
Hexacoordinate Silicon Compounds with a Dianionic Tetradentate (N,N′,N′,N)-Chelating Ligand

Abstract
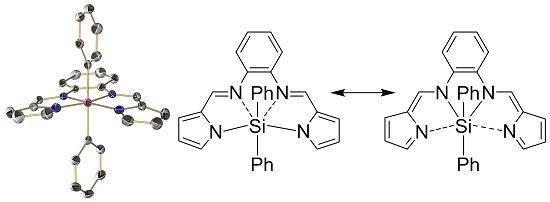
:
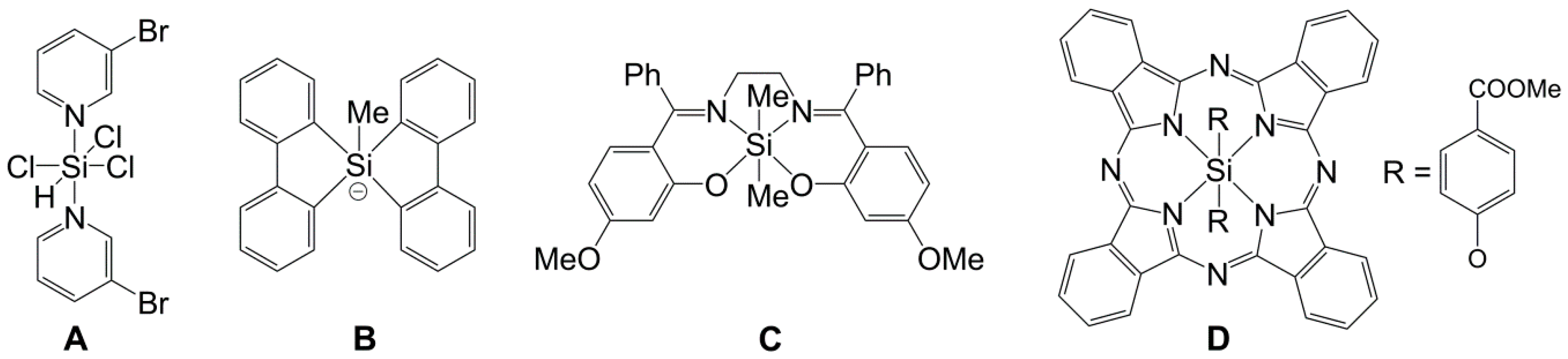
1. Introduction
2. Results and Discussion
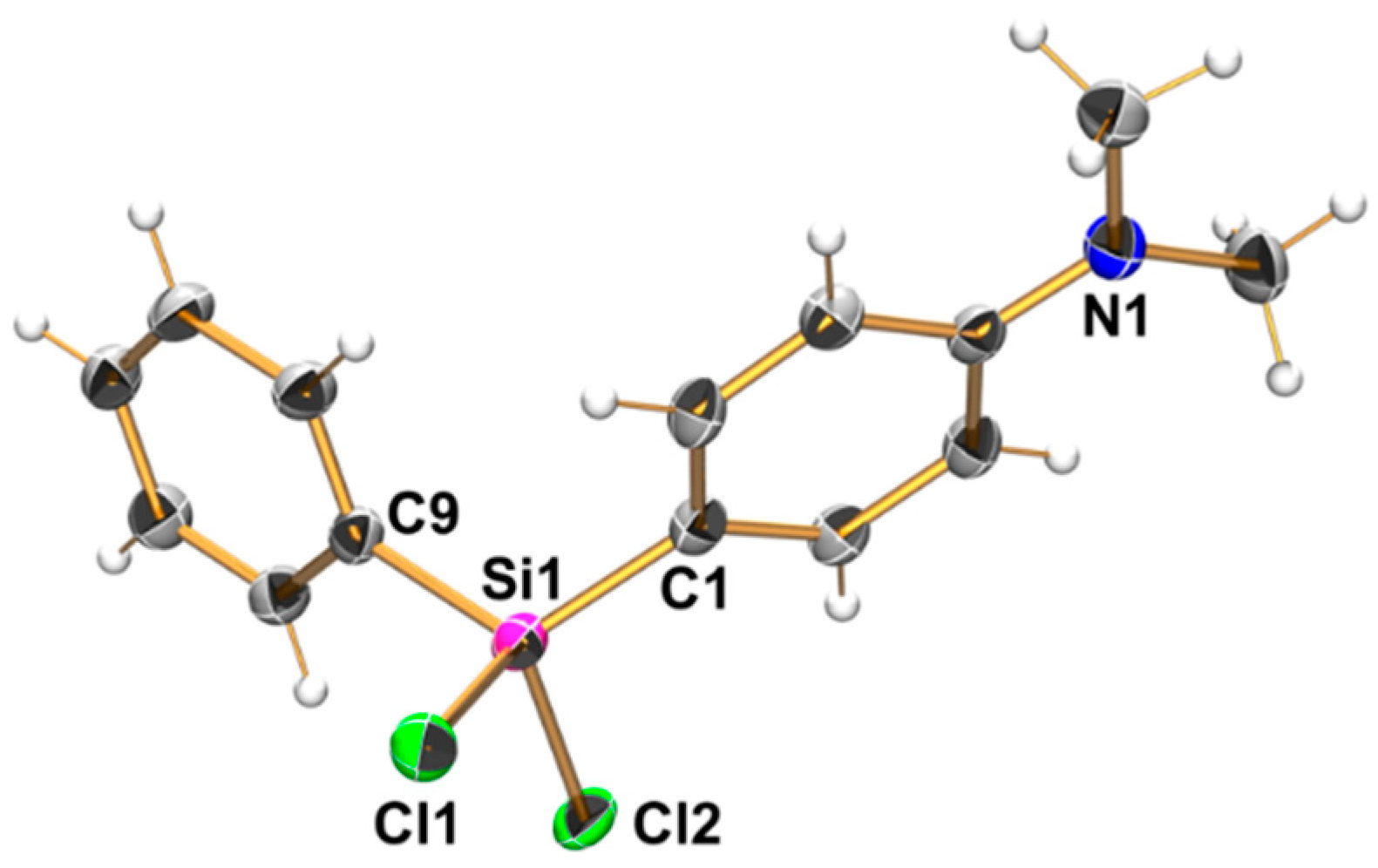
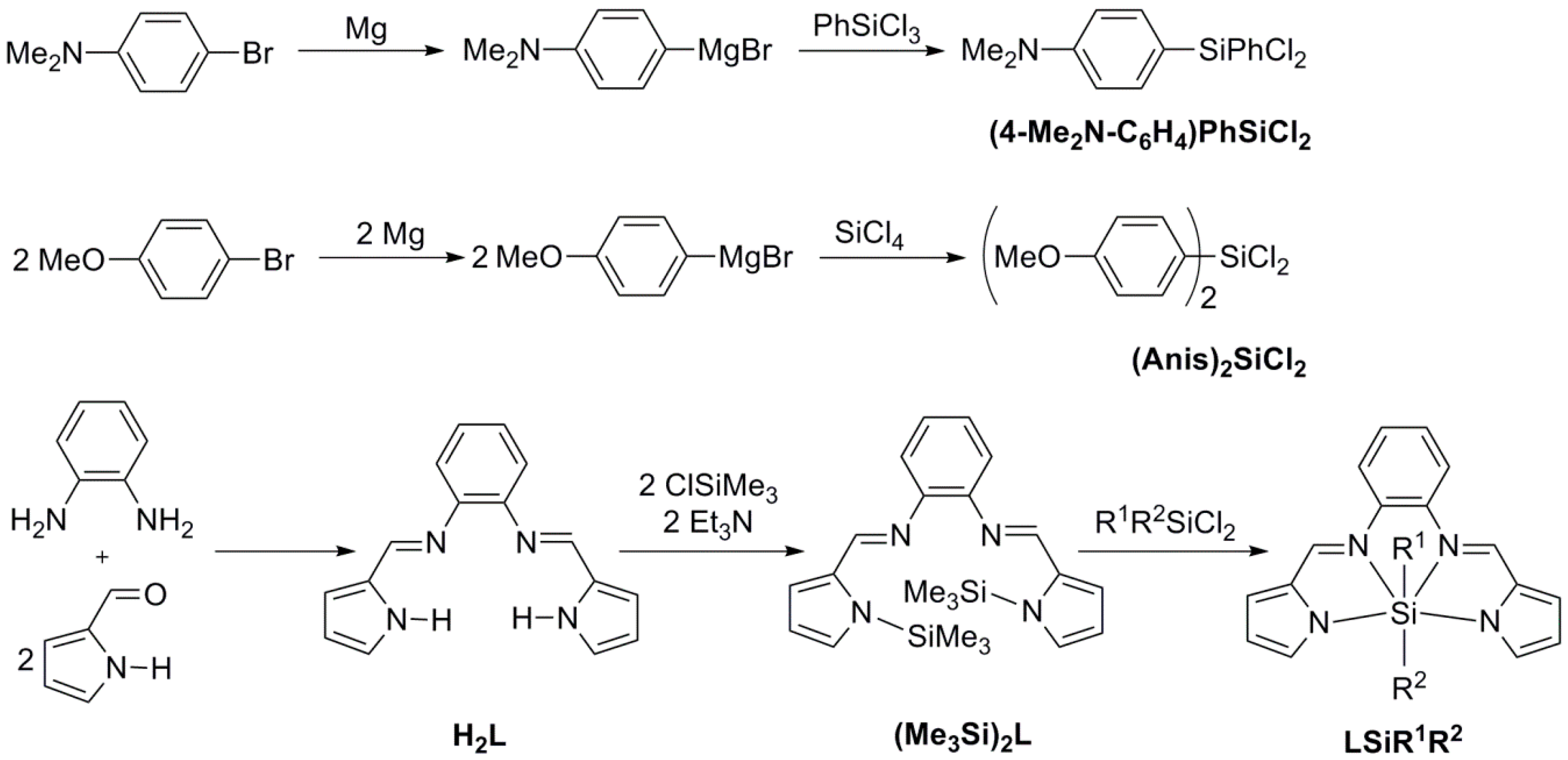
2.1. Syntheses
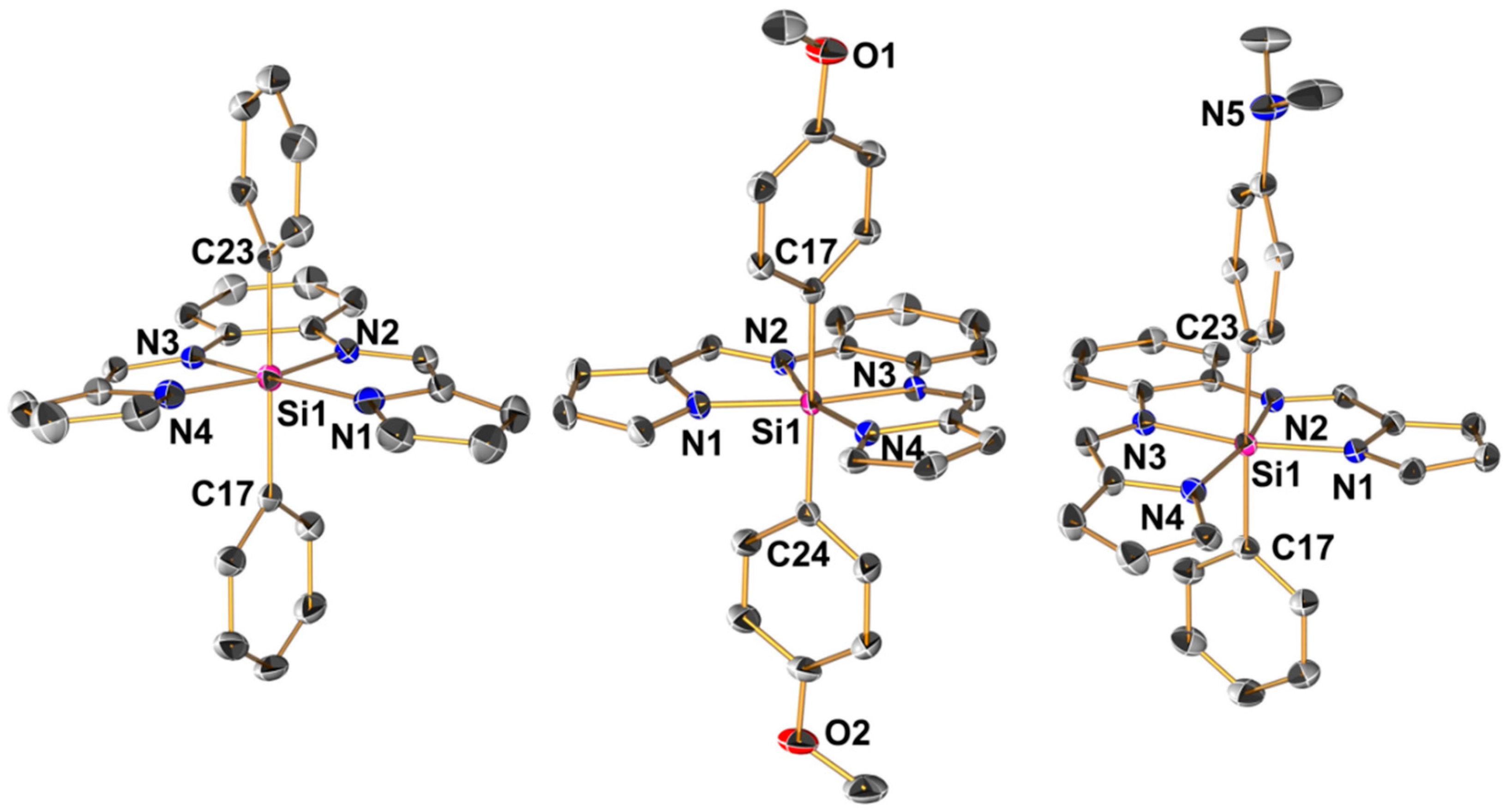
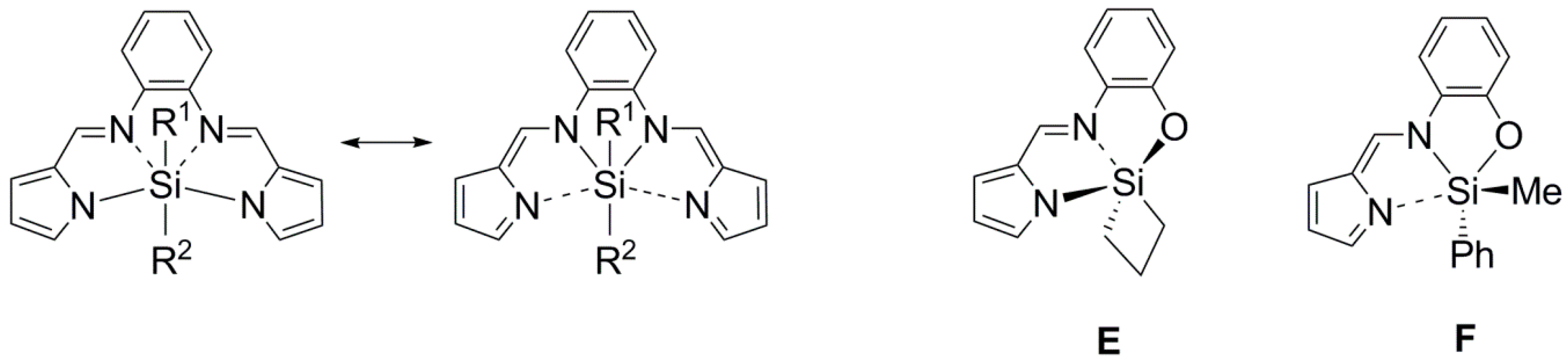
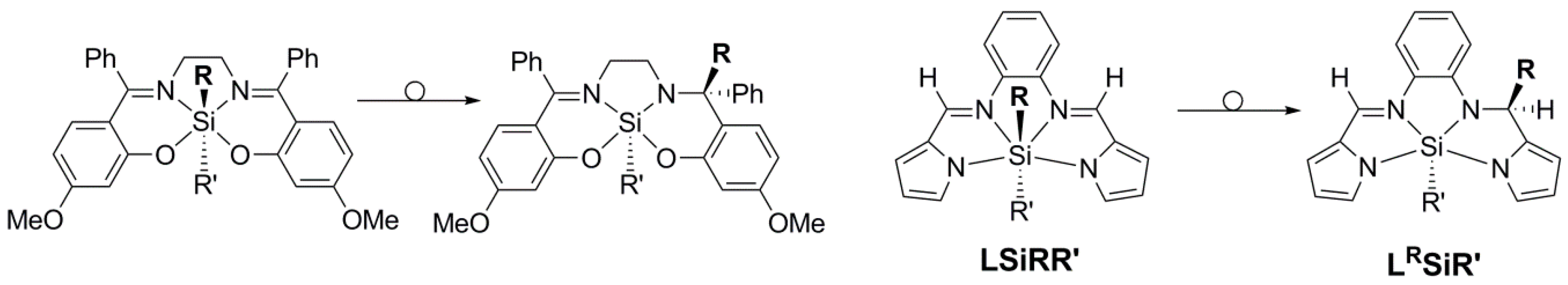
2.2. Molecular Structures
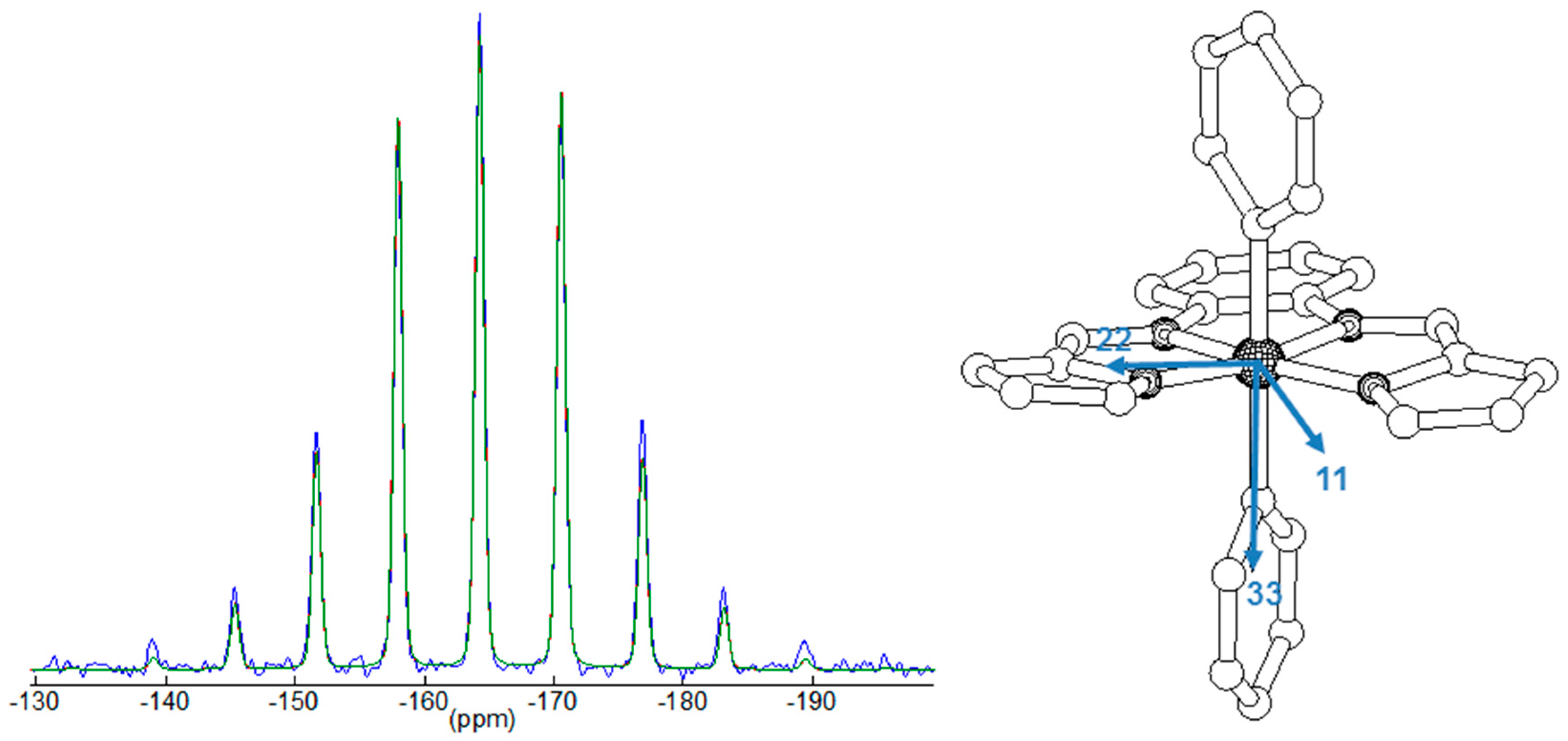
2.3. 29Si NMR
2.4. Light Sensitivity
2.5. Charge Distribution
3. Experimental Section
3.1. General Considerations
3.2. Syntheses
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fester, G.W.; Wagler, J.; Brendler, E.; Kroke, E. Stable trichlorosilane-pyridine adducts. Eur. J. Inorg. Chem. 2008, 2008, 5020–5023. [Google Scholar] [CrossRef]
- Fester, G.W.; Wagler, J.; Brendler, E.; Böhme, U.; Roewer, G.; Kroke, E. Octahedral adducts of dichlorosilane with substituted pyridines: Synthesis, reactivity and a comparison of their structures and 29Si NMR chemical shifts. Chem. Eur. J. 2008, 14, 3164–3176. [Google Scholar] [CrossRef] [PubMed]
- Fester, G.W.; Wagler, J.; Brendler, E.; Böhme, U.; Gerlach, D.; Kroke, E. Octahedral HSiCl3 and HSiCl2Me adducts with pyridines. J. Am. Chem. Soc. 2009, 131, 6855–6864. [Google Scholar] [CrossRef] [PubMed]
- Couzijn, E.P.A.; Slootweg, J.C.; Ehlers, A.W.; Lammertsma, K. Pentaorganosilicates. Z. Anorg. Allg. Chem. 2009, 635, 1273–1278. [Google Scholar] [CrossRef]
- Couzijn, E.P.A.; Ehlers, A.W.; Schakel, M.; Lammertsma, K. Electronic structure and stability of pentaorganosilicates. J. Am. Chem. Soc. 2006, 128, 13634–13639. [Google Scholar] [CrossRef] [PubMed]
- Deerenberg, S.; Schakel, M.; de Keijzer, A.H.J.F.; Kranenburg, M.; Lutz, M.; Spek, A.L.; Lammertsma, K. Tetraalkylammonium pentaorganosilicates: The first highly stable silicates with five hydrocarbon ligands. Chem. Commun. 2002, 348–349. [Google Scholar] [CrossRef]
- Wagler, J.; Böhme, U.; Brendler, E.; Blaurock, S.; Roewer, G. Novel hexacoordinate diorganosilanes with salen-type ligands: Molecular structure versus 29Si NMR chemical shifts. Z. Anorg. Allg. Chem. 2005, 631, 2907–2913. [Google Scholar] [CrossRef]
- Wagler, J.; Böhme, U.; Roewer, G. Activation of Si–Si bond by hypercoordination—Cleavage of a disilane and formation of a Si–C bond. Organometallics 2004, 23, 6066–6069. [Google Scholar] [CrossRef]
- Wagler, J.; Doert, T.; Roewer, G. Synthesis of amines from imines in the coordination sphere of silicon—Surprising photo-rearrangement of hexacoordinate organosilanes. Angew. Chem. Int. Ed. 2004, 43, 2441–2444. [Google Scholar] [CrossRef] [PubMed]
- Wagler, J.; Roewer, G. Syntheses of allyl- and 3-silylpropyl-substituted salen-like tetradentate ligands via Hypercoordinate silicon complexes. Z. Naturforschung B 2006, 61, 1406–1412. [Google Scholar]
- Wagler, J.; Roewer, G.; Gerlach, D. Photo-driven Si–C bond cleavage in hexacoordinate silicon complexes. Z. Anorg. Allg. Chem. 2009, 635, 1279–1287. [Google Scholar] [CrossRef]
- Kano, N.; Yamamura, M.; Kawashima, T. Reactivity control of an allylsilane bearing a 2-(phenylazo)phenyl group by photoswitching of the coordination number of silicon. J. Am. Chem. Soc. 2004, 126, 6250–6251. [Google Scholar] [CrossRef] [PubMed]
- Kalikhman, I.; Gostevskii, B.; Kertsnus, E.; Deuerlein, S.; Stalke, D.; Botoshansky, M.; Kost, D. Competing reactions of hypercoordinate silicon dichelates. J. Phys. Org. Chem. 2008, 21, 1029–1034. [Google Scholar] [CrossRef]
- Kertsnus-Banchik, E.; Kalikhman, I.; Gostevskii, B.; Deutsch, Z.; Botoshansky, M.; Kost, D. Hydride migration from silicon to an adjacent unsaturated imino carbon: Intramolecular hydrosilylation. Organometallics 2008, 27, 5285–5294. [Google Scholar] [CrossRef]
- Novak, M.; Dostal, L.; Turek, J.; Alonso, M.; de Proft, F.; Ruzicka, A.; Jambor, R. Spontaneous double hydrometallation induced by N→M coordination in organometallic hydrides of Group 14 elements. Chem. Eur. J. 2016. in print. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.; Dostal, L.; Alonso, M.; de Proft, F.; Ruzicka, A.; Lycka, A.; Jambor, R. Hydrosilylation induced by N→Si intramolecular coordination: Spontaneous transformation of organosilane into 1-aza-silole-type molecules in the absence of a catalyst. Chem. Eur. J. 2014, 20, 2542–2550. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, M.; Kano, N.; Kawashima, T.; Matsumoto, T.; Harada, J.; Ogawa, K. Crucial role of N···Si interactions in the solid-state coloration of disilylazobenzenes. J. Org. Chem. 2008, 73, 8244–8249. [Google Scholar] [CrossRef] [PubMed]
- Wagler, J.; Gerlach, D.; Böhme, U.; Roewer, G. Intramolecular inter-ligand charge transfer in hexacoordinate silicon complexes. Organometallics 2006, 25, 2929–2933. [Google Scholar] [CrossRef]
- Wagler, J.; Roewer, G. Intramolecular interligand charge transfer and a red hexacoordinate Si-complex with salen-type ligand vs. colorless tetracoordinate salen-Si-complexes with similar substituents. Inorg. Chim. Acta 2007, 360, 1717–1724. [Google Scholar] [CrossRef]
- Gerlach, D.; Ehlers, A.W.; Lammertsma, K.; Wagler, J. Silicon(IV) chelates of an (ONN′)-tridentate pyrrole-2-carbaldimine ligand: Syntheses, structures and UV/Vis properties. Z. Naturforschung B 2009, 64, 1571–1579. [Google Scholar] [CrossRef]
- Wagler, J.; Brendler, E. Metallasilatranes: Palladium(II) and platinum(II) as lone-pair donors to silicon(IV). Angew. Chem. Int. Ed. 2010, 49, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Truflandier, L.A.; Brendler, E.; Wagler, J.; Autschbach, J. 29Si DFT/NMR observation of spin-orbit effect in metallasilatrane sheds some light on the strength of the metal→Si interaction. Angew. Chem. Int. Ed. 2011, 50, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Autschbach, J.; Sutter, K.; Truflandier, L.A.; Brendler, E.; Wagler, J. Atomic contributions from spin-orbit coupling to 29Si NMR chemical shifts in metallasilatrane complexes. Chem. Eur. J. 2012, 18, 12803–12813. [Google Scholar] [CrossRef] [PubMed]
- Wahlicht, S.; Brendler, E.; Heine, T.; Zhechkov, L.; Wagler, J. 7-Azaindol-1-yl(organo)silanes and their PdCl2 complexes: Pd-capped tetrahedral silicon coordination spheres and paddlewheels with a Pd–Si axis. Organometallics 2014, 33, 2479–2488. [Google Scholar] [CrossRef]
- Gualco, P.; Mallet-Ladeira, S.; Kameo, H.; Nakazawa, H.; Mercy, M.; Maron, L.; Amgoune, A.; Bourissou, D. Coordination of a triphosphine-silane to gold: Formation of a trigonal pyramidal complex featuring Au→Si interaction. Organometallics 2015, 34, 1449–1453. [Google Scholar] [CrossRef]
- Kameo, H.; Kawamoto, T.; Bourissou, D.; Sakaki, S.; Nakazawa, H. Evaluation of the σ-donation from group 11 metals (Cu, Ag, Au) to silane, germane, and stannane based on the experimental/theoretical systematic approach. Organometallics 2015, 34, 1440–1448. [Google Scholar] [CrossRef]
- Gualco, P.; Mercy, M.; Ladeira, S.; Coppel, Y.; Maron, L.; Amgoune, A.; Bourissou, D. Hypervalent silicon compounds by coordination of diphosphine-silanes to gold. Chem. Eur. J. 2010, 16, 10808–10817. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ou, C.; Wang, C.; Uchiyama, M.; Deng, L. Silane-functionalized N-heterocyclic carbene-cobalt complexes containing a five-coordinate silicon with a covalent Co–Si bond. Organometallics 2015, 34, 1546–1551. [Google Scholar] [CrossRef]
- Junold, K.; Baus, J.A.; Burschka, C.; Vent-Schmidt, T.; Riedel, S.; Tacke, R. Five-coordinate silicon(II) compounds with Si–M bonds (M = Cr, Mo, W, Fe): Bis[N,N′-diisopropylbenzamidinato(−)]silicon(II) as a ligand in transition-metal complexes. Inorg. Chem. 2013, 52, 11593–11599. [Google Scholar] [CrossRef] [PubMed]
- Wagler, J.; Brendler, E.; Heine, T.; Zhechkov, L. Disilicon complexes with two hexacoordinate Si atoms: Paddlewheel-shaped isomers with (ClN4)Si-Si(S4Cl) and (ClN2S2)Si-Si(S2N2Cl) skeletons. Chem. Eur. J. 2013, 19, 14296–14303. [Google Scholar] [CrossRef] [PubMed]
- Wagler, J.; Brendler, E.; Langer, T.; Pöttgen, R.; Heine, T.; Zhechkov, L. Ylenes in the MII→SiIV (M = Si, Ge, Sn) coordination mode. Chem. Eur. J. 2010, 16, 13429–13434. [Google Scholar] [CrossRef] [PubMed]
- Wächtler, E.; Gericke, R.; Kutter, S.; Brendler, E.; Wagler, J. Molecular structures of pyridinethiolato complexes of Sn(II), Sn(IV), Ge(IV), and Si(IV). Main Group Met. Chem. 2013, 36, 181–191. [Google Scholar] [CrossRef]
- Levason, W.; Pugh, D.; Reid, G. Phosphine and diphosphine complexes of silicon(IV) halides. Inorg. Chem. 2013, 52, 5185–5193. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Theis, B.; Baus, J.A.; Burschka, C.; Bertermann, R.; Tacke, R. Neutral pentacoordinate silicon(IV) complexes with a tridentate dianionic O,N,O or N,N,O ligand, an anionic PhX ligand (X = O, S, Se), and a phenyl group: Synthesis and structural characterization in the solid state and in solution. Z. Anorg. Allg. Chem. 2014, 640, 300–309. [Google Scholar] [CrossRef]
- Baus, J.A.; Burschka, C.; Bertermann, R.; Fonseca Guerra, C.; Bickelhaupt, F.M.; Tacke, R. Neutral six-coordinate and cationic five-coordinate silicon(IV) complexes with two bidentate monoanionic N,S-pyridine-2-thiolato(−) ligands. Inorg. Chem. 2013, 52, 10664–10676. [Google Scholar] [CrossRef] [PubMed]
- Junold, K.; Baus, J.A.; Burschka, C.; Auerhammer, D.; Tacke, R. Stable five-coordinate silicon(IV) complexes with SiN4X skeletons (X = S, Se, Te) and Si:X double bonds. Chem. Eur. J. 2012, 18, 16288–16291. [Google Scholar] [CrossRef] [PubMed]
- Kobelt, C.; Burschka, C.; Bertermann, R.; Fonseca Guerra, C.; Bickelhaupt, F.M.; Tacke, R. Synthesis and structural characterisation of neutral pentacoordinate silicon(IV) complexes with a tridentate dianionic N,N,S chelate ligand. Dalton Trans. 2012, 41, 2148–2162. [Google Scholar] [CrossRef] [PubMed]
- Marckwordt, A.; Rajasekharan-Nair, R.; Steel, G.; Kennedy, A.R.; Reglinski, J.; Spicer, M.D. The first structurally characterised example of silicon in an S6 coordination sphere. Eur. J. Inorg. Chem. 2016. in print. [Google Scholar] [CrossRef] [Green Version]
- Mutneja, R.; Singh, R.; Kaur, V.; Wagler, J.; Fels, S.; Kroke, E. Schiff base tailed silatranes for the fabrication of functionalized silica based magnetic nano-cores possessing active sites for the adsorption of copper ions. New J. Chem. 2016, 40, 1640–1648. [Google Scholar] [CrossRef]
- Liberman-Martin, A.L.; Bergman, R.G.; Tilley, T.D. Lewis acidity of bis(perfluorocatecholato)silane: Aldehyde hydrosilylation catalyzed by a neutral silicon compound. J. Am. Chem. Soc. 2015, 137, 5328–5331. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Y.; Purushothaman, I.; de, S.; Li, B.; Zhu, H.; Parameswaran, P.; Ye, Q.; Liu, W. Fine tuning of the substituents on the N-geminal phosphorus/silicon-based lewis pairs for the synthesis of Z-Type silyliminophosphoranylalkenes. Organometallics 2015, 34, 4209–4217. [Google Scholar] [CrossRef]
- Ignatyev, I.S.; Kochina, T.A.; Avrorin, V.V.; Gurzhiy, V.V.; Fundamensky, V.S. Molecular and crystal structures of 2-phenyl-2-hydro-6-methyl-1,3-dioxa-6-aza-2-silacyclooctane. J. Mol. Struct. 2015, 1094, 169–173. [Google Scholar] [CrossRef]
- Sterkhova, I.V.; Lazarev, I.M.; Smirnov, V.I.; Lazareva, N. 1-(Methylaminomethyl)silatrane: Synthesis, characterization and reactivity. J. Organomet. Chem. 2015, 775, 27–32. [Google Scholar] [CrossRef]
- Maguylo, C.; Chukwu, C.; Aun, M.; Monroe, T.B.; Ceccarelli, C.; Jones, D.S.; Merkert, J.W.; Donovan-Merkert, B.T.; Schmedake, T.A. Exploring the structure and redox activity of hexacoordinate bis(bipyridyl)silicon(IV) complexes. Polyhedron 2015, 94, 52–58. [Google Scholar] [CrossRef]
- Waerder, B.; Steinhauer, S.; Bader, J.; Neumann, B.; Stammler, H.-G.; Vishnevskiy, Y.V.; Hoge, B.; Mitzel, N.W. Pentafluoroethyl-substituted α-silanes: Model compounds for new insights. Dalton Trans. 2015, 44, 13347–13358. [Google Scholar] [CrossRef] [PubMed]
- Mueck, F.M.; Baus, J.A.; Nutz, M.; Burschka, C.; Poater, J.; Bickelhaupt, F.M.; Tacke, R. Reactivity of the donor-stabilized silylenes [iPrNC(Ph)NiPr]2Si and [iPrNC(NiPr2)NiPr]2Si: Activation of CO2 and CS2. Chem. Eur. J. 2015, 21, 16665–16672. [Google Scholar] [CrossRef] [PubMed]
- Mueck, F.M.; Kloss, D.; Baus, J.A.; Burschka, C.; Bertermann, R.; Poater, J.; Fonseca Guerra, C.; Bickelhaupt, F.M.; Tacke, R. Stable four-coordinate guanidinatosilicon(IV) complexes with SiN3El skeletons (El = S, Se, Te) and Si=El double bonds. Chem. Eur. J. 2015, 21, 14011–14021. [Google Scholar] [CrossRef] [PubMed]
- Tacke, R.; Kobelt, C.; Baus, J.A.; Bertermann, R.; Burschka, C. Synthesis, structure and reactivity of a donor-stabilized silylene with a bulky bidentate benzamidinato ligand. Dalton Trans. 2015, 44, 14959–14974. [Google Scholar] [CrossRef] [PubMed]
- Duggal, P. Synthesis, characterization and quantum mechanical calculations of five coordinated anionic silicates. J. Chem. Biol. Phys. Sci. 2016, 6, 153–164. [Google Scholar]
- Singh, G.; Promila; Saroa, A.; Singh, J.; Sharm, R.P.; Ferretti, V. Schiff base functionalized Organopropylsilatranes: Synthesis and structural characterization. J. Chem. Sci. 2016, 128, 193–200. [Google Scholar] [CrossRef]
- Chipanina, N.N.; Lazareva, N.F.; Oznobikhina, L.P.; Lazarev, I.M.; Shainyan, B.A. The hydrolysis of (O–Si)-chelate [N-(acetamido)methyl]dimethylchlorosilanes. DFT and MP2 study, QTAIM and NBO analysis. Comput. Theor. Chem. 2015, 1070, 162–173. [Google Scholar] [CrossRef]
- Singh, G.; Rani, S.; Saroa, A.; Girdhar, S.; Singh, J.; Arora, A.; Aulakh, D.; Wriedt, M. Organosilatranes with thioester-anchored heterocyclic ring assembly: Cu2+ ion binding and fabrication of hybrid silica nanoparticles. RSC Adv. 2015, 5, 65963–65974. [Google Scholar] [CrossRef]
- Doronina, E.P.; Sidorkin, V.F.; Lazareva, N.F. Analysis of the hypersensitivity of the 29Si NMR chemical shift of the pentacoordinate silicon compounds to the temperature effect. N-(Silylmethyl)acetamides. J. Phys. Chem. A 2015, 119, 3663–3673. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, D.; Nishigaichi, Y. Allyl-transfer reaction from photoexcited hypervalent allylsilicon reagent toward dicyanobenzenes. Chem. Lett. 2015, 44, 163–165. [Google Scholar] [CrossRef]
- Kano, N.; Miyake, H.; Sasaki, K.; Kawashima, T. Oxidative bond cleavage of the silicon–silicon bond of a pentacoordinated disilicate. Heteroat. Chem. 2015, 26, 183–186. [Google Scholar] [CrossRef]
- Schwarzer, A.; Fels, S.; Boehme, U. Two reversible enantiotropic phase transitions in a pentacoordinate silicon complex with an O,N,O′-tridentate valinate ligand. Acta Crystallogr. C 2015, 71, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Aghazadeh Meshgi, M.; Baumgartner, J.; Marschner, C. Oligosilanylsilatranes. Organometallics 2015, 34, 3721–3731. [Google Scholar] [CrossRef] [PubMed]
- Boettcher, T.; Steinhauer, S.; Lewis-Alleyne, L.C.; Neumann, B.; Stammler, H.-G.; Bassil, B.S.; Roeschenthaler, G.-V.; Hoge, B. NHC→SiCl4: An ambivalent carbene-transfer reagent. Chem. Eur. J. 2015, 21, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Kost, D.; Kalikhman, I. Hypercoordinate silicon complexes based on hydrazide ligands. A remarkably flexible molecular system. Acc. Chem. Res. 2009, 42, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Levason, W.; Reid, G.; Zhang, W. Coordination complexes of silicon and germanium halides with neutral ligands. Coord. Chem. Rev. 2011, 255, 1319–1341. [Google Scholar] [CrossRef]
- Wagler, J.; Böhme, U.; Kroke, E. Higher-coordinated molecular silicon compounds. In Structure and Bonding; Scheschkewitz, D., Ed.; Springer: Berlin, Germany, 2014; Volume 155, pp. 29–105. [Google Scholar]
- Korlyukov, A.A. Coordination compounds of tetravalent silicon, germanium and tin: The structure, chemical bonding and intermolecular interactions in them. Russ. Chem. Rev. 2015, 84, 422–440. [Google Scholar] [CrossRef]
- Peloquin, D.M.; Schmedake, T.A. Recent advances in hexacoordinate silicon with pyridine-containing ligands: Chemistry and emerging applications. Coord. Chem. Rev. 2016. in print. [Google Scholar] [CrossRef]
- Gerlach, D.; Brendler, E.; Heine, T.; Wagler, J. Dianion of pyrrole-2-N-(o-hydroxyphenyl)carbaldimine as an interesting tridentate (ONN) ligand system in hypercoordinate silicon complexes. Organometallics 2007, 26, 234–240. [Google Scholar] [CrossRef]
- Wagler, J. A Disilane with a hypercoordinate silicon atom: Coordination of an imine ligand versus Si–Si bond splitting. Organometallics 2007, 26, 155–159. [Google Scholar] [CrossRef]
- Wagler, J.; Brendler, E. Hypercoordinate diorganosilanes featuring an (ONO) tridentate ligand. A surprising equilibrium between penta- and tetracoordination. Z. Naturforschung B 2007, 62, 225–234. [Google Scholar]
- Wagler, J.; Gerlach, D.; Roewer, G. 2-N-(Quinoline-8-yl)iminomethylphenolate—A (ONN)-tridentate ligand system in silicon complex chemistry. Inorg. Chim. Acta 2007, 360, 1935–1942. [Google Scholar] [CrossRef]
- Wagler, J.; Hill, A.F. Templated rearrangement of silylated benzoxazolin-2-ones: A novel tridentate (ONO)2− chelating ligand system. Organometallics 2007, 26, 3630–3632. [Google Scholar] [CrossRef]
- Lippe, K.; Gerlach, D.; Kroke, E.; Wagler, J. N-(o-Aminophenyl)-2-oxy-4-methoxybenzophenoneimine—Si-chelation by a tridentate ONN ligand system versus benzimidazoline formation. Inorg. Chem. Commun. 2008, 11, 492–496. [Google Scholar] [CrossRef]
- Kämpfe, A.; Kroke, E.; Wagler, J. Hypercoordinate silicon complexes of (ONN′ vs. ONO′) Schiff base type N-(2-carbamidophenyl)imines: Examples for exclusively O-silylated carbamides. Eur. J. Inorg. Chem. 2009, 2009, 1027–1035. [Google Scholar] [CrossRef]
- Gericke, R.; Gerlach, D.; Wagler, J. Ring-strain-formation Lewis-acidity? A pentacoordinate silacyclobutane comprising exclusively equatorial Si–C bonds. Organometallics 2009, 28, 6831–6834. [Google Scholar] [CrossRef]
- Schwarz, D.; Brendler, E.; Kroke, E.; Wagler, J. Pentacoordinate silicon complexes with N-(2-pyridylmethyl)salicylamide as a dianionic (ONN′) tridentate chelator. Z. Anorg. Allg. Chem. 2012, 638, 1768–1775. [Google Scholar] [CrossRef]
- Wagler, J.; Böhme, U.; Roewer, G. Silicon-enamine complexes: Pentacoordinate silicon compounds. Angew. Chem. Int. Ed. 2002, 41, 1732–1734. [Google Scholar] [CrossRef]
- Wagler, J.; Böhme, U.; Brendler, E.; Roewer, G. First X-ray structure of a cationic silicon complex with salen-type ligand: An unusual compound with two different Si–N dative bonds. Z. Naturforschung B 2004, 59, 1348–1352. [Google Scholar]
- Wagler, J.; Roewer, G. First X-ray structures of ethylene bridged neutral dimeric hexacoordinate silicon complexes with tetradentate salen-type ligands. Z. Naturforschung B 2005, 60, 709–714. [Google Scholar]
- Wagler, J.; Böhme, U.; Brendler, E.; Thomas, B.; Goutal, S.; Mayr, H.; Kempf, B.; Remennikov, B.Y.; Roewer, G. Switching between penta- and hexacoordination with salen-silicon-complexes. Inorg. Chim. Acta 2005, 358, 4270–4286. [Google Scholar] [CrossRef]
- Lippe, K.; Gerlach, D.; Kroke, E.; Wagler, J. Hypercoordinate organosilicon complexes of an ONN′O′ chelating ligand: Regio- and diastereoselectivity of rearrangement reactions in Si-salphen-systems. Organometallics 2009, 28, 621–629. [Google Scholar] [CrossRef]
- Kane, K.M.; Lorenz, C.R.; Heilman, D.M.; Lemke, F.R. Substituent effects on the spectroscopic properties and reactivity of hexacoordinate silicon(IV) porphyrin complexes. Inorg. Chem. 1998, 37, 669–673. [Google Scholar] [CrossRef]
- Zheng, J.-Y.; Konishi, K.; Aida, T. Crystallographic studies of organosilicon porphyrins: Stereoelectronic effects of axial groups on the nonplanarity of the porphyrin ring. Inorg. Chem. 1998, 37, 2591–2594. [Google Scholar] [CrossRef]
- Ishida, S.; Yoshimura, K.; Matsumoto, H.; Kyushin, S. Selective Si–C bond cleavage on a diorganosilicon porphyrin complex bearing different axial ligands. Chem. Lett. 2009, 38, 362–363. [Google Scholar] [CrossRef]
- Taskin, G.C.; Durmus, M.; Yüksel, F.; Mantareva, V.; Kussovski, V.; Angelov, I.; Atilla, D. Axially paraben substituted silicon(IV) phthalocyanines towards dental pathogen Streptococcus mutans: Synthesis, photophysical, photochemical and in vitro properties. J. Photochem. Photobiol. A 2015, 306, 31–40. [Google Scholar] [CrossRef]
- Kämpfe, A.; Kroke, E.; Wagler, J. Silicon compounds of 1,1-bis(pyrrol-2-yl)ethenes: Molecular structures and chemical and spectroscopic properties. Organometallics 2014, 33, 112–120. [Google Scholar] [CrossRef]
- Kämpfe, A.; Brendler, E.; Kroke, E.; Wagler, E. 2-Acylpyrroles as mono-anionic O,N-chelating ligands in silicon coordination chemistry. Chem. Eur. J. 2014, 20, 9409–9418. [Google Scholar] [CrossRef] [PubMed]
- Kämpfe, A.; Brendler, E.; Kroke, E.; Wagler, J. Tp*Cu(I)–CN–SiL2–NC–Cu(I)Tp*—A hexacoordinate Si-complex as connector for redox active metals via π-conjugated ligands. Dalton Trans. 2015, 44, 4744–4750. [Google Scholar] [CrossRef] [PubMed]
- Munro, O.Q.; Joubert, S.D.; Grimmer, C.D. Molecular recognition: Preorganization of a bis(pyrrole) Schiff base derivative for tight dimerization by hydrogen bonding. Chem. Eur. J. 2006, 12, 7987–7999. [Google Scholar] [CrossRef] [PubMed]
- Wächtler, E.; Kämpfe, A.; Krupinski, K.; Gerlach, D.; Kroke, E.; Brendler, E.; Wagler, J. New Insights into hexacoordinated silicon complexes with 8-oxyquinolinato ligands: 1,3-Shift of Si-bound hydrocarbyl substituents and the influence of Si-bound halides on the 8-oxyquinolinate coordination features. Z. Naturforschung B 2014, 69, 1402–1418. [Google Scholar] [CrossRef]
- Kost, D.; Kingston, V.; Gostevskii, B.; Ellern, A.; Stalke, D.; Walfort, B.; Kalikhman, I. Donor-stabilized silyl cations. 3. Ionic dissociation of hexacoordinate silicon complexes to pentacoordinate siliconium salts driven by ion solvation. Organometallics 2002, 21, 2293–2305. [Google Scholar] [CrossRef]
- Mason, J. Conventions for the reporting of nuclear magnetic shielding (or shift) tensors suggested by participants in the NATO ARW on NMR Shielding Constants at the University of Maryland, College Park, July 1992. Solid State Nucl. Magn. Reson. 1993, 2, 285–288. [Google Scholar] [CrossRef]
- Herzfeld, J.; Berger, A.E. Sideband intensities in NMR spectra of sample spinning at the magic angle. J. Chem. Phys. 1980, 73, 6021–6030. [Google Scholar] [CrossRef]
- Brendler, E.; Wächtler, E.; Wagler, J. Hypercoordinate silacycloalkanes: Step-by-step tuning of N→Si Interactions. Organometallics 2009, 28, 5459–5465. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Shelxs-97, Program for the Solution of Crystal Structures, version WinGX© (release 97-2) 1993–1997; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. Shelxl-97, Program for the Refinement of Crystal Structures, version WinGX© (release 97-2) 1993–1997; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. ORTEP-3 for windows—A version of ORTEP-III with a graphical user interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- POV-RAY, version 3.6.2; Trademark of Persistence of Vision Raytracer Pty. Ltd.: Williamstown, Victoria, Australia Copyright Hallam Oaks Pty. Ltd., 1994–2004. Available online: http://www.povray.org/download/ (accessed on 22 December 2011).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998, 108, 664–675. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Raghavachari, K.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar]
- Foster, J.P.; Weinhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural bond orbital analysis of near-Hartree-Fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F. Natural localized molecular orbitals. J. Chem. Phys. 1985, 83, 1736–1740. [Google Scholar] [CrossRef]
- Carpenter, J.E.; Weinhold, F. Analysis of the geometry of the hydroxymethyl radical by the “different hybrids for different spins” natural bond orbital procedure. J. Mol. Struct. THEOCHEM 1988, 46, 41–62. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor–acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Weinhold, F.; Carpenter, J.E. The Structure of Small Molecules and Ions; Naaman, R., Vager, Z., Eds.; Plenum: New York, NY, USA, 1988; pp. 227–236. [Google Scholar]
- Head-Gordon, M.; Pople, J.A.; Frisch, M.J. MP2 energy evaluation by direct methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Saebø, S.; Almlöf, J. Avoiding the integral storage bottleneck in LCAO calculations of electron correlation. Chem. Phys. Lett. 1989, 154, 83–89. [Google Scholar] [CrossRef]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. A direct MP2 gradient method. Chem. Phys. Lett. 1990, 166, 275–280. [Google Scholar] [CrossRef]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. Semidirect algorithms for the MP2 energy and gradient. Chem. Phys. Lett. 1990, 166, 281–289. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Head-Gordon, T. Analytic MP2 frequencies without fifth-order storage. Theory and application to bifurcated hydrogen bonds in the water hexamer. Chem. Phys. Lett. 1994, 220, 122–128. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Peterson, K.A.; Woon, D.E.; Dunning, T.H., Jr. Benchmark calculations with correlated molecular wave functions. IV. The classical barrier height of the H + H2 → H2 + H reaction. J. Chem. Phys. 1994, 100, 7410–7415. [Google Scholar] [CrossRef]
- Wilson, A.K.; van Mourik, T.; Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. VI. Sextuple zeta correlation consistent basis sets for boron through neon. J. Mol. Struct. THEOCHEM 1996, 388, 339–349. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Popp, F.; Nätscher, J.B.; Daiss, J.O.; Burschka, C.; Tacke, R. The 2,4,6-trimethoxyphenyl unit as a unique protecting group for silicon in synthesis and the silylation potential of (2,4,6-trimethoxyphenyl)silanes. Organometallics 2007, 26, 6014–6028. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | (4-Me2N-C6H4) PhSiCl2 | LSiPh2·THF | [LSi(Anis)2]2·THF | LSiPh(4-Me2N-C6H4)·THF |
|---|---|---|---|---|
| Formula | C14H15Cl2NSi | C32H30N4OSi | C64H60N8O5Si2 | C34H35N5OSi |
| Mr | 296.26 | 514.69 | 1077.38 | 557.76 |
| T (K) | 150(2) | 150(2) | 150(2) | 150(2) |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | monoclinic | triclinic | monoclinic | monoclinic |
| Space group | P21/c | P-1 | P21/c | Cc |
| a (Å) | 16.6233(11) | 9.7135(3) | 14.3173(4) | 14.5651(7) |
| b (Å) | 7.5451(3) | 14.9827(4) | 18.2897(8) | 22.8720(8) |
| c (Å) | 12.2941(8) | 18.4932(4) | 21.3671(7) | 10.4529(5) |
| α (°) | 90 | 82.491(1) | 90 | 90 |
| β (°) | 110.965(5) | 83.162(1) | 101.839(2) | 124.370(3) |
| γ (°) | 90 | 88.353(1) | 90 | 90 |
| V (Å3) | 1439.90(15) | 2649.10(12) | 5476.1(3) | 2874.2(2) |
| Z | 4 | 4 | 4 | 4 |
| ρcalc (g·cm−1) | 1.367 | 1.290 | 1.307 | 1.289 |
| μMoKα (mm−1) | 0.516 | 0.122 | 0.125 | 0.119 |
| F(000) | 616 | 1088 | 2272 | 1184 |
| θmax(°), Rint | 30.0, 0.0500 | 27.0, 0.0384 | 28.0, 0.0496 | 30.0, 0.0350 |
| Completeness | 100% | 98.6% | 99.9% | 99.9% |
| Reflns collected | 19,787 | 45,819 | 52,823 | 27,745 |
| Reflns unique | 4203 | 11,420 | 13,197 | 7879 |
| Restraints | 0 | 0 | 0 | 4 |
| Parameters | 165 | 685 | 716 | 377 |
| GoF | 1.044 | 1.060 | 1.047 | 1.052 |
| R1, wR2 [I > 2σ (I)] | 0.0348, 0.0771 | 0.0463, 0.1140 | 0.0441, 0.1013 | 0.0331, 0.0800 |
| R1, wR2 (all data) | 0.0535, 0.0841 | 0.0790, 0.1241 | 0.0744, 0.1129 | 0.0381, 0.0831 |
| Largest peak/hole (e·Å−3) | 0.428, −0.305 | 0.564, −0.410 | 0.678, −0.590 | 0.307, −0.190 |
| Bond | LSiPh(4-Me2N-C6H4) | LSi(Anis)2 1 | LSiPh2 1 | LSiPh2 2 | LSiPhCl 2 | LSiCl2 2 |
|---|---|---|---|---|---|---|
| Si–C | 1.994(1) 3 | 1.995(2) | 1.983(2) | 1.967 | 1.949 | - |
| 1.964(1) 4 | 1.974(2) | 1.977(2) | 1.967 | - | - | |
| Si–Npyrrole | 1.900(1) | 1.904(1) | 1.882(2) | 1.948 | 1.877 | 1.851 |
| 1.890(1) | 1.896(1) | 1.890(1) | 1.948 | 1.877 | 1.852 | |
| Si–Nimine | 1.919(1) | 1.923(2) | 1.906(1) | 1.933 | 1.906 | 1.892 |
| 1.905(1) | 1.915(1) | 1.908(1) | 1.933 | 1.906 | 1.892 | |
| Si–Cl | - | - | - | - | 2.294 | 2.227 |
| - | - | - | - | - | 2.228 |
| Parameter | LSiPh2exp | LSiPh2calc | (O,N,N,O)SiPh2 |
|---|---|---|---|
| δiso (ppm) | −164.3 | −166.9 | −177.7 |
| δ11 (ppm) | −147.3 | −149.9 | −162.8 |
| δ22 (ppm) | −164.6 | −168.1 | −174.3 |
| δ33 (ppm) | −181.0 | −182.9 | −196.0 |
| Ω (ppm) 1 | 33.7 | 33.0 | 33.2 |
| κ 2 | −0.03 | −0.11 | 0.31 |
| Moiety | LSiPh(4-Me2N-C6H4) | LSi(Anis)2 | LSiPh2 | LSiPhCl | LSiCl2 |
|---|---|---|---|---|---|
| Si | 2.216 | 2.209 | 2.215 | 2.186 | 2.102 |
| L | −1.110 | −1.107 | −1.100 | −1.051 | −1.023 |
| R | −0.559 | −0.556 | −0.557 | −0.562 | −0.539 |
| (Ph) | (Anis) | (Ph) | (Ph) | (Cl) | |
| R′ | −0.547 | −0.546 | −0.557 | −0.572 | −0.540 |
| (4-Me2N-C6H4) | (Anis) | (Ph) | (Cl) | (Cl) |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerlach, D.; Brendler, E.; Wagler, J. Hexacoordinate Silicon Compounds with a Dianionic Tetradentate (N,N′,N′,N)-Chelating Ligand. Inorganics 2016, 4, 8. https://doi.org/10.3390/inorganics4020008
Gerlach D, Brendler E, Wagler J. Hexacoordinate Silicon Compounds with a Dianionic Tetradentate (N,N′,N′,N)-Chelating Ligand. Inorganics. 2016; 4(2):8. https://doi.org/10.3390/inorganics4020008
Chicago/Turabian StyleGerlach, Daniela, Erica Brendler, and Jörg Wagler. 2016. "Hexacoordinate Silicon Compounds with a Dianionic Tetradentate (N,N′,N′,N)-Chelating Ligand" Inorganics 4, no. 2: 8. https://doi.org/10.3390/inorganics4020008
APA StyleGerlach, D., Brendler, E., & Wagler, J. (2016). Hexacoordinate Silicon Compounds with a Dianionic Tetradentate (N,N′,N′,N)-Chelating Ligand. Inorganics, 4(2), 8. https://doi.org/10.3390/inorganics4020008