Cationic Protic Imidazolylidene NHC Complexes of Cp*IrCl+ and Cp*RhCl+ with a Pyridyl Tether Formed at Ambient Temperature

Abstract
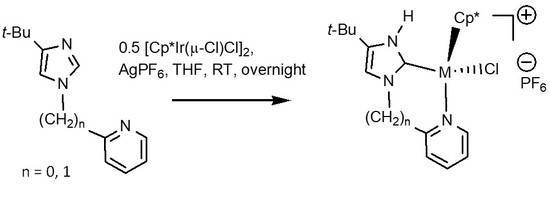
:
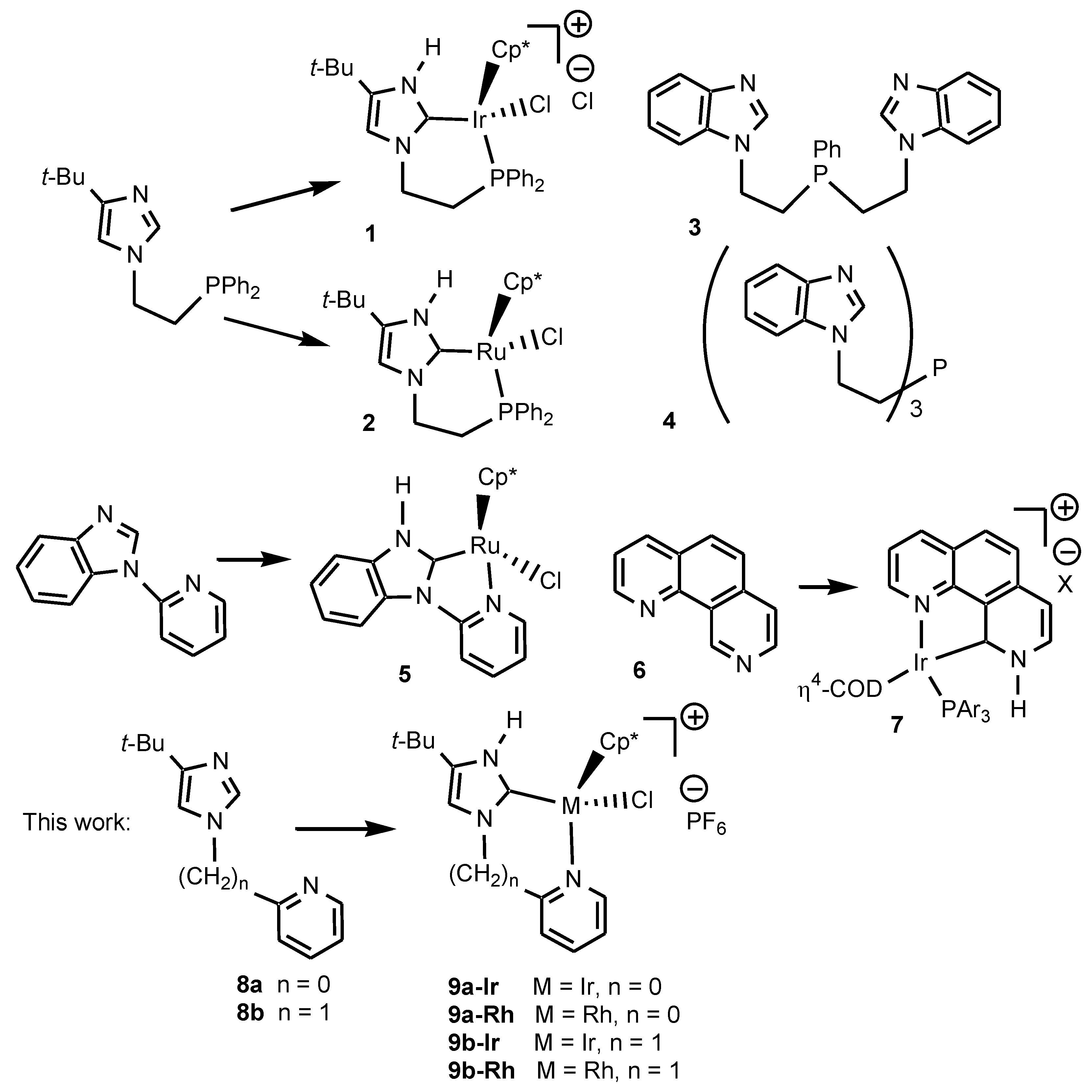
1. Introduction
2. Results
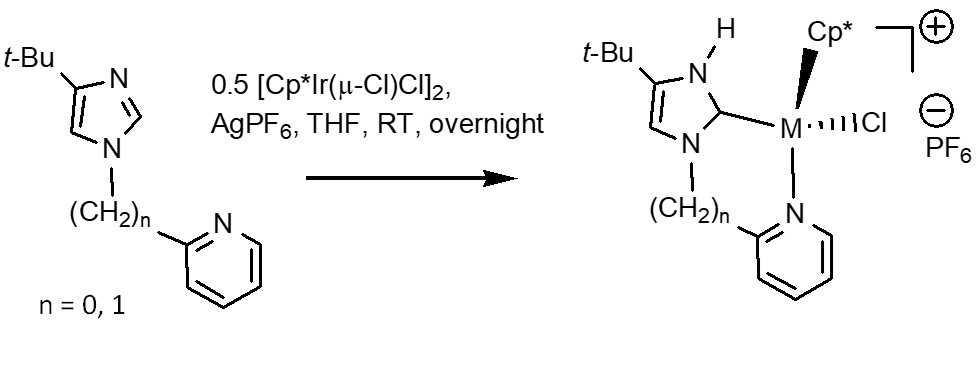
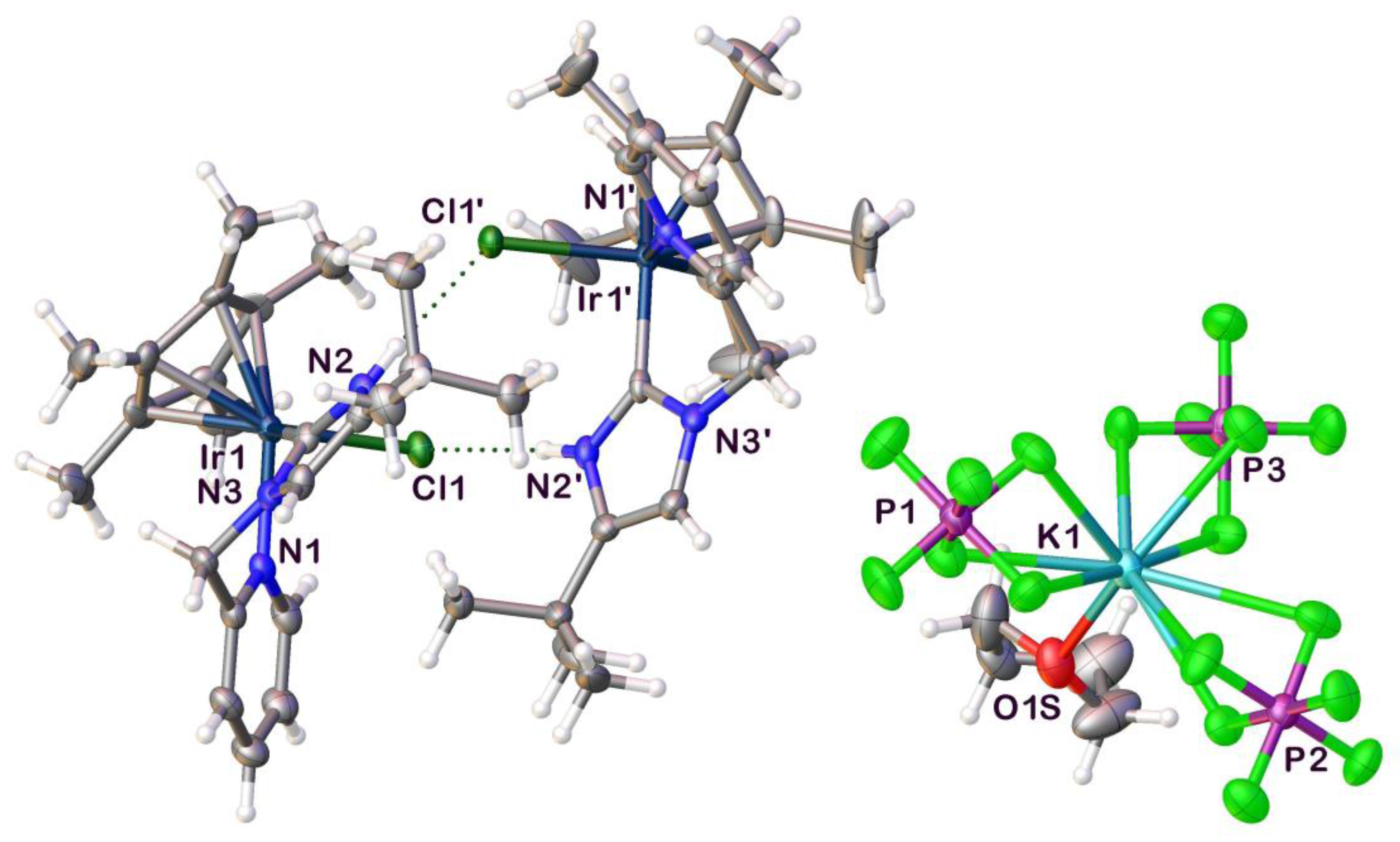
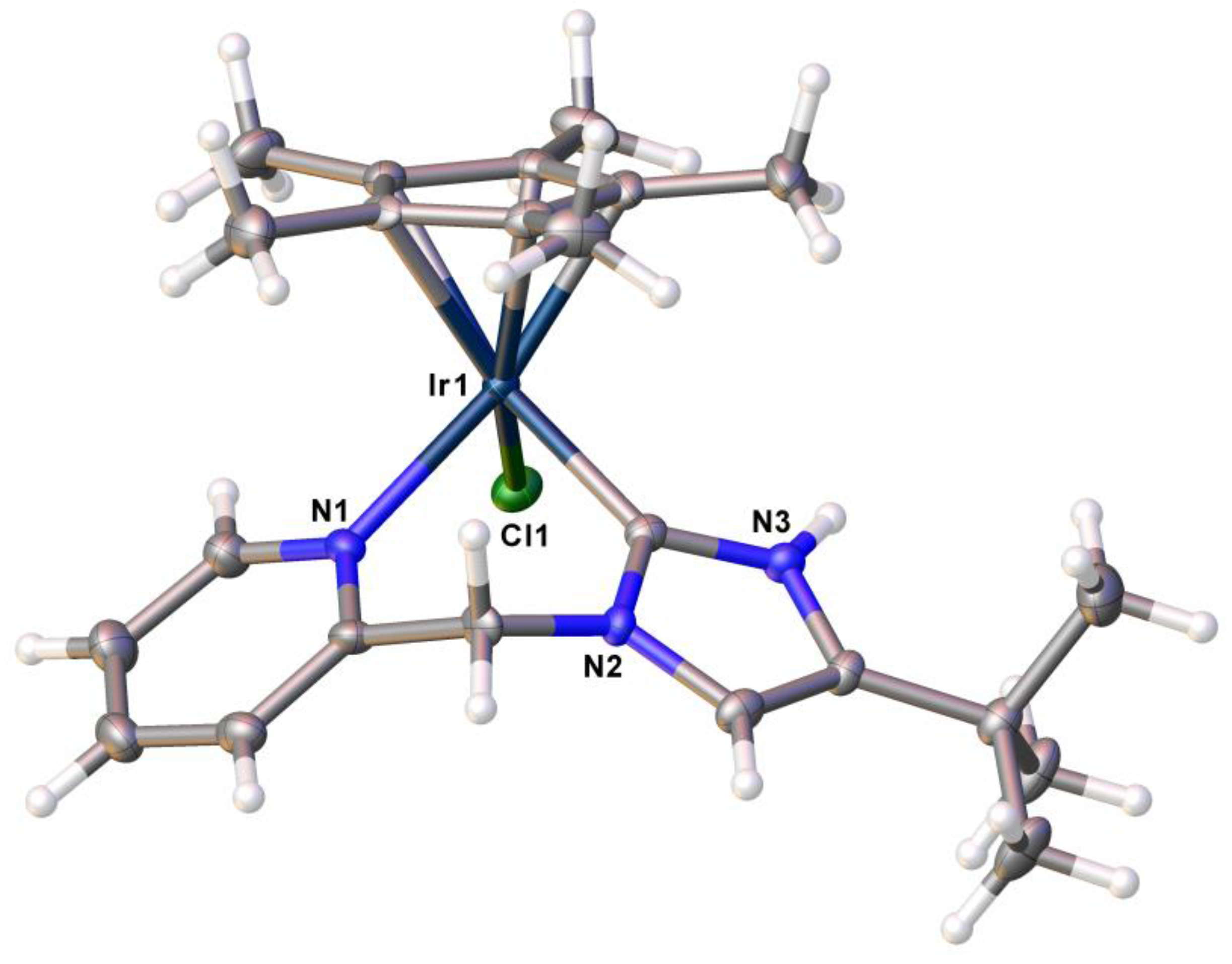
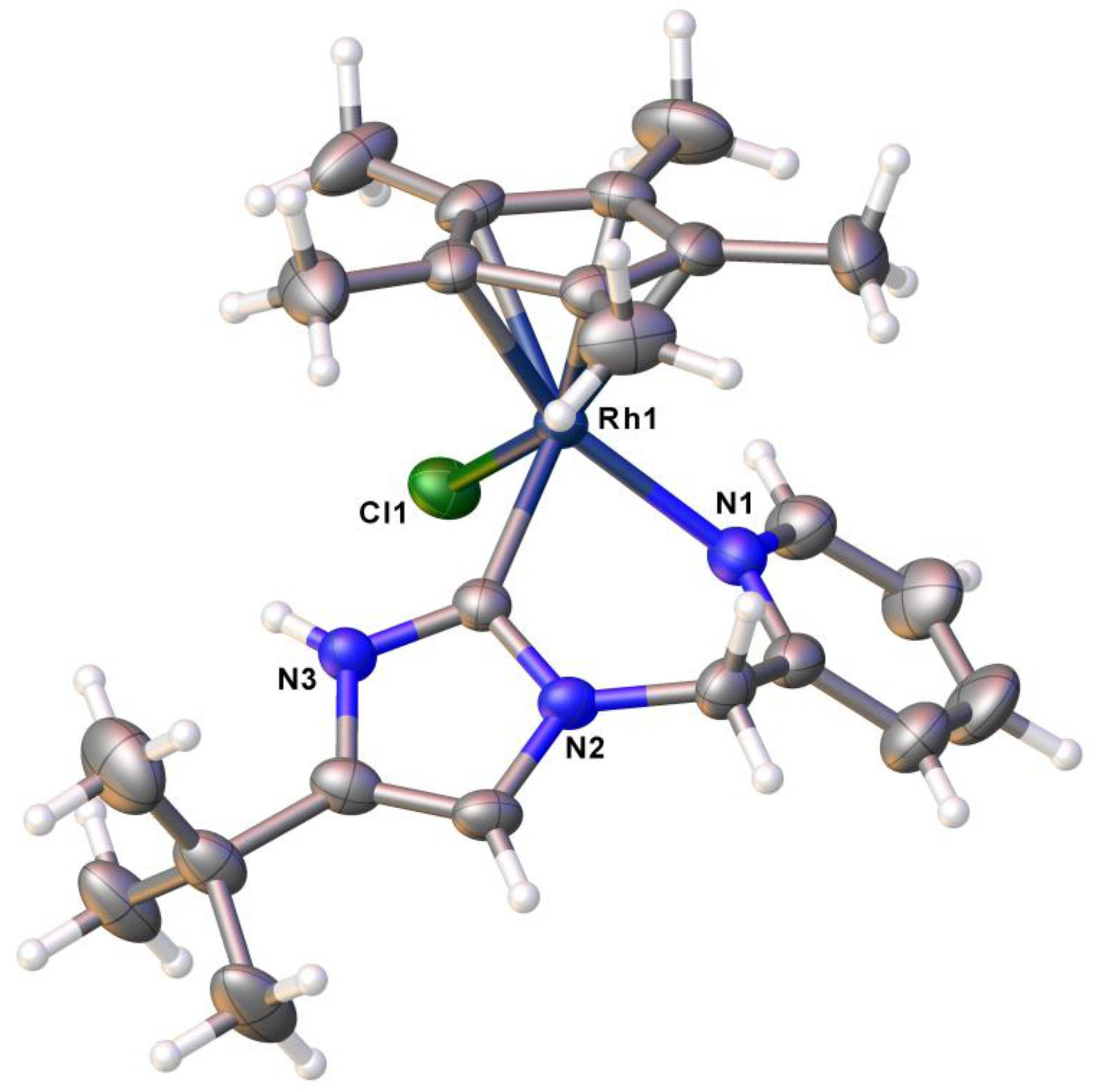
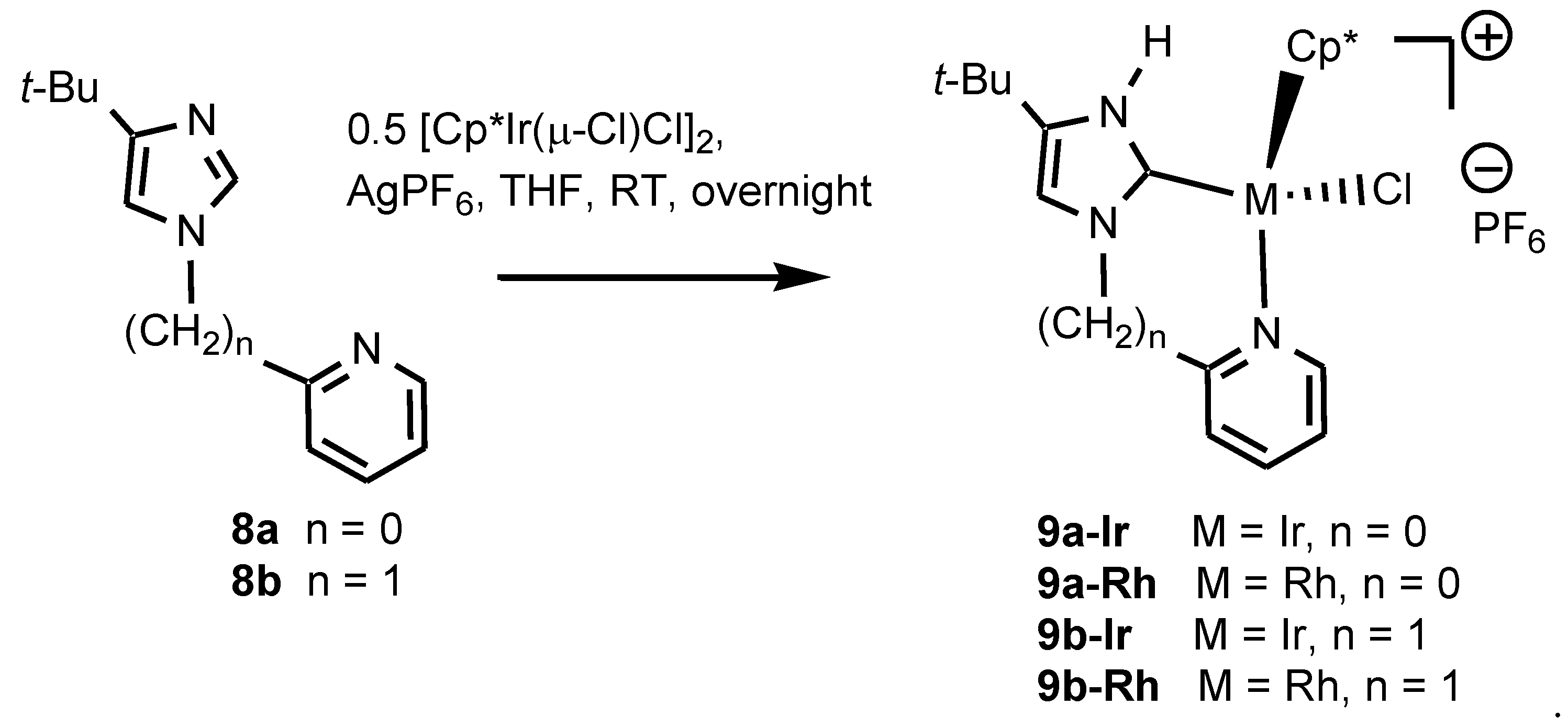
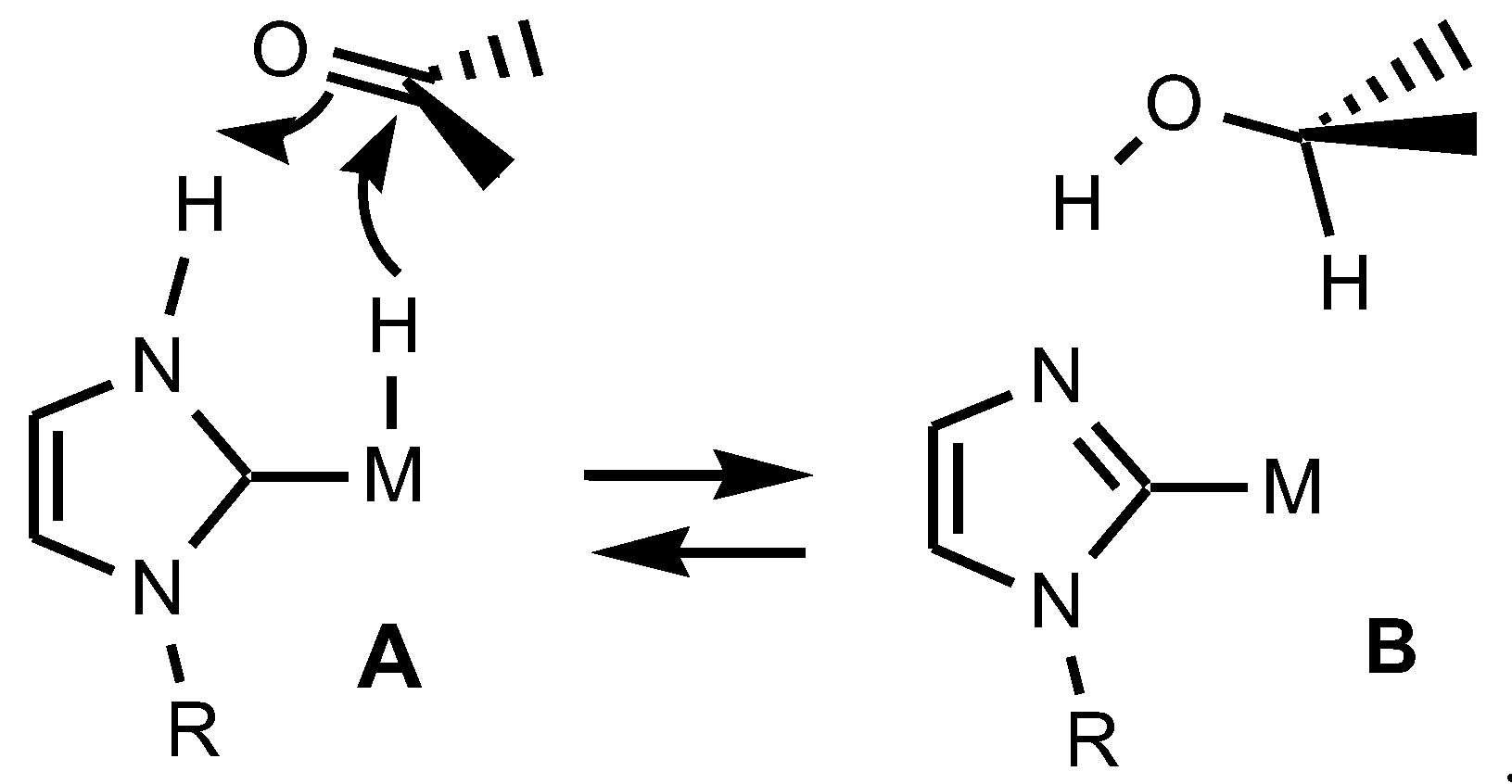
2.1. Syntheses and Characterization by NMR and X-ray Diffraction
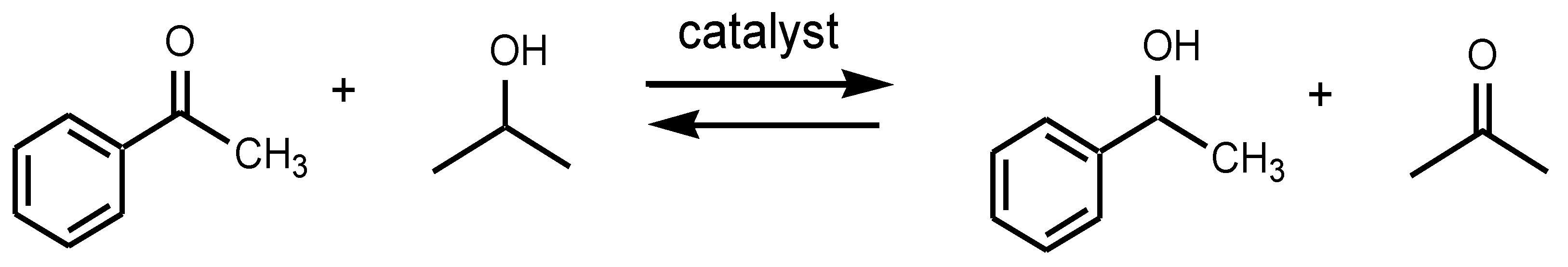
2.2. Catalysis
3. Discussion
4. Materials and Methods
4.1. Synthesis of 8a Using NaH
4.2. Synthesis of 8a [2-(4-tert-butyl-1H-imidazol-1-yl)pyridine] Using CuI
4.3. Synthesis of 9a-Ir with the Aid of KPF6
4.4. Synthesis of 9a-Ir with the Aid of AgPF6
4.5. Synthesis of 9a-Rh with the Aid of KPF6
4.6. Synthesis of 9a-Rh with the Aid of AgPF6
4.7. Synthesis of 9b-Ir with the Aid of KPF6
4.8. Synthesis of 9b-Ir with the Aid of AgPF6
4.9. Synthesis of 9b-Rh with the Aid of KPF6
4.10. Synthesis of 9b-Rh with the Aid of AgPF6
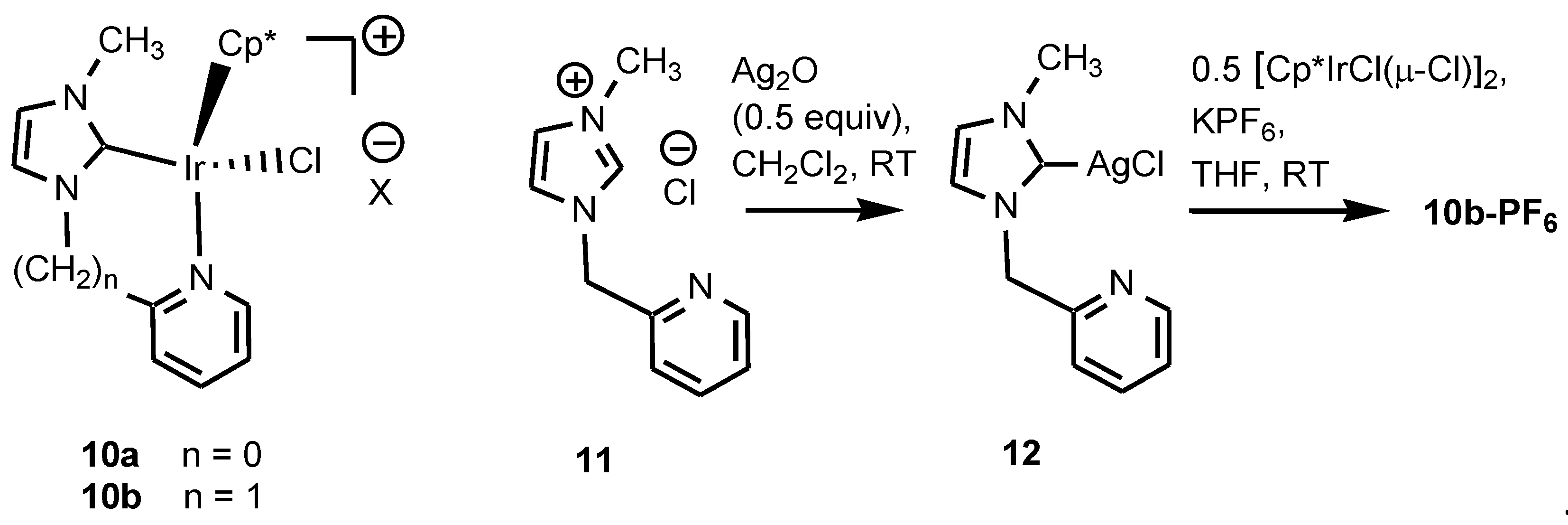
4.11. Synthesis of 10b-PF6
4.12. General Catalytic Procedures for the Reduction of Acetophenone to 1-Phenylethanol
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jahnke, M.C.; Hahn, F.E. Complexes with protic (NH,NH and NH,NR) N-heterocyclic carbene ligands. Coord. Chem. Rev. 2015, 293–294, 95–115. [Google Scholar] [CrossRef]
- Kuwata, S.; Ikariya, T. Metal-ligand bifunctional reactivity and catalysis of protic N-heterocyclic carbene and pyrazole complexes featuring β-NH units. Chem. Commun. 2014, 50, 14290–14300. [Google Scholar] [CrossRef] [PubMed]
- Huertos, M.A.; Perez, J.; Riera, L.; Menendez-Velazquez, A. From N-alkylimidazole ligands at a rhenium center: Ring opening or formation of NHC complexes. J. Am. Chem. Soc. 2008, 130, 13530–13531. [Google Scholar] [CrossRef] [PubMed]
- Huertos, M.A.; Perez, J.; Riera, L.; Diaz, J.; Lopez, R. From bis(N-alkylimidazole) to bis(NH-NHC) in rhenium carbonyl complexes. Angew. Chem. Int. Ed. 2010, 49, 6409–6412. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, J.; Perandones, B.F. Base-promoted tautomerization of imidazole ligands to N-heterocyclic carbenes and subsequent transmetalation reaction. J. Am. Chem. Soc. 2007, 129, 9298–9299. [Google Scholar] [CrossRef] [PubMed]
- Bonati, F.; Burini, A.; Pietroni, B.R.; Bovio, B. Reactions of C-imidazolyllithium derivatives with group Ib compounds: Tris[μ-(1-alkylimidazolato-N3,C2)]trigold(I) and -silver(I). Crystal structure of bis(1-benzylimidazolin-2-ylidene)gold(I) chloride. J. Organomet. Chem. 1989, 375, 147–160. [Google Scholar] [CrossRef]
- Raubenheimer, H.G.; Cronje, S. Carbene complexes derived from lithiated heterocycles, mainly azoles, by transmetalation. J. Organomet. Chem. 2001, 617–618, 170–181. [Google Scholar] [CrossRef]
- Meier, N.; Hahn, F.E.; Pape, T.; Siering, C.; Waldvogel, S.R. Molecular recognition utilizing complexes with NH,NH-stabilized carbene ligands. Eur. J. Inorg. Chem. 2007, 1210–1214. [Google Scholar] [CrossRef]
- Dobereiner, G.E.; Chamberlin, C.A.; Schley, N.D.; Crabtree, R.H. Acyl protection strategy for synthesis of a protic NHC complex via N-acyl methanolysis. Organometallics 2010, 29, 5728–5731. [Google Scholar] [CrossRef]
- Isobe, K.; Kai, E.; Nakamura, Y.; Nishimoto, K.; Miwa, T.; Kawaguchi, S.; Kinoshita, K.; Nakatsu, K. Trans-bromo(2-, 3-, and 4-pyridyl)bis(triethylphosphine)palladium(II) complexes. J. Am. Chem. Soc. 1980, 102, 2475–2476. [Google Scholar] [CrossRef]
- Isobe, K.; Kawaguchi, S. Organopalladium(II) complexes containing carbon-bonded pyridine and picoline as a ligand: Preparation, structures, and reactions. Heterocycles 1981, 16, 1603–1612. [Google Scholar]
- Crociani, B.; Di Bianca, F.; Giovenco, A.; Scrivanti, A. Protonation and methylation reactions of 2-pyridyl-palladium(II) and platinumu(II) complexes. J. Organomet. Chem. 1983, 251, 393–411. [Google Scholar] [CrossRef]
- Miranda-Soto, V.; Grotjahn, D.B.; DiPasquale, A.G.; Rheingold, A.L. Imidazol-2-yl complexes of Cp*Ir as bifunctional ambident reactants. J. Am. Chem. Soc. 2008, 130, 13200–13201. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Soto, V.; Grotjahn, D.B.; Cooksy, A.L.; Golen, J.A.; Moore, C.E.; Rheingold, A.L. A labile and catalytically active imidazol-2-yl fragment system. Angew. Chem. Int. Ed. 2011, 50, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Marelius, D.C.; Moore, C.E.; Rheingold, A.L.; Grotjahn, D.B. Reactivity studies of pincer bis-protic N-heterocyclic carbene complexes of platinum and palladium under basic conditions. Beilstein J. Org. Chem. 2016, 12, 1334–1339. [Google Scholar] [CrossRef] [PubMed]
- Marelius, D.C.; Darrow, E.H.; Moore, C.E.; Rheingold, A.L.; Grotjahn, D.B. Hydrogen-bonding pincers with two protic N-heterocyclic carbenes from direct metalation of a 1,8-bis(imidazol-1-yl)carbazole by platinum, palladium, as well as nickel. Chem. Eur. J. 2015, 21, 10988–10992. [Google Scholar] [CrossRef] [PubMed]
- Flowers, S.E.; Cossairt, B.M. Mono- and dimetalation of a tridentate bisimidazole-phosphine ligand. Organometallics 2014, 33, 4341–4344. [Google Scholar] [CrossRef]
- Araki, K.; Kuwata, S.; Ikariya, T. Isolation and interconversion of protic N-heterocyclic carbene and imidazolyl complexes: Application to catalytic dehydrative condensation of N-(2-pyridyl)benzimidazole and allyl alcohol. Organometallics 2008, 27, 2176–2178. [Google Scholar] [CrossRef]
- Song, G.; Su, Y.; Periana, R.A.; Crabtree, R.H.; Han, K.; Zhang, H.; Li, X. Anion-exchange-triggered 1,3-shift of an NH proton to iridium in protic N-heterocyclic carbenes: Hydrogen-bonding and ion-pairing effects. Angew. Chem., Int. Ed. 2010, 49, 912–917. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.; Breinbauer, R. A simple synthesis of functionalized 3-methyl-1-pyridinyl-1H-imidazolium salts as bidentate N-heterocyclic-carbene precursors and their application in ir-catalyzed arene borylation. Tetrahedron Lett. 2010, 51, 6622–6625. [Google Scholar] [CrossRef]
- Chiu, P.L.; Lai, C.-L.; Chang, C.-F.; Hu, C.-H.; Lee, H.M. Synthesis, structural characterization, computational study, and catalytic activity of metal complexes based on tetradentate pyridine/N-heterocyclic carbene ligand. Organometallics 2005, 24, 6169–6178. [Google Scholar] [CrossRef]
- Xiao, X.-Q.; Jin, G.-X. Functionalized N-heterocyclic carbene iridium complexes: Synthesis, structure and addition polymerization of norbornene. J. Organomet. Chem. 2008, 693, 3363–3368. [Google Scholar] [CrossRef]
- Gnanamgari, D.; Sauer, E.L.O.; Schley, N.D.; Butler, C.; Incarvito, C.D.; Crabtree, R.H. Iridium and ruthenium complexes with chelating N-heterocyclic carbenes: Efficient catalysts for transfer hydrogenation, β-alkylation of alcohols, and N-alkylation of amines. Organometallics 2009, 28, 321–325. [Google Scholar] [CrossRef]
- Hintermair, U.; Campos, J.; Brewster, T.P.; Pratt, L.M.; Schley, N.D.; Crabtree, R.H. Hydrogen-transfer catalysis with Cp*IrIII complexes: The influence of the ancillary ligands. ACS Catal. 2014, 4, 99–108. [Google Scholar] [CrossRef]
- Navarro, M.; Smith, C.A.; Albrecht, M. Enhanced catalytic activity of iridium(III) complexes by facile modification of C,N-bidentate chelating pyridylideneamide ligands. Inorg. Chem. 2017, 56, 11688–11701. [Google Scholar] [CrossRef] [PubMed]
- Mazloomi, Z.; Pretorius, R.; Pamies, O.; Albrecht, M.; Dieguez, M. Triazolylidene iridium complexes for highly efficient and versatile transfer hydrogenation of C=O, C=N, and C=C bonds and for acceptorless alcohol oxidation. Inorg. Chem. 2017, 56, 11282–11298. [Google Scholar] [CrossRef] [PubMed]
- Corberán, R.; Peris, E. An unusual example of base-free catalyzed reduction of C=O and C=NR bonds by transfer hydrogenation and some useful implications. Organometallics 2008, 27, 1954–1958. [Google Scholar] [CrossRef]
- Moore, C.M.; Szymczak, N.K. 6,6’-dihydroxy terpyridine: A proton-responsive bifunctional ligand and its application in catalytic transfer hydrogenation of ketones. Chem. Commun. 2013, 49, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Nieto, I.; Livings, M.S.; Sacci, J.B.; Reuther, L.E.; Zeller, M.; Papish, E.T. Transfer hydrogenation in water via a ruthenium catalyst with OH groups near the metal center on a bipy scaffold. Organometallics 2011, 30, 6339–6342. [Google Scholar] [CrossRef]
- Larsen, C.R.; Erdogan, G.; Grotjahn, D.B. General catalyst control of the monoisomerization of 1-alkenes to trans-2-alkenes. J. Am. Chem. Soc. 2014, 136, 1226–1229. [Google Scholar] [CrossRef] [PubMed]
- Grotjahn, D.B.; Kraus, J.E.; Amouri, H.; Rager, M.-N.; Cortes-Llamas, S.A.; Mallari, A.A.; DiPasquale, A.G.; Liable-Sands, L.M.; Golen, J.A.; Zakharov, L.N.; et al. Multimodal study of secondary interactions in Cp*Ir complexes of imidazolylphosphines bearing an NH group. J. Am. Chem. Soc. 2010, 132, 7919–7934. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | 9b-Ir | 9b-Rh | Angles | 9b-Ir | 9b-Rh |
|---|---|---|---|---|---|
| M1–C1 | 2.017(3) | 2.020(4) | Cl1–M1–C1 | 89.33(11) | 90.66(12) |
| H1N–Cl1 | 3.436 | 3.432 | Cl1–M1–N3 | 86.46(9) | 88.08(11) |
| M1–N3 | 2.108(2) | 2.111(4) | C1–Ir1–N3 | 85.01(12) | 85.49(16) |
| C11–M1 | 2.4154(8) | 2.407(1) | - | - | - |
| Time (h) | [Cp*IrIIICl(μ-Cl)]2 | 9a-Ir | 9b-Ir | 10b-PF6 |
|---|---|---|---|---|
| 0 | 0 | 0 | 0 | 0 |
| 1 | 58.7 | 11.0 | 36.9 | 2.7 |
| 3 | 66.2 | 34.9 | 75.4 | 7.6 |
| 6 | 66.6 | 15.7 | ||
| 20 | 91.2 | 36.1 | ||
| 48 | 80.8 | 54.2 | ||
| 72 | 80.7 | 61.3 |
| Time (min) | 9a-Rh | 9b-Rh | [Cp*RhIIICl(μ-Cl)]2 |
|---|---|---|---|
| 0 | 0 | 0 | 0 |
| 5 | 13.9 | 19.8 | 2.5 |
| 10 | - | 54.8 | 4.0 |
| 15 | 18.4 | 73.8 | 3.8 |
| 20 | - | 84.5 | 4.1 |
| 25 | 22.9 | 92.0 | 4.2 |
| 30 | - | 92.7 | 4.5 |
| 35 | 24.7 | 95.4 | 4.3 |
| 40 | - | 92.1 | 5.1 |
| 45 | 32.4 | 94.5 | 5.5 |
| 50 | - | 91.6 | 5.8 |
| 55 | - | 97.0 | 5.0 |
| 60 | 49.1 | 96.3 | 5.7 |
| 180 | 99.7 | 94.8 | 9.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grotjahn, D.B.; Martin, J.K.; Tom, T.N.; Rheingold, A.L. Cationic Protic Imidazolylidene NHC Complexes of Cp*IrCl+ and Cp*RhCl+ with a Pyridyl Tether Formed at Ambient Temperature. Inorganics 2018, 6, 27. https://doi.org/10.3390/inorganics6010027
Grotjahn DB, Martin JK, Tom TN, Rheingold AL. Cationic Protic Imidazolylidene NHC Complexes of Cp*IrCl+ and Cp*RhCl+ with a Pyridyl Tether Formed at Ambient Temperature. Inorganics. 2018; 6(1):27. https://doi.org/10.3390/inorganics6010027
Chicago/Turabian StyleGrotjahn, Douglas B., Jessica K. Martin, Taylon N. Tom, and Arnold L. Rheingold. 2018. "Cationic Protic Imidazolylidene NHC Complexes of Cp*IrCl+ and Cp*RhCl+ with a Pyridyl Tether Formed at Ambient Temperature" Inorganics 6, no. 1: 27. https://doi.org/10.3390/inorganics6010027
APA StyleGrotjahn, D. B., Martin, J. K., Tom, T. N., & Rheingold, A. L. (2018). Cationic Protic Imidazolylidene NHC Complexes of Cp*IrCl+ and Cp*RhCl+ with a Pyridyl Tether Formed at Ambient Temperature. Inorganics, 6(1), 27. https://doi.org/10.3390/inorganics6010027