
On the Dehydrocoupling of Alkenylacetylenes Mediated by Various Samarocene Complexes: A Charming Story of Metal Cooperativity Revealing a Novel Dual Metal σ-Bond Metathesis Type of Mechanism (DM|σ-BM)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

2. Results and Discussion
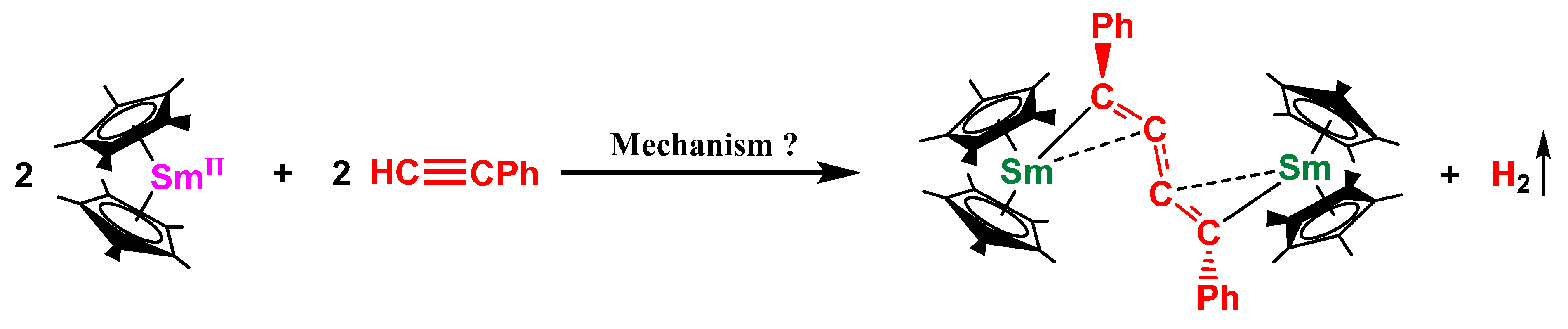
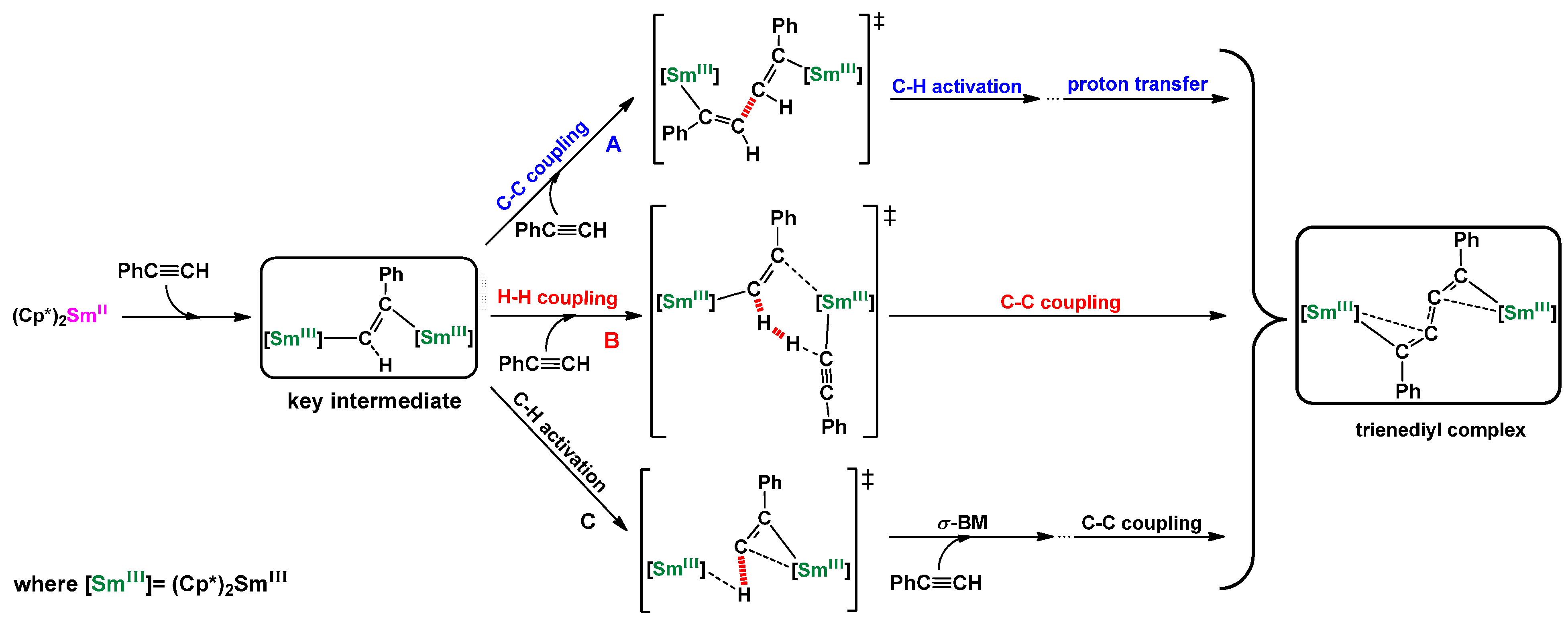
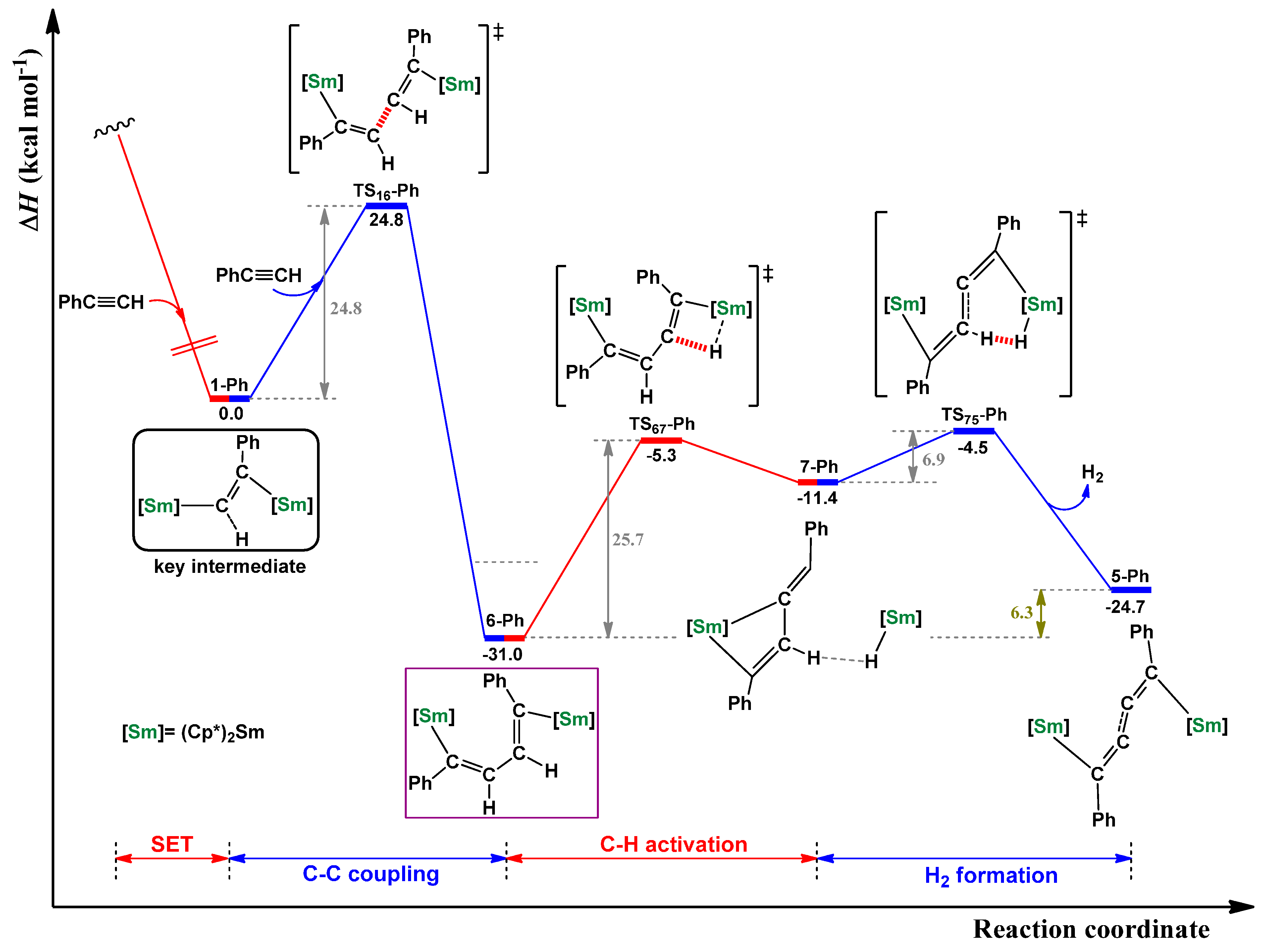
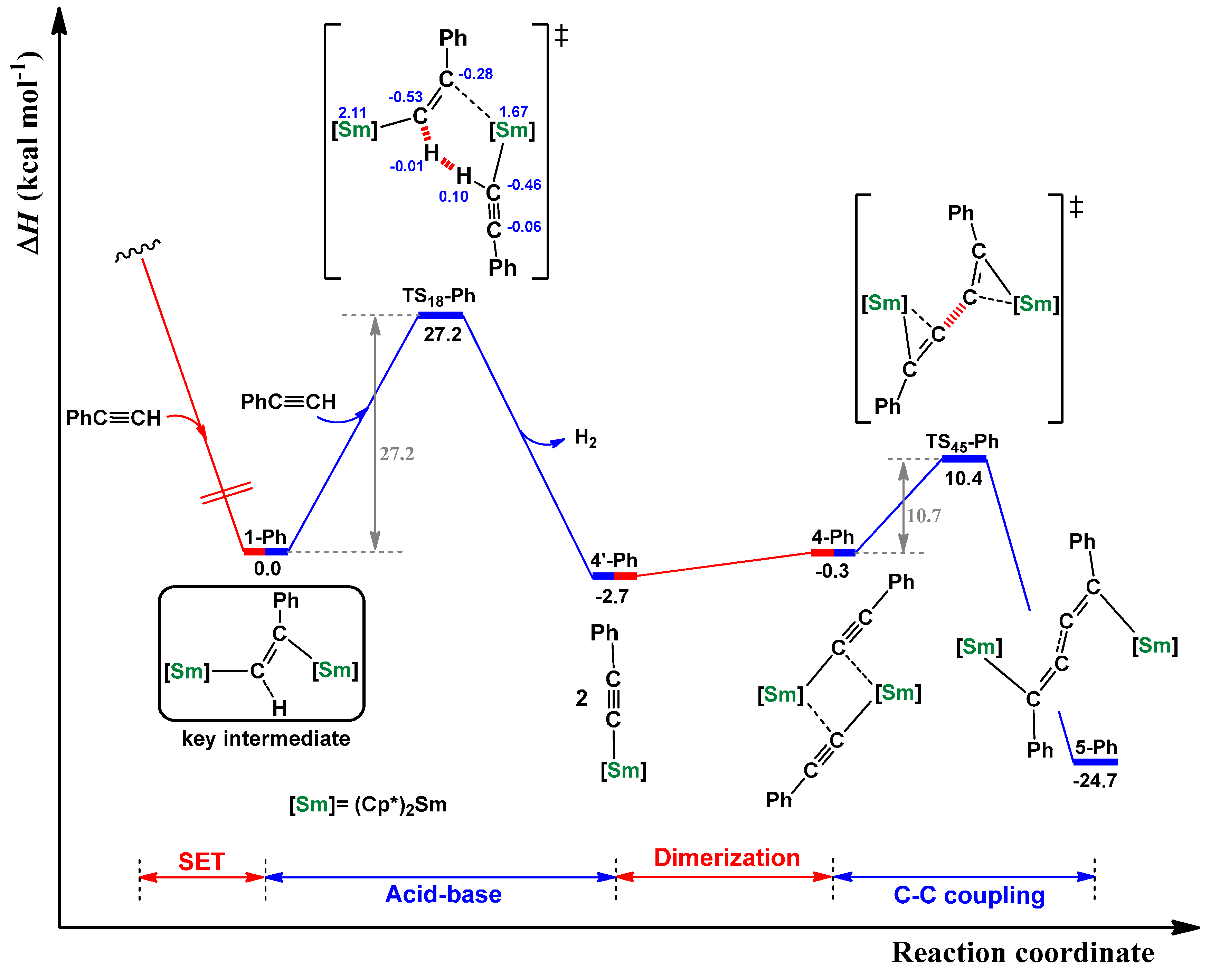
2.1. Dehydrocoupling of PhC≡CH Using Cp*2Sm




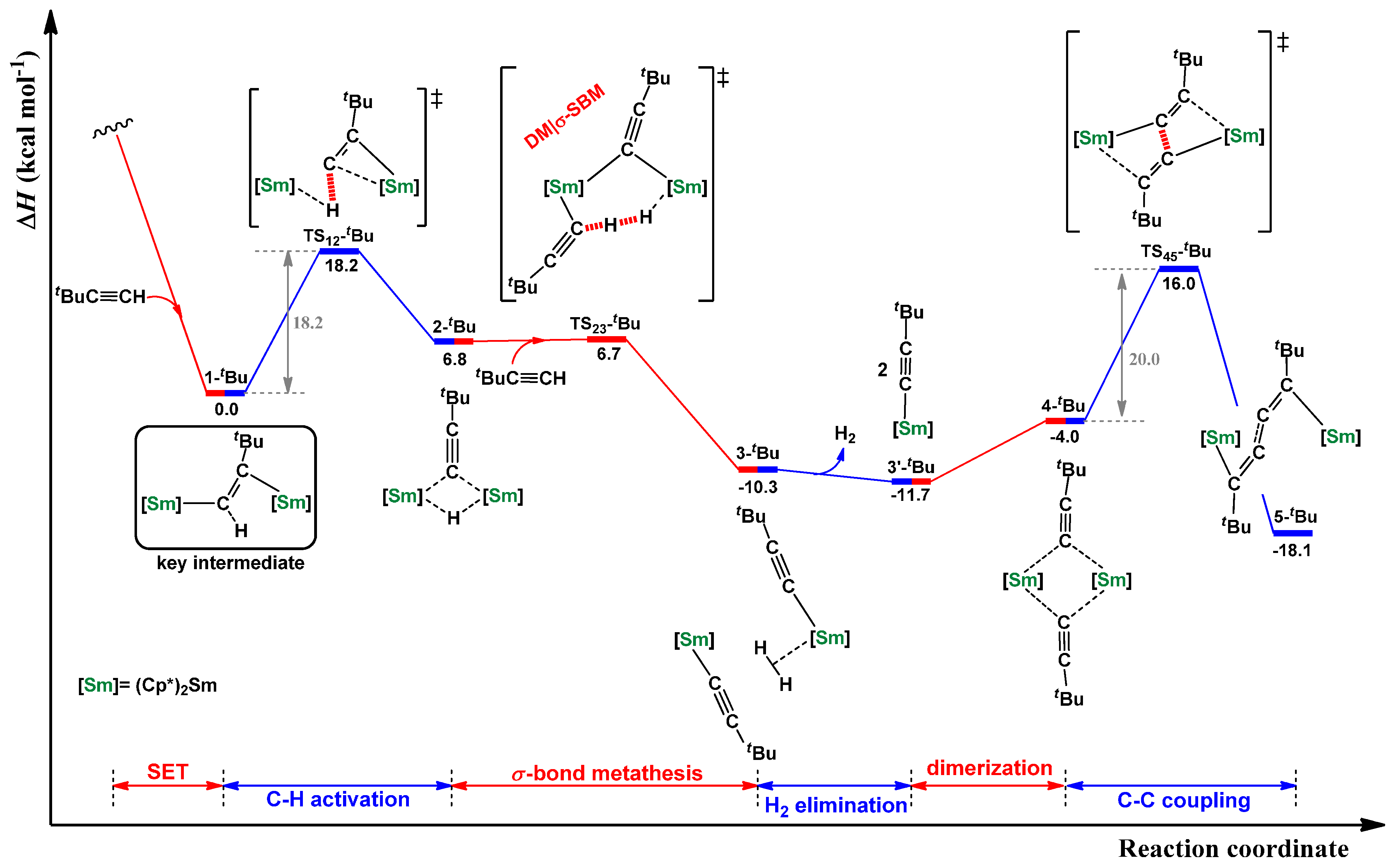
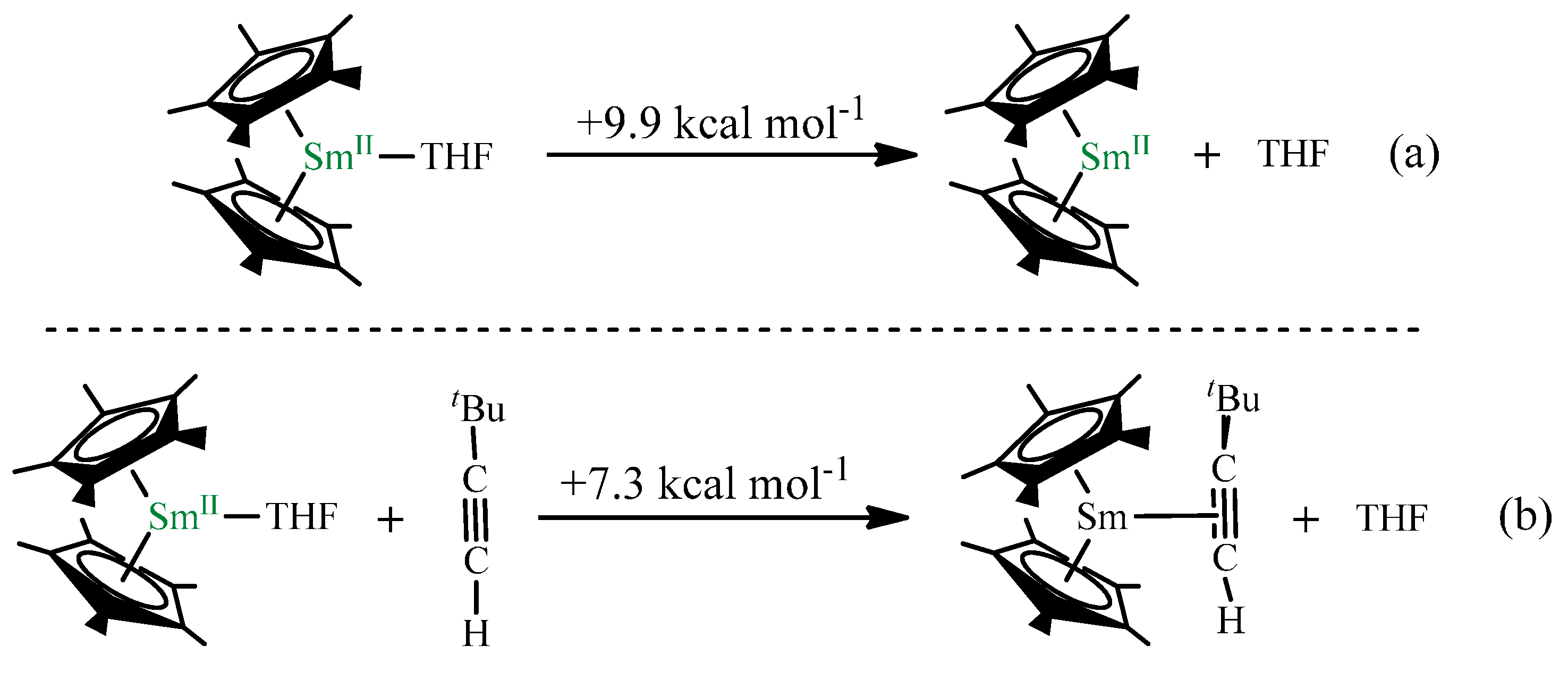
2.2. Dehydrocoupling of tBuC≡CH Using Cp*2Sm


2.3. Dehydrocoupling of Hex-1-yne Using Cp*2Sm

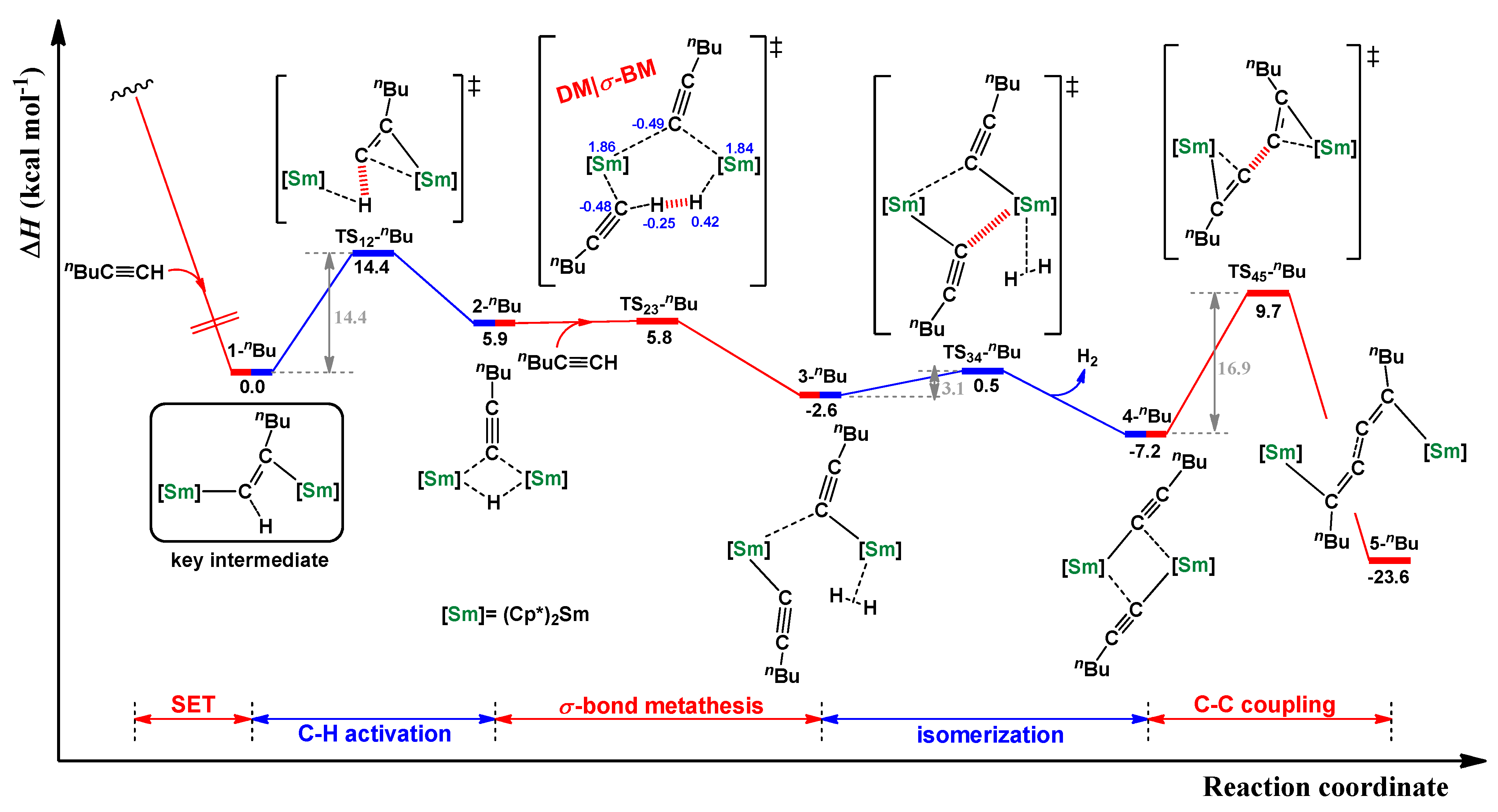
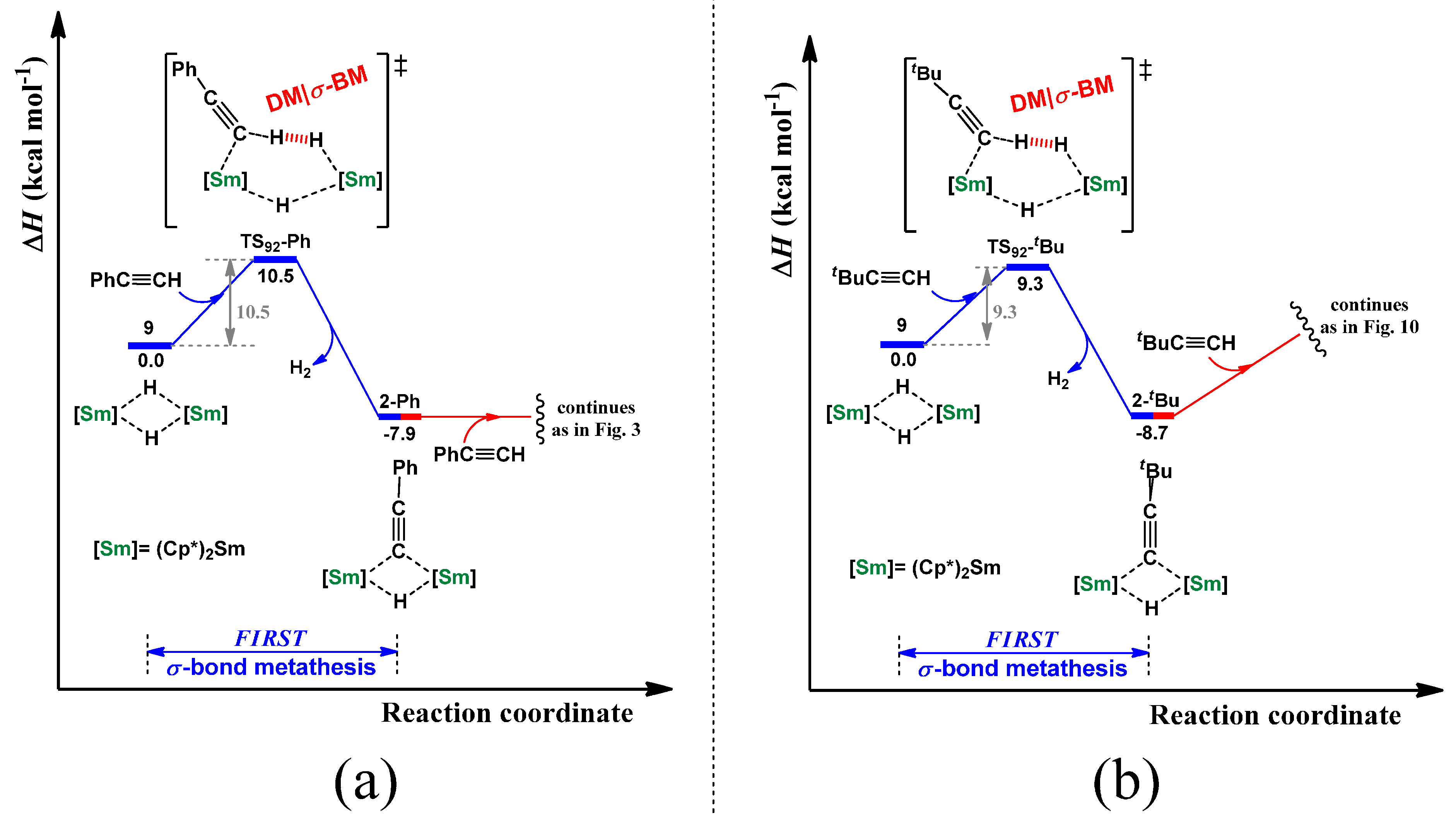
2.4. Dehydrocoupling of Various Terminal Alkynes Using [Cp*2Sm(μ-H)2]2 Complex

3. Computational Section
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Krief, A.; Laval, A.-C. Coupling of organic halides with carbonyl compounds promoted by SmI2, the Kagan reagent. Chem. Rev. 1999, 99, 745–777. [Google Scholar] [CrossRef] [PubMed]
- Procter, D.J.; Flowers, R.A., II; Skrydstrup, T. Organic Synthesis Using Samarium Diiodide: A Practical Guide; Royal Society of Chemistry Publishing: Cambridge, UK, 2010. [Google Scholar]
- Evans, W.J.; Chamberlain, L.R.; Ulibarri, T.A.; Ziller, J.W. Reactivity of trimethylaluminum with (C5Me5)2Sm(THF)2: Synthesis, structure, and reactivity of the samarium methyl complexes (C5Me5)2Sm[(μ-Me)AlMe2(μ-Me)]2Sm(C5Me5)2 and (C5Me5)2SmMe(THF). J. Am. Chem. Soc. 1988, 110, 6423–6432. [Google Scholar] [CrossRef]
- Nolan, S.P.; Stern, C.L.; Marks, T.J. Organo-f-element thermochemistry. Absolute metal-ligand bond disruption enthalpies in bis(pentamethylcyclopentadienyl)samarium hydrocarbyl, hydride, dialkylamide, alkoxide, halide, thiolate, and phosphide complexes. Implications for organolanthanide bonding and reactivity. J. Am. Chem. Soc. 1989, 111, 7844–7853. [Google Scholar]
- Evans, W.J.; Ulibarri, T.A.; Ziller, J.W. Reactivity of (C5Me5)2Sm with aryl-substituted alkenes: Synthesis and structure of a bimetallic styrene complex that contains an η2-arene lanthanide interaction. J. Am. Chem. Soc. 1990, 112, 219–223. [Google Scholar] [CrossRef]
- Evans, W.J.; Ulibarri, T.A.; Ziller, J.W. Reactivity of (C5Me5)2Sm and related species with alkenes: Synthesis and structural characterization of a series of organosamarium allyl complexes. J. Am. Chem. Soc. 1990, 112, 2314–2324. [Google Scholar] [CrossRef]
- Evans, W.J.; Keyer, R.A.; Ziller, J.W. Investigation of organolanthanide-based carbon–carbon bond formation: Synthesis, structure, and coupling reactivity of organolanthanide alkynide complexes, including the unusual structures of the trienediyl complex [(C5Me5)2Sm]2[μ-η2:η2-Ph(CH2)2C=C=C=C–(CH2)2Ph] and the unsolvated alkynide [(C5Me5)2Sm(C≡CCMe3)]2. Organometallics 1993, 12, 2618–2633. [Google Scholar]
- Evans, W.J.; Davis, B.L. Chemistry of tris(pentamethylcyclopentadienyl) f-element complexes, (C5Me5)3M. Chem. Rev. 2002, 102, 2119–2136. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Keyer, R.A.; Ziller, J.W. Carbon–carbon bond formation by coupling of two phenylethynyl ligands in an organolanthanide system. Organometallics 1990, 9, 2628–2631. [Google Scholar] [CrossRef]
- Wood, G.L.; Knobler, C.B.; Hawthorne, M.F. Synthesis of Cp2Ti[C≡CSi(CH3)3]2 and the synthesis and molecular structure of [Cp2TiC≡CSi(CH3)3]2. Inorg. Chem. 1989, 28, 382–384. [Google Scholar] [CrossRef]
- Sekutowski, D.G.; Stucky, G.D. Oxidative coupling of the phenylethynyl group in μ-(1-3η:2-4η-trans,trans-1,4-diphenylbutadiene)-bis(bis(η5-methylcyclopentadienyl)titanium) and the reaction of 1,4-diphenyl-1,3-butadiene with bis(cyclopentadienyl)titanium(II). J. Am. Chem. Soc. 1976, 98, 1376–1382. [Google Scholar] [CrossRef]
- Heeres, H.J.; Nijhoff, J.; Teuben, J.H. Reversible carbon–carbon bond formation in organolanthanide systems. Preparation and properties of lanthanide acetylides [Cp*2LnC≡CR]n and their rearrangement products [Cp*2Ln]2-μ-η2:η2-RC4R) (Ln = La, Ce; R = alkyl). Organometallics 1993, 12, 2609–2617. [Google Scholar] [CrossRef]
- Forsyth, C.M.; Nolan, S.P.; Stern, C.L.; Marks, T.J.; Rheingold, A.L. Alkyne coupling reactions mediated by organolanthanides. Probing the mechanism by metal and alkyne variation. Organometallics 1993, 12, 3618–3623. [Google Scholar] [CrossRef]
- Kefalidis, C.E.; Perrin, L.; Maron, L. Computational insights into carbon–carbon homocoupling reactions mediated by organolanthanide(III) complexes. Dalton Trans. 2014, 43, 4520–4529. [Google Scholar] [CrossRef] [PubMed]
- Kosog, B.; Kefalidis, C.E.; Heinemann, F.W.; Maron, L.; Meyer, K. Uranium(III)-mediated C–C-coupling of terminal alkynes: Formation of dinuclear uranium(IV) vinyl complexes. J. Am. Chem. Soc. 2012, 134, 12792–12797. [Google Scholar] [CrossRef] [PubMed]
- Kefalidis, C.E.; Essafi, S.; Perrin, L.; Maron, L. Qualitative estimation of the single-electron transfer step energetics mediated by samarium(II) complexes: A “SOMO−LUMO gap” approach. Inorg. Chem. 2014, 53, 3427–3433. [Google Scholar] [CrossRef] [PubMed]
- Labouille, S.; Nief, F.; Maron, L. Theoretical treatment of redox processes involving lanthanide(II) compounds: Reactivity of organosamarium(II) and organothulium(II) complexes with CO2 and pyridine. J. Phys. Chem. A 2011, 115, 8295–8301. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Bloom, I.; Hunter, W.E.; Atwood, J.L. Organolanthanide hydride chemistry. 3. Reactivity of low-valent samarium with unsaturated hydrocarbons leading to a structurally characterized samarium hydride complex. J. Am. Chem. Soc. 1983, 105, 1401–1403. [Google Scholar] [CrossRef]
- Ortiz, J.V.; Hoffmann, R. Hydride bridges between LnCp2 centers. Inorg. Chem. 1985, 24, 2095–2104. [Google Scholar] [CrossRef]
- Schaverien, C.J. Alkoxides as ancillary ligands in organolanthanide chemistry: Synthesis of, reactivity of, and olefin polymerization by the μ-hydride-μ-alkyl compounds [Y(C5Me5)(OC6H3tBu2)]2(μ-H)(μ-alkyl). Organometallics 1994, 13, 69–82. [Google Scholar] [CrossRef]
- Duchateau, R.; van Wee, C.T.; Meetsma, A.; van Duijnen, P.T.; Teuben, J.H. Insertion and C–H bond activation of unsaturated substrates by bis(benzamidinato)yttrium alkyl, [PhC(NSiMe3)2]2YR (R = CH2Ph·THF, CH(SiMe3)2), and hydrido, {[PhC(NSiMe3)2]2Y(μ-H)}2, compounds. Organometallics 1996, 15, 2291–2302. [Google Scholar] [CrossRef]
- Steigerwald, M.L.; Goddard, W.A. The 2s + 2s reactions at transition metals. 1. The reactions of D2 with Cl2TiH+, Cl2TiH, and Cl2ScH. J. Am. Chem. Soc. 1984, 106, 308–311. [Google Scholar] [CrossRef]
- Ziegler, T.; Folga, E.; Berces, A. A density functional study on the activation of hydrogen–hydrogen and hydrogen–carbon bonds by Cp2Sc–H and Cp2Sc–CH3. J. Am. Chem. Soc. 1993, 115, 636–646. [Google Scholar] [CrossRef]
- Maron, L.; Eisenstein, O. DFT study of H−H activation by Cp2LnH d0 complexes. J. Am. Chem. Soc. 2001, 123, 1036–1039. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Hu, J.; Lin, Y.; Xing, Y.; Shen, Q. The reactivity of lanthanide alkyl compounds with phenylacetylene: Synthesis and structure of [(ButCp)2LnC≡CPh]2 (Ln= Nd, Gd). Polyhedron 1996, 15, 2165–2169. [Google Scholar] [CrossRef]
- Cuenca, T.; Gómez, R.; Gómez-Sal, P.; Rodriguez, G.M.; Royo, P. Reactions of titanium- and zirconium(III) complexes with unsaturated organic systems. X-ray structure of {[(η5-C5H5)Zr(CH3)]2[μ-η1-η2-CN(Me2C6H3)] (μ-η5-η5-C10H8)}. Organometallics 1992, 11, 1229–1234. [Google Scholar] [CrossRef]
- Atwood, J.L.; Hunter, W.E.; Evans, W.J. Synthesis and crystallographic characterization of a dimeric alkynide-bridged organolanthanide: [(C5H5)2ErC≡CC(CH3)3]2. Inorg. Chem. 1981, 20, 4115–4119. [Google Scholar] [CrossRef]
- Evans, W.J.; Bloom, I.; Hunter, W.E.; Atwood, J.L. Synthesis of organosamarium complexes containing Sm–C and Sm–P bonds. Crystallographic characterization of [(MeC5H4)2SmC≡CCMe3]2. Organometallics 1983, 2, 709–714. [Google Scholar] [CrossRef]
- Shen, Q.; Zheng, D.; Lin, L.; Lin, Y. Synthesis of (t-C4H9C5H4)2Sm(DME) and its reactivity with phenylacetylene: Synthesis and structure of ((t-C4H9C5H4)2SmC≡CPh)2. J. Organomet. Chem. 1990, 391, 307–312. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Gaussian 09, Revision A.02. Gaussian Inc.: Wallingford, CT, USA, 2009.
- Dolg, M.; Stoll, H.; Savin, A.; Preuss, H. Energy-adjusted pseudopotentials for the rare earth elements. Theor. Chim. Acta 1989, 75, 173–194. [Google Scholar] [CrossRef]
- Dolg, M.; Stoll, H.; Preuss, H. A combination of quasirelativistic pseudopotential and ligand field calculations for lanthanoid compounds. Theor. Chim. Acta 1993, 85, 441–450. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–223. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor–acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Weinhold, F. Natural bond orbital methods. In Encyclopedia of Computational Chemistry; Schleyer, P.V.R., Allinger, N.L., Clark, T., Eds.; John Wiley & Sons: Chichester, UK, 1998; Volume 3, pp. 1792–1811. [Google Scholar]
- Waterman, R. σ-Bond metathesis: A 30-year retrospective. Organometallics 2013, 32, 7249–7263. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kefalidis, C.E.; Maron, L. On the Dehydrocoupling of Alkenylacetylenes Mediated by Various Samarocene Complexes: A Charming Story of Metal Cooperativity Revealing a Novel Dual Metal σ-Bond Metathesis Type of Mechanism (DM|σ-BM). Inorganics 2015, 3, 573-588. https://doi.org/10.3390/inorganics3040573
Kefalidis CE, Maron L. On the Dehydrocoupling of Alkenylacetylenes Mediated by Various Samarocene Complexes: A Charming Story of Metal Cooperativity Revealing a Novel Dual Metal σ-Bond Metathesis Type of Mechanism (DM|σ-BM). Inorganics. 2015; 3(4):573-588. https://doi.org/10.3390/inorganics3040573
Chicago/Turabian StyleKefalidis, Christos E., and Laurent Maron. 2015. "On the Dehydrocoupling of Alkenylacetylenes Mediated by Various Samarocene Complexes: A Charming Story of Metal Cooperativity Revealing a Novel Dual Metal σ-Bond Metathesis Type of Mechanism (DM|σ-BM)" Inorganics 3, no. 4: 573-588. https://doi.org/10.3390/inorganics3040573
APA StyleKefalidis, C. E., & Maron, L. (2015). On the Dehydrocoupling of Alkenylacetylenes Mediated by Various Samarocene Complexes: A Charming Story of Metal Cooperativity Revealing a Novel Dual Metal σ-Bond Metathesis Type of Mechanism (DM|σ-BM). Inorganics, 3(4), 573-588. https://doi.org/10.3390/inorganics3040573