fe-S cluster assembly and s
2C32 and ms
2i
6A37 modifications were restored, but not mnm
5s
2U34 thiomodifications. Furthermore, we tried to complement the
E. coli Δ
mnmA mutant strain with the MnmA homologs from humans, CTU1 and MTU1, that either bind Fe-S clusters or also belong to the D-type thiouridylases, respectively. While MTU1 is also Fe-S cluster-independent like MnmA [
33,
34], CTU1 was shown previously to carry a [3Fe-4S] cluster [
20]. Nevertheless, both proteins were not able to reconstitute the
E. coli Δ
mnmA mutant strain based on their inability to bind and transfer sulfur to
E. coli tRNA. We show that
E. coli tRNA appears not to be bound to MTU1. The reason for the difference in tRNA remains elusive since MnmA was also unable to bind to human tRNA. We show that the sidechain mnm
5 modification does not hinder the binding to MTU1 since the tRNA without this modification was also not bound. There must be conclusively some structural differences between
E. coli and human tRNA. The reason why some thiouridylases are Fe-S cluster-dependent while others are not remains also elusive. It might depend on the growth condition of the respective organism. When the s
2U34 thiomodification is synthesized under aerobic conditions, the thiomodification might easily become oxidized and exchanged by oxygen. Building an Fe-S cluster on MnmA would produce an inactive thiouridylase in the presence of oxygen in this organism; thus, the s
2U34 modification is inactive under aerobiosis. The cell might save energy in this way.
4. Materials and Methods
4.1. Aerobic Purification MnmA
LB medium supplemented with 50 μg/mL of kanamycin was inoculated with either BL21 DE3 pJD69-mnmA or CL100 pJD69-mnmA. Cultures were grown overnight at 37 °C and 180 rpm. In total, 20 mL of the preculture per liter of the main culture was added to the LB medium supplemented with 50 μg/mL of kanamycin. Cultivation continued at 37 °C and 160 rpm until OD600 = 0.6 was reached, and IPTG was added to a final concentration of 200 μM. The cultures were further incubated for 4 h at 30 °C and 160 rpm. The cells were harvested by centrifugation (5000× g, 4 °C, 5 min) and were resuspended in 50 mM HEPES, 150 mM KCl, 8.6% (v/v) glycerol, 10 mM imidazole, pH 7.5 buffer. The cell suspension was frozen at −20 °C and stored. After thawing the cells, deoxyribonuclease I (DNase I) was added at a final concentration of 1 μg/mL. The cell suspension was treated twice with a high-pressure homogenizer (CF Cell Disrupter; Constant Systems Ltd., Daventry, UK) at a pressure of 1.35 kbar. Cell debris was removed by centrifugation (20,000× g, 4 °C, 30 min). The supernatant was loaded on a column packed with a nickel–nitrilotriacetic acid (Ni-NTA) agarose resin (Macherey-Nagel GmbH; Düren, Germany) (with 0.5 mL of the resin per L culture) for affinity chromatography. The column was washed first with a 10 × column volume of 50 mM HEPES, 150 mM KCl, 8.6% (v/v) glycerol, 10 mM imidazole, pH 7.5 buffer, followed by a wash with a 20 × column volume of the buffer containing 20 mM imidazole. For elution, 40 mL of a 150 mM imidazole buffer was applied to the column, and fractions of 2 mL were collected. Samples of the fractions were analyzed with SDS-PAGE. After pooling the fractions containing the protein of interest, the solution was concentrated with centrifugal filters (Amicon™ Ultra-4 Centrifugal Filter Unit, molecular weight cut-off (MWCO) 30 kDa; Merck Millipore, Molsheim, France). Afterward, the buffer of the protein solution was changed to 50 mM HEPES, 150 mM KCl, 8.6% (v/v) glycerol, pH 7.5 buffer using a PD-10 column (Sephadex™ G-25 M, Cytiva, Marlborough, MA, USA). The resulting eluent was concentrated and loaded on a Superose 12 16/50 size exclusion column (Cytiva) equilibrated in 10 mM Tris-HCl, 50 mM KCl, 12 mM Mg(OAc)2, pH 7.5 buffer for final purification. Fractions containing MnmA were collected. The protein concentration was determined photometrically. Aliquots of the MnmA solution were frozen in liquid nitrogen and stored at −80 °C.
4.2. Anaerobic Purification MnmA
The anaerobic purification of MnmA was performed as described above in an anaerobic chamber (vinyl chamber, Coy Laboratory Products Inc.; Grass Lake, MI, USA) using anaerobic buffers.
4.3. Reconstitution of MnmA with Iron–Sulfur Clusters
Reconstitution was carried out under anaerobic conditions. Reaction mixtures contained 3 µM IscS, 1 mM L-cysteine, 200 µM DTT, 1 mM FeCl3, 25 µM PLP, and 100 µM MnmA in 1 mL. Briefly, 100 µM E. coli MnmA and 200 µM DTT were incubated for 30 min at 4 °C in 10 mM Tris-HCl, 300 mM NaCl, pH 9 buffer. Afterwards, 25 µM PLP was added to the mixture, followed by the addition of 1 mM L-cysteine and 1 mM FeCl3; the reaction was continued for 4 h at 4 °C. Reconstituted E. coli MnmA was concentrated using 30 kDa Amicon ultrafilters. The protein was then desalted into buffer containing 50 mM HEPES, 150 mM KCl, 8.6% glycerol at pH 7.5 using PD-10 filtration columns, and the protein samples with an Fe-S cofactor and a shoulder were determined by a UV spectrophotometer.
4.4. Purification of E. coli IscS
E. coli IscS was purified, as reported previously [
35].
4.5. tRNA Extraction
RNA was extracted from E. coli WT and ΔiscS cells using phenol–chloroform phase separation. Chloroform was added to E. coli samples resuspended in a TriFast reagent at a ratio of 1:5 and was vortexed and incubated for 5 min. This was followed by centrifugation at 13,000× g for 30 min at 4 °C for phase separation. The aqueous phase containing RNA was precipitated with isopropanol overnight at −20 °C. The RNA pellet was recovered by centrifugation at 13,000× g at 4 °C for 90 min and was then washed twice with 75% cold ethanol. Finally, the recovered RNA was dissolved in 0.3 M NaOAc at pH 4.5. RNA samples were separated using 5% urea-acrylamide gel for 1 h at 200 V. RNA bands were gels visualized using a Biorad gel camera following ethidium bromide staining. Corresponding tRNA bands on the gel were cut and were incubated with an RNA diffusion buffer (50 mM NaOAc, 150 mM NaCl, pH 7.0) at 4 °C overnight. tRNA samples were filtered with a 45 µM filter and precipitated with isopropanol overnight at 20 °C. Samples were then centrifuged at 13,000× g at 4 °C for 90 min. Pellets were then washed twice with 75% cold ethanol and were dissolved in 0.3 M NaOAc at pH 4.5. tRNA concentrations were then quantified using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific; Wilmington, DE, USA).
4.6. Surface Plasmon Resonance (SPR) Measurements
For the binding experiments using the SPR-based method, a BiacoreTM T200 instrument was employed using CM5 sensor chips at a temperature of 25 °C and a flow rate of 30 mL/min. For data evaluation, the Biacore control T200 software and evaluation T200 software were used (Cytiva; Marlborough, MA, USA). The autosampler rack was at 8 °C to cool the samples. Proteins for each immobilization were obtained from independent purifications. The running buffer containing 20 mM phosphate, 150 mM NaCl, 0.005% (v/v) Tween 20, pH 7.4 was employed. Analytes with concentrations of 0.16, 0.31, 0.63, 1.25, 2.5, 5, 10, and 20 μM were injected for 4.5 min at a flow rate of 30 mL/min followed by a 15 min dissociation and regeneration of the sensor surface with 50 mM of HCl for 1 min. Bovine serum albumin (BSA) served as a negative control ligand. Binding curves for both flow cells were corrected by the subtraction of buffer injection curves.
4.7. In Vitro tRNA Thiolation Assay
For in vitro s2U formation, 1 mM of L-cysteine, 100 µM IscS and 150 µM E. coli MnmA were used in the presence of 1 mM DTT, 1 mM ATP, 20 µM PLP and 300 µg tRNA in a total reaction volume of 500 µL with 10 mM Tris-HCl, 50 mM KCl, 12 mM Mg(OAc)2, and pH 7.5 buffer. The mixture was incubated at 37 °C for 2 h. The same procedure was repeated anaerobically in the glove box for anaerobically purified MnmA. After the completion of the reactions, tRNA was recovered by the tRNA extraction procedure described above.
4.8. HPLC Analysis
In total, 200 µg of tRNA in 50 mM NaOAc, 30 mM ZnOAc, pH 5.3 buffer was digested with P1 nuclease (1 µL per 50 µg of tRNA) and was incubated at 42 °C overnight. In total, 13 µL of 1 M of unbuffered Tris was added, followed by FastAP (1 µL/50 µg), and incubation was continued at 42 °C overnight. Samples were loaded in HPLC vials and analyzed using the LiChrospher
® RP-18 column, following a stepwise gradient program as described by Gehrke to quantify the nucleosides. [
36].
4.9. Trypsinolysis Assay
The method was adopted with modifications from Kambampati and Lauhon [
15]. The proteolytic digestion of MnmA and MTU1 with trypsin was conducted in 10mM Tris-HCl, 50 mM KCl, 12 mM Mg(OAc)
2, pH 7.5 buffer with a final volume of 100 μL. A total of 0.13 mg/mL MnmA (3.17 μM) was preincubated with a 1.5 mg/mL bulk of tRNA purified from Δ
iscS or Δ
MOCS3 knock-out cells [
37] (human embryonic kidney cells (HEK) 293T) and 1 mM of ATP at 37 °C for 5 min. Cleavage was started by adding trypsin (TPCK treated from bovine pancreas with a 12,500 U/mg protein, Sigma) with a final concentration of 2.6 μg/mL or 6.5 μg/mL, which corresponds to a MnmA/trypsin ratio of either 50:1 (
w/
w) or 20:1 (
w/
w), respectively. Cleavage samples were incubated at 37 °C for 1 h. At 0, 5, 10, 20, 30, and 60 min, 15 μL aliquots containing approximately 2 μg MnmA were taken from the reaction mixtures, and a 5 μL 4 × SDS loading buffer was added to stop the reaction. Afterward, the samples were analyzed by SDS-PAGE (method 2.4.10) with 10% acrylamide gels.
4.10. In Vitro AMP Formation
To demonstrate the adenylation of the tRNA by His10-MnmA and thus the indirect binding of the ribonucleic acid to the protein, 20 μM tRNA (isolated from ΔiscS) was incubated with 20 μM MnmA as well as 250 μM ATP, 250 μM MgCl2 and 2 U PPi in a volume of 300 μL with 100 mM of Tris-HCl at pH 7.2 for a period of 2 h at room temperature. The reaction was stopped by adding 1% (w/v) SDS. The samples were then heat-denatured for 15 min at 95 °C and were then centrifuged in Amicon Ultra 0.5 concentrators for 1 h at 14,000× g and 0 °C. The AMP formed during the reaction was applied in 50 mM of (NH4)2HPO4 at pH 2.5 with 2% MeOH on a HPLC C18 RP column (Hypersil-ODS-Particle, 5 μm, 250 × 4.6 mm). Detection was performed at 260 nm.
4.11. In Vitro Thiolation Labeling of tRNA
Purified tRNA from ΔiscS was radiolabeled with L-35S-cysteine for sulfur transfer to tRNA. Briefly, 10 µCi L-35S-cysteine, 100 µM IscS and 150 µM of E. coli MnmA were incubated in the presence of 1 mM DTT, 1mM ATP, 20 µM PLP and 300 µg tRNA in a total reaction volume of 50 µL of 10 mM Tris, 50 mM KCl, 12 mM Mg(OAc)2, pH 7.5 buffer. The mixture was incubated at 37 °C for 2 h. The tRNA was isolated using phenol–chloroform phase separation (chloroform/Trifast (1:5)), which was vortexed and incubated for 5 min. This was followed by centrifugation at 13,000× g for 30 min at 4 °C for phase separation. The aqueous phase containing RNA was precipitated with isopropanol overnight at −20 °C. The RNA pellet was recovered by centrifugation at 13,000× g at 4 °C for 90 min and was then washed twice with 75% cold ethanol. Finally, the recovered RNA was dissolved in 0.3 M NaOAc, pH 4.5 buffer. RNA samples were separated into 5% urea-acrylamide gels for 1 h at 200 V. The radioactivity on the gel was transferred onto a membrane overnight, and a picture of radiolabeled tRNA was taken.
4.12. Electron Paramagnetic Resonance (EPR) Spectroscopy
To characterize the Fe-S cluster bound to MnmA, EPR samples were prepared anaerobically in a Coy chamber (O
2 < 50 ppm) at 4 °C in a thermoblock in a 10 mM Tris-HCl, 50 mM KCl, 12 mM MgCl
2, buffer at pH 8.5. To 72 µL of 275 µM MnmA-rec (as obtained with a light, yellowish-black hue), 8 µL of 100 mM Na
2S
2O
4 was added, which, upon mixing, resulted in a very slight but observable color change. The sample was incubated for 30 min with a reductant at 4 °C before loading the mixture into a quartz EPR capillary (3.9 mm outer diameter; QSIL SE, Langewiesen, Germany) and freezing in a liquid N
2-cooled ethanol bath, and before complete freezing in liquid N
2. Continuous wave X-band EPR spectra were obtained on a laboratory-built spectrometer equipped with a Bruker SHQ resonator with an ESR 910 helium flow cryostat and an ITC503 temperature controller (Oxford Instruments; Abingdon, UK). Instrument components of the laboratory-built spectrometer included an ER041MR microwave bridge (Bruker), an SR810 lock-in amplifier (Stanford Research Systems; Sunnyvale, CA, USA), and a 53181A microwave counter (Agilent Technologies; Santa Clara, CA, USA). A Cu(II)-EDTA standard was used as a reference for the spin quantitation of the MnmA Fe-S cluster, while routine magnetic field calibrations were used to compensate field offsets between the Hall probe and the sample position was performed by measuring a reference N@C
60 sample at ambient temperature [
38,
39]. Spin quantitation was performed via double integration using the utility ‘spincounting’ (
https://github.com/lcts/spincounting, accessed on 23 July 2022) in Matlab R2023a. The spectral simulation of the bound Fe-S cluster was performed using the EasySpin simulation package (version 6.0.0-dev.48) in Matlab R2023a (The Mathworks; Natick, MA, USA) [
31,
40]. An analysis of the power dependence of the MnmA-reduced Fe-S cluster was performed by plotting log (S/√P) vs. log P using the intensity of the signal feature at 3530 G. Paramagnetic signals that do not show power saturation effects result in plots that are parallel to the abscissa with power saturation effects that exhibit a slope toward the abscissa with increasing power [
31].
PM,
P1/2, and
b were estimated by fitting the plot to the equation
S = √
P/(1 +
P/
P1/2)
0.5b.
4.13. Quantification of Fe Content
The iron content was determined for purified MnmA proteins by inductively coupled plasma–optical emission spectroscopy (ICP-OES) after published procedures [
41].














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}