Synthesis and Characterization of N-Heterocyclic Carbene-Coordinated Silicon Compounds Bearing a Fused-Ring Bulky Eind Group


 , ,
, , 
Abstract
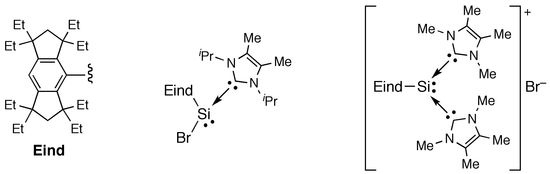
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussions
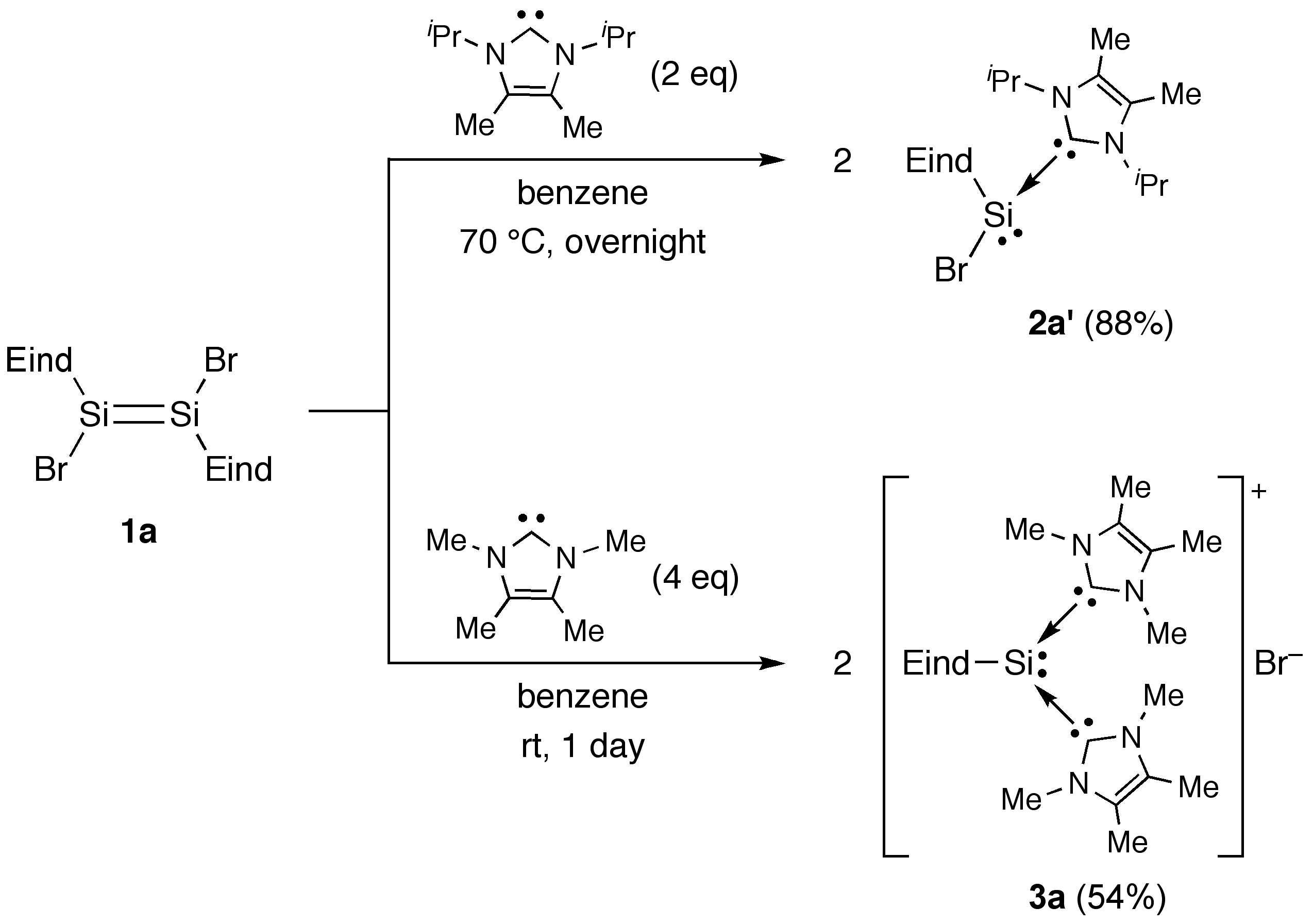
2.1. Reactions of (Eind)BrSi=SiBr(Eind) (1a) with NHCs
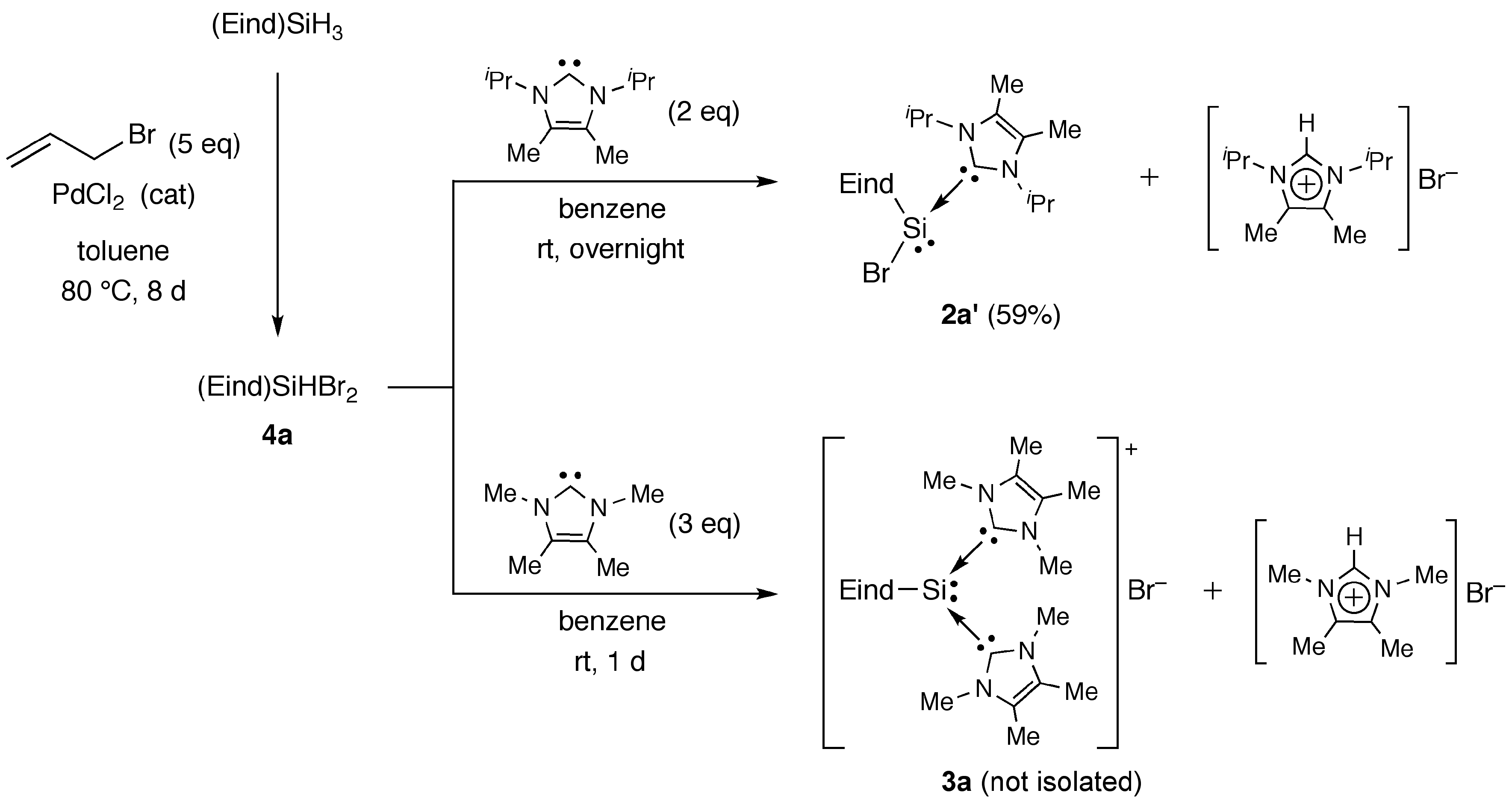
2.2 Reactions of (Eind)SiHBr2 (4a) with NHCs
3. Materials and Methods
3.1. General Procedures
3.1.1. Synthesis of (Eind)SiHBr2 (4a)
3.1.2. Synthesis of (Im-iPr2Me2)→SiBr(Eind) (2a′)
(Method A) Reaction of (Eind)BrSi=SiBr(Eind) (1a) with Im-iPr2Me2
(Method B) Reaction of (Eind)SiHBr2 (4a) with Im-iPr2Me2
3.1.3. Synthesis of [(Im-Me4)2→Si(Eind)]+[Br]– (3a)
(Method A) Reaction of (Eind)BrSi=SiBr(Eind) (1a) with Im-Me4
(Method B) Reaction of (Eind)SiHBr2 (4a) with Im-Me4
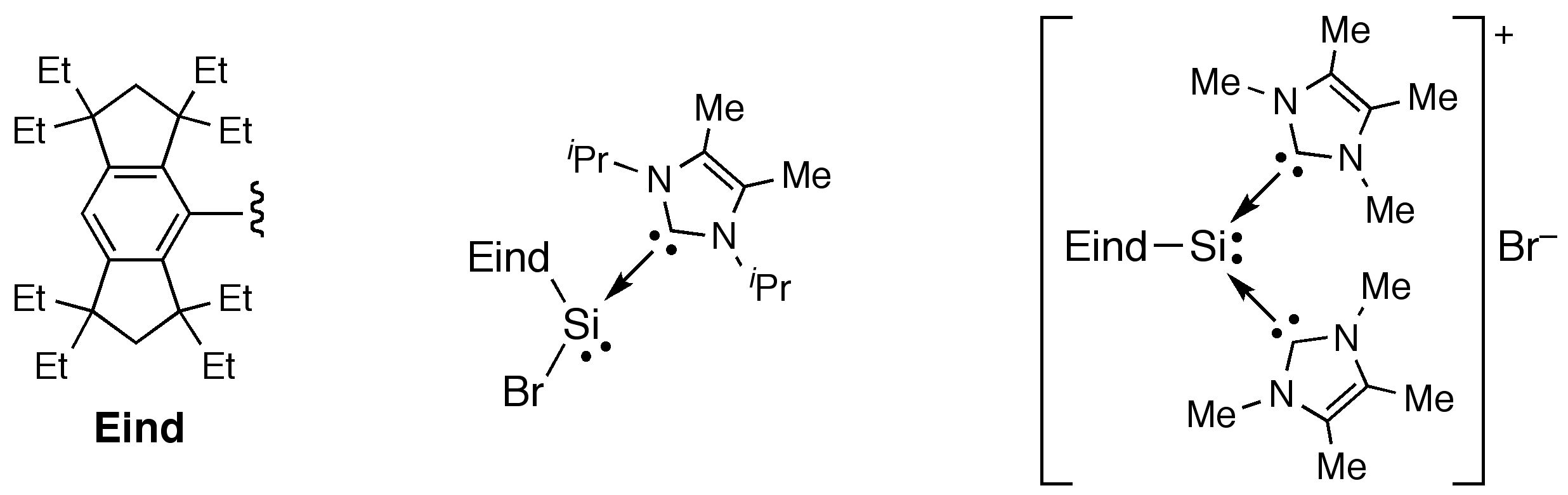
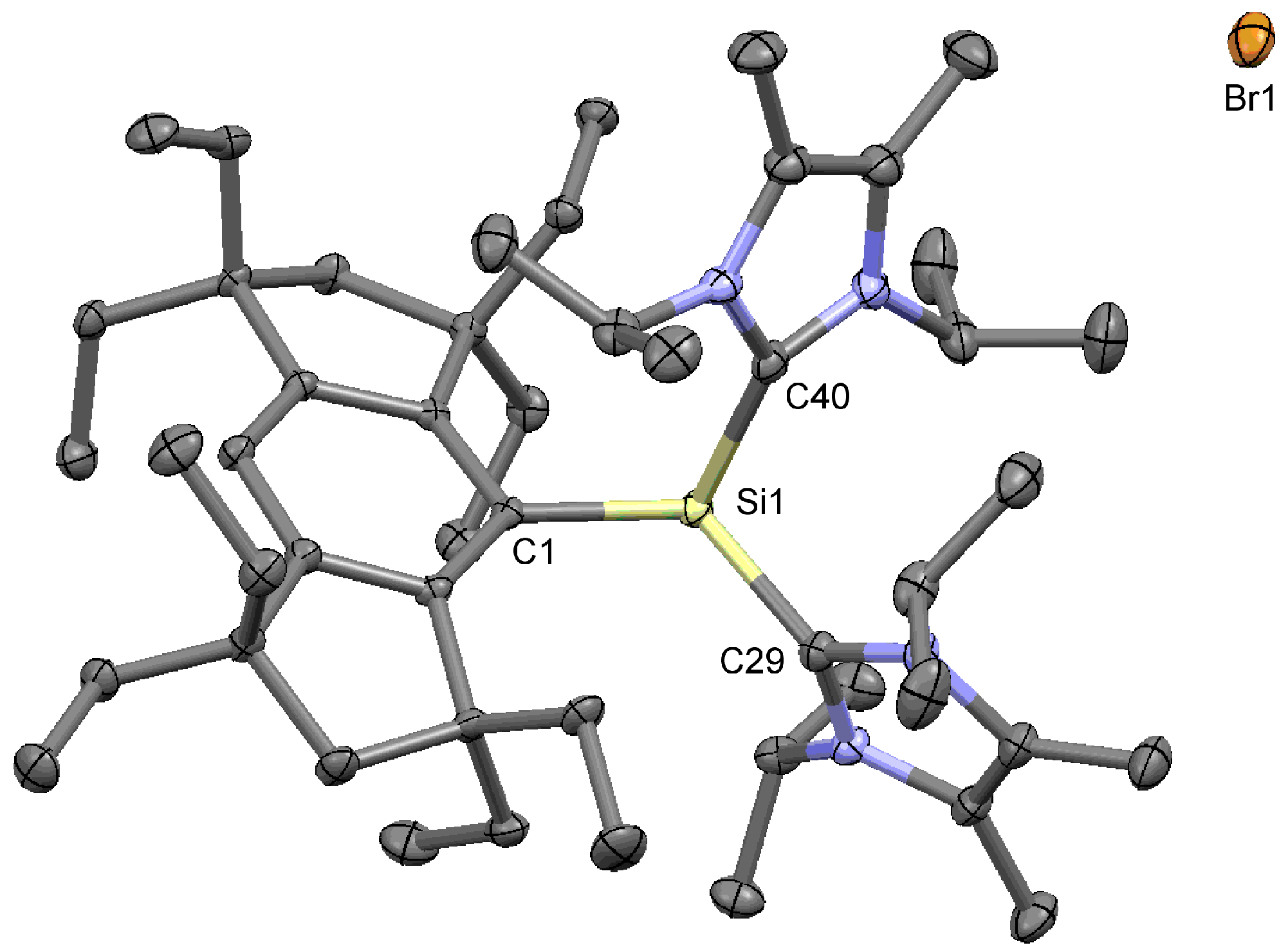
3.2. X-ray Crystallographic Studies of 3a′ and 4a
3.2.1. [(Im-iPr2Me2)2→Si(Eind)]+[Br−] (3a′)
3.2.2. (Eind)SiHBr2 (4a)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kira, M.; Iwamoto, T. Progress in the chemistry of stable disilenes. Adv. Organomet. Chem. 2006, 54, 73–148. [Google Scholar]
- Wang, Y.; Robinson, G.H. Unique homonuclear multiple bonding in main group compounds. Chem. Commun. 2009, 5201–5213. [Google Scholar] [CrossRef] [PubMed]
- Mizuhata, Y.; Sasamori, T.; Tokitoh, N. Stable heavier carbene analogues. Chem. Rev. 2009, 109, 3479–3511. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.C.; Power, P.P. π-Bonding and lone pair effect in multiple bonds involving heavier main group elements: Developments in the new millennium. Chem. Rev. 2010, 110, 3877–3923. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.Y.; Sekiguchi, A. Organometallic Compounds of Low-Coordinate Si, Ge, Sn and Pb: From Phantom Species to Stable Compounds; John Wiley & Sons Ltd.: Chichester, UK, 2010; ISBN 978-0-470-72543-6. [Google Scholar]
- Asay, M.; Jones, C.; Driess, M. N-Heterocyclic carbene analogues with low-valent group 13 and group 14 elements: Syntheses, structures, and reactivities of a new generation of multitalented ligands. Chem. Rev. 2011, 111, 354–396. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Xiong, Y.; Driess, M. Zwitterionic and donor-stabilized N-heterocyclic silylenes (NHSis) for metal-free activation of small molecules. Organometallics 2011, 30, 1748–1767. [Google Scholar] [CrossRef]
- Kira, M. Bonding and structure of disilenes and related unsaturated Group-14 element compounds. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 167–191. [Google Scholar] [CrossRef] [PubMed]
- Scheschkewitz, D. Functional Molecular Silicon Compounds II: Low Oxidation States; Springer: Basel, Switzerland, 2014; ISBN 978-3-319-03734-9. [Google Scholar]
- Wang, Y.; Robinson, G.H. N-heterocyclic carbene—Main-group chemistry: A rapidly evolving field. Inorg. Chem. 2014, 53, 11815–11832. [Google Scholar] [CrossRef] [PubMed]
- Ghadwal, R.S.; Azhakar, R.; Roesky, H.W. Dichlorosilylene: A high temperature transient species to an indispensable building block. Acc. Chem. Res. 2013, 46, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Ghadwal, R.S.; Roesky, H.W.; Merkel, S.; Henn, J.; Stalke, D. Lewis base stabilized dichlorosilylene. Angew. Chem. Int. Ed. 2009, 48, 5683–5686. [Google Scholar] [CrossRef] [PubMed]
- Filippou, A.C.; Chernov, O.; Schnakenburg, G. SiBr2(Idipp): A stable N-heterocyclic carbene adduct of dibromosilylene. Angew. Chem. Int. Ed. 2009, 48, 5687–5690. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Stollberg, P.; Herbst-Irmer, R.; Stalke, D.; Andrada, D.M.; Frenking, G.; Roesky, H.W. Carbene-dichlorosilylene stabilized phosphinidenes exhibiting strong intramolecular charge transfer transition. J. Am. Chem. Soc. 2015, 137, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Hickox, H.P.; Wang, Y.; Xie, Y.; Wei, P.; Schaefer, H.F., III; Robinson, G.H. Push-pull stabilization of parent monochlorosilylenes. J. Am. Chem. Soc. 2016, 138, 9799–9802. [Google Scholar] [CrossRef] [PubMed]
- Geiβ, D.; Arz, M.I.; Straβmann, M.; Schnakenburg, G.; Filippou, A.C. Si=P double bonds: experimental and theoretical study of an NHC-stabilized phosphasilenylidene. Angew. Chem. Int. Ed. 2015, 54, 2739–2744. [Google Scholar]
- Ghana, P.; Arz, M.I.; Das, U.; Schnakenburg, G.; Filippou, A.C. Si=Si double bonds: Synthesis of an NHC-stabilized disilavinylidene. Angew. Chem. Int. Ed. 2015, 54, 9980–9985. [Google Scholar] [CrossRef] [PubMed]
- Filippou, A.C.; Chernov, O.C.; Blom, B.; Stumpf, K.W.; Schnakenburg, G. Stable N-heterocyclic carbene adducts of arylchlorosilylenes and their germanium homologues. Chem. Eur. J. 2010, 16, 2866–2872. [Google Scholar] [CrossRef] [PubMed]
- Filippou, A.C.; Chernov, O.C.; Stumpf, K.W.; Schnakenburg, G. Metal-silicon triple bonds: The molybdrnumsilylidyne complex [Cp(CO)2Mo≡Si-Ar]. Angew. Chem. Int. Ed. 2010, 49, 3296–3300. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Matsuo, T.; Hashizume, D.; Tamao, K. Room-temperature dissociation of 1,2-dibromodisilenes to bromosilylenes. J. Am. Chem. Soc. 2011, 133, 19710–19713. [Google Scholar] [CrossRef] [PubMed]
- Agou, T.; Sasamori, T.; Tokitoh, N. Synthesis of an arylbromosilylene-platinum complex by using a 1,2-dibromodisilene as a silylene source. Organometallics 2012, 31, 1150–1154. [Google Scholar] [CrossRef]
- Agou, T.; Hayakawa, N.; Sasamori, T.; Matsuo, T.; Hashizume, D.; Tokitoh, N. Reactions of diaryldibromodisilenes with N-heterocyclic carbenes: Formation of formal bis-NHC adducts of silyliumylidene cations. Chem. Eur. J. 2014, 20, 9246–9249. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.U.; Szilvási, T.; Inoue, S. A facile access to a novel NHC-stabilized silyliumylidene ion and C–H activation of phenylacetylene. Chem. Commun. 2014, 50, 12619–12622. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.U.; Szilvási, T.; Irran, E.; Inoue, S. An NHC-stabilized silicon analogue of acylium ion: Synthesis, structure, reactivity, and theoretical studies. J. Am. Chem. Soc. 2015, 137, 5828–5836. [Google Scholar] [CrossRef] [PubMed]
- Mork, B.V.; Tilley, T.D. Multiple bonding between silicon and molybdenum: A transition-metal complex with considerable silylyne character. Angew. Chem. Int. Ed. 2003, 42, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Suzuki, K.; Fukawa, T.; Li, B.L.; Ito, M.; Shoji, Y.; Otani, T.; Li, L.C.; Kobayashi, M.; Hachiya, M.; et al. Synthesis and structures of a series of bulky “Rind-Br” based on a rigid fused-ring s-hydrindacene skeleton. Bull. Chem. Soc. Jpn. 2011, 84, 1178–1191. [Google Scholar] [CrossRef]
- Matsuo, T.; Tamao, K. Fused-ring bulky “Rind” groups producing new possibilities in elemento-organic chemistry. Bull. Chem. Soc. Jpn. 2015, 88, 1201–1220. [Google Scholar] [CrossRef]
- Cui, H.; Cui, C. Silylation of N-heterocyclic carbene with aminochlorosilane and -disilane: Dehydrohalogenation vs. Si–Si bond cleavage. Dalton Trans. 2011, 40, 11937–11940. [Google Scholar] [CrossRef] [PubMed]
- Hadlington, T.J.; Szilvási, T.; Driess, M. Silylene-nichkel promoted cleavage of B–O bonds: From catechol borane to the hydroborylene ligand. Angew. Chem. Int. Ed. 2017, 56, 7470–7474. [Google Scholar] [CrossRef] [PubMed]
- Sasamoti, T.; Hironaka, K.; Sugiyama, Y.; Takagi, N.; Nagase, S.; Hosoi, Y.; Furukawa, Y.; Tokitoh, N. Synthesis and reactions of a stable 1,2-diaryl-1,2-dibromodisilene: A precursor for substituted disilenes and a 1,2-diaryldisilyne. J. Am. Chem. Soc. 2008, 130, 13856–13857. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.-H.; Ho, P.-Y.; Su, M.-D. Mechanisms for the reactions of Group 10 transition metal complexes with metal-group 14 element bonds, Bbt(Br)E=M(PCy3)2 (E = C, Si, Ge, Sn, Pb; M = Pd and Pt). Inorg. Chem. 2013, 52, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Filippou, A.C.; Hoffmann, D.; Schnakenburg, G. Triple bonds of niobium with silicon, germanium and tin: The tetrylidyne complexes [(κ3-tmps)(CO)2Nb≡E–R] (E = Si, Ge, Sn; tmps = MeSi(CH2OMe2)3; R = aryl). Chem. Sci. 2017, 8, 6290–6299. [Google Scholar] [CrossRef]
- Blom, B. Reactivity of Ylenes at Late Transition Metal Centers. Dissertation, University of Bonn, Göttingen, Germany, 2011. [Google Scholar]
- Papazoglou, I. Unprecedented Tetrylidyne Complexes of Group 6 and Group 10 Metals. Dissertation, University of Bonn, Dr. Hut Verlag, München, Germany, 28 May 2017. [Google Scholar]
- Filippou, A.C.; Lebedev, Y.N.; Chernov, O.; Straβmann, M.; Schnakenburg, G. Silicon(II) coordination chemistry: N-Heterocyclic carbene complexes of Si2+ and SiI+. Angew. Chem. Int. Ed. 2013, 52, 6974–6978. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, D.; Wendel, D.; Ahmad, S.U.; Szilvási, T.; Pöthig, A.; Inoue, S. Chalcogen-atom transfer and exchange reactions of NHC-stabilized heavier silaacylium ions. Dalton Trans. 2017, 46, 16014–16018. [Google Scholar] [CrossRef] [PubMed]
- Fukazawa, A.; Li, Y.; Yamaguchi, S.; Tsuji, H.; Tamao, K. Coplanar oligo(p-phenylenedisilenylene)s based on the octaethyl-substituted s-hydrindacenyl groups. J. Am. Chem. Soc. 2007, 129, 14164–14165. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Hayakawa, N.; Nakabayashi, K.; Matsuo, T.; Hashizume, D.; Fueno, H.; Tanaka, K.; Tamao, K. Highly coplanar (E)-1,2-di(1-naphthyl)disilene involving a distinct CH–π interaction with the perpendicularly oriented protecting Eind group. Chem. Lett. 2014, 43, 432–434. [Google Scholar] [CrossRef]
- Iwata, A.; Toyoshima, Y.; Hayashida, T.; Ochi, T.; Kunai, A.; Ohshita, J. PdCl2 and NiCl2-catalyzed hydrogen-halogen exchange for the convenient preparation of bromo- and iodosilanes and germanes. J. Organomet. Chem. 2003, 667, 90–95. [Google Scholar] [CrossRef]
- Kunai, A.; Ochi, T.; Iwata, A.; Ohshita, J. Synthesis of bromohydrosilanes: Reactions of hydrosilanes with CuBr2 in the presence of CuI. Chem. Lett. 2001, 1228–1229. [Google Scholar] [CrossRef]
- Agou, T.; Sugiyama, Y.; Sasamori, T.; Sakai, H.; Furukawa, Y.; Takagi, N.; Guo, J.-D.; Nagasa, S.; Hashizume, D.; Tokitoh, N. Synthesis of Kinetically Stabilized 1,2-Dihydrodisilenes. J. Am. Chem. Soc. 2012, 134, 4120–4123. [Google Scholar] [CrossRef] [PubMed]
- Simons, R.S.; Haubrich, S.T.; Mork, B.V.; Niemeyer, M.; Power, P.P. The Syntheses and Characterization of the Bulky Terphenylsilanes and Chlorosilanes 2,6-Mes2C6H3SiCl3, 2,6-Trip2C6H3SiCl3, 2,6-Mes2C6H3SiHCl2, 2,6-Trip2C6H3SiHCl2, 2,6-Mes2C6H3SiH3, 2,6-Trip2C6H3SiH3 and 2,6-Mes2C6H3SiC12SiCl3. Main Group Chem. 1998, 2, 275–283. [Google Scholar] [CrossRef]
- Weidemann, N.; Schnakenburg, G.; Filippou, A.C. Novel silanes with sterically demanding aryl substituents. Z. Anorg. Allg. Chem. 2009, 635, 253–259. [Google Scholar] [CrossRef]
- Li, B.; Tsujimoto, S.; Li, Y.; Tsuji, H.; Tamao, K.; Hashizume, D.; Matsuo, T. Synthesis and characterization of diphosphenes bearing fused-ring bulky Rind groups. Heteroat. Chem. 2014, 25, 612–618. [Google Scholar] [CrossRef]
- Kuhn, S.; Kratz, T. Synthesis of imidazol-2-ylidenes by reduction of imidazole-2(3H)-thiones. Synthesis 1993, 561–562. [Google Scholar] [CrossRef]
- CrysAlisPro; Agilent Technologies Ltd.: Yarnton, Oxfordshire, UK, 2014.
- CrystalClear; Rigaku/MSC. Inc.: The Woodlands, TX, USA, 2005.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, C71, 3–8. [Google Scholar]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayakawa, N.; Sadamori, K.; Mizutani, S.; Agou, T.; Sugahara, T.; Sasamori, T.; Tokitoh, N.; Hashizume, D.; Matsuo, T. Synthesis and Characterization of N-Heterocyclic Carbene-Coordinated Silicon Compounds Bearing a Fused-Ring Bulky Eind Group. Inorganics 2018, 6, 30. https://doi.org/10.3390/inorganics6010030
Hayakawa N, Sadamori K, Mizutani S, Agou T, Sugahara T, Sasamori T, Tokitoh N, Hashizume D, Matsuo T. Synthesis and Characterization of N-Heterocyclic Carbene-Coordinated Silicon Compounds Bearing a Fused-Ring Bulky Eind Group. Inorganics. 2018; 6(1):30. https://doi.org/10.3390/inorganics6010030
Chicago/Turabian StyleHayakawa, Naoki, Kazuya Sadamori, Shinsuke Mizutani, Tomohiro Agou, Tomohiro Sugahara, Takahiro Sasamori, Norihiro Tokitoh, Daisuke Hashizume, and Tsukasa Matsuo. 2018. "Synthesis and Characterization of N-Heterocyclic Carbene-Coordinated Silicon Compounds Bearing a Fused-Ring Bulky Eind Group" Inorganics 6, no. 1: 30. https://doi.org/10.3390/inorganics6010030
APA StyleHayakawa, N., Sadamori, K., Mizutani, S., Agou, T., Sugahara, T., Sasamori, T., Tokitoh, N., Hashizume, D., & Matsuo, T. (2018). Synthesis and Characterization of N-Heterocyclic Carbene-Coordinated Silicon Compounds Bearing a Fused-Ring Bulky Eind Group. Inorganics, 6(1), 30. https://doi.org/10.3390/inorganics6010030