Atomic Layer Deposition on Porous Materials: Problems with Conventional Approaches to Catalyst and Fuel Cell Electrode Preparation




Abstract
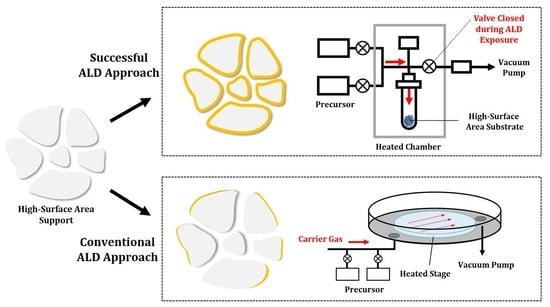
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Opportunities for Using ALD
1.2. Problems with Conventional ALD Approaches to Catalyst and Electrode Preparation
1.3. Design Considerations for Semiconductor Applications
1.4. Design Considerations for Catalysts and SOFC Electrodes
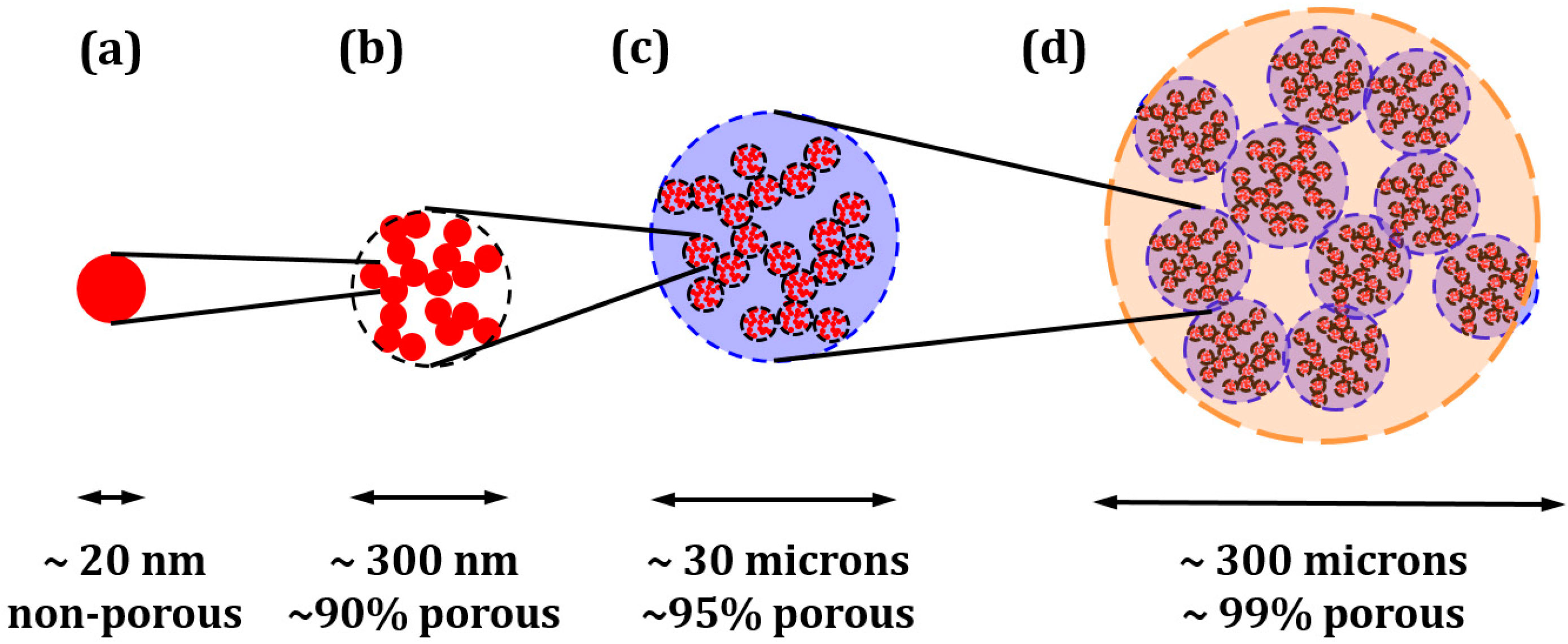
2. Considerations for Performing ALD in Catalysts and SOFC Electrodes
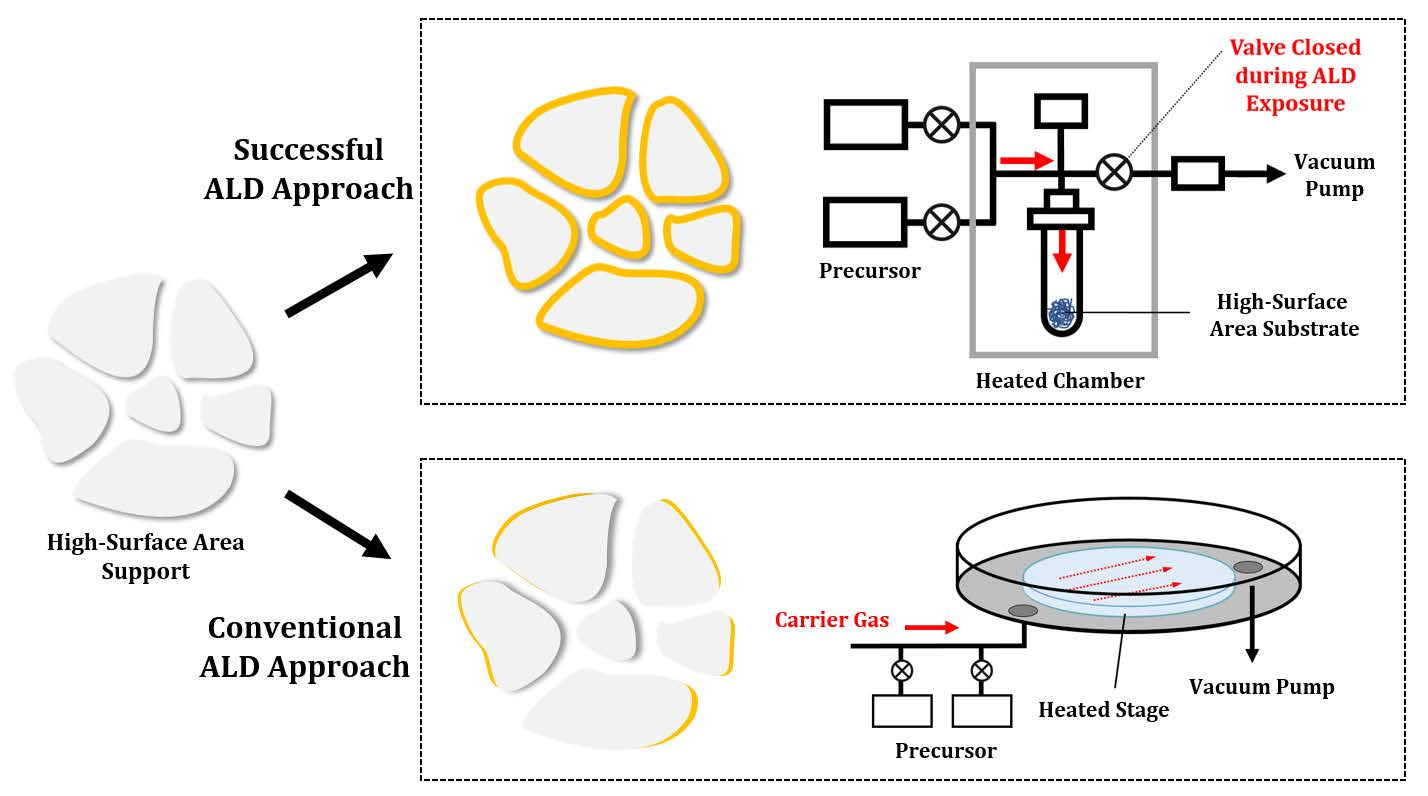
2.1. Adsorption of the Gaseous Precursor onto the Surface of the Substrate
2.2. Purging the Excess Precursor and Reaction Byproducts from the Sample
2.3. Gaseous Reactants to Remove Ligands and to Regenerate Sites
2.4. Purging the Excess Reactants and Reaction Byproducts
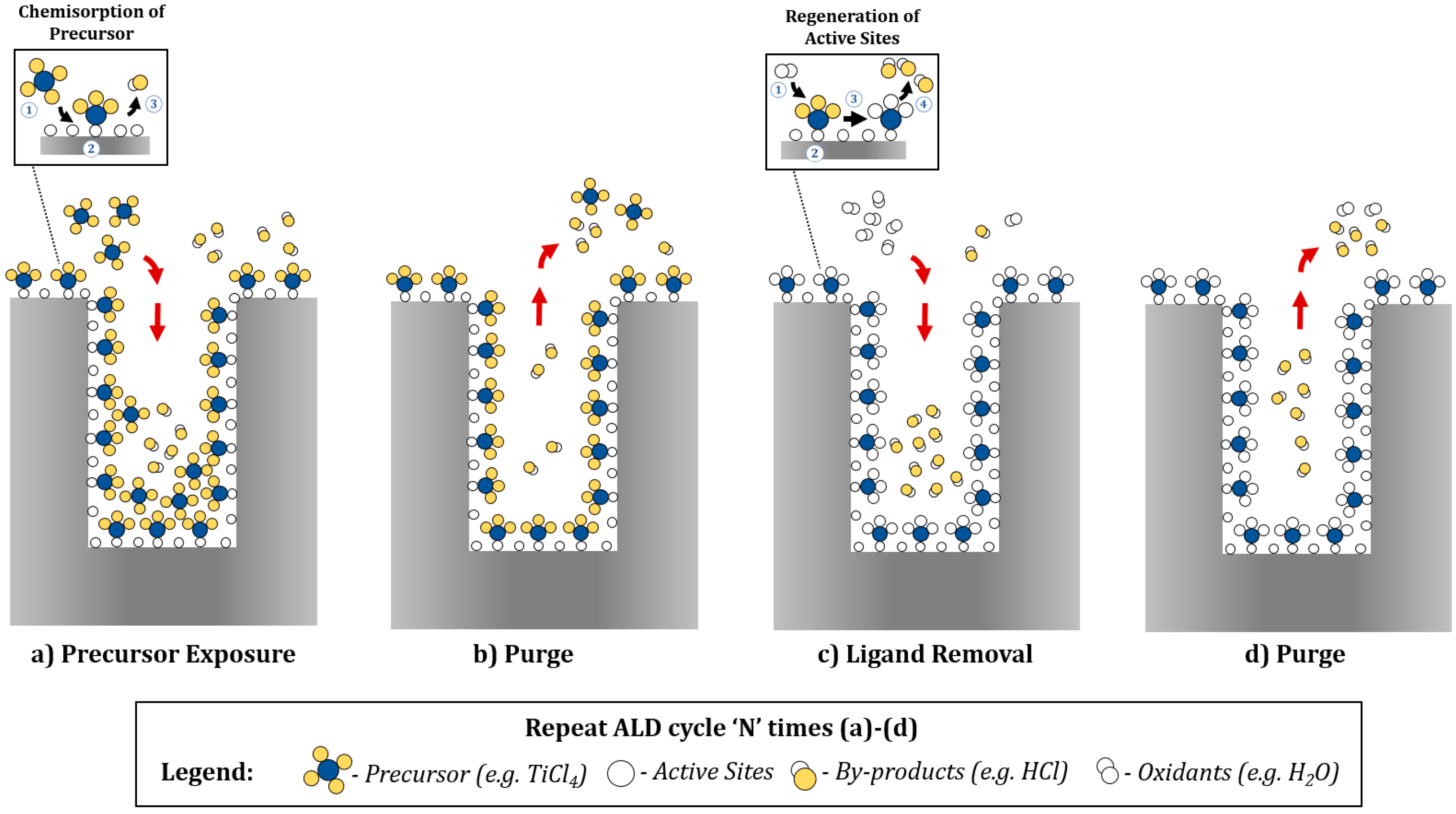
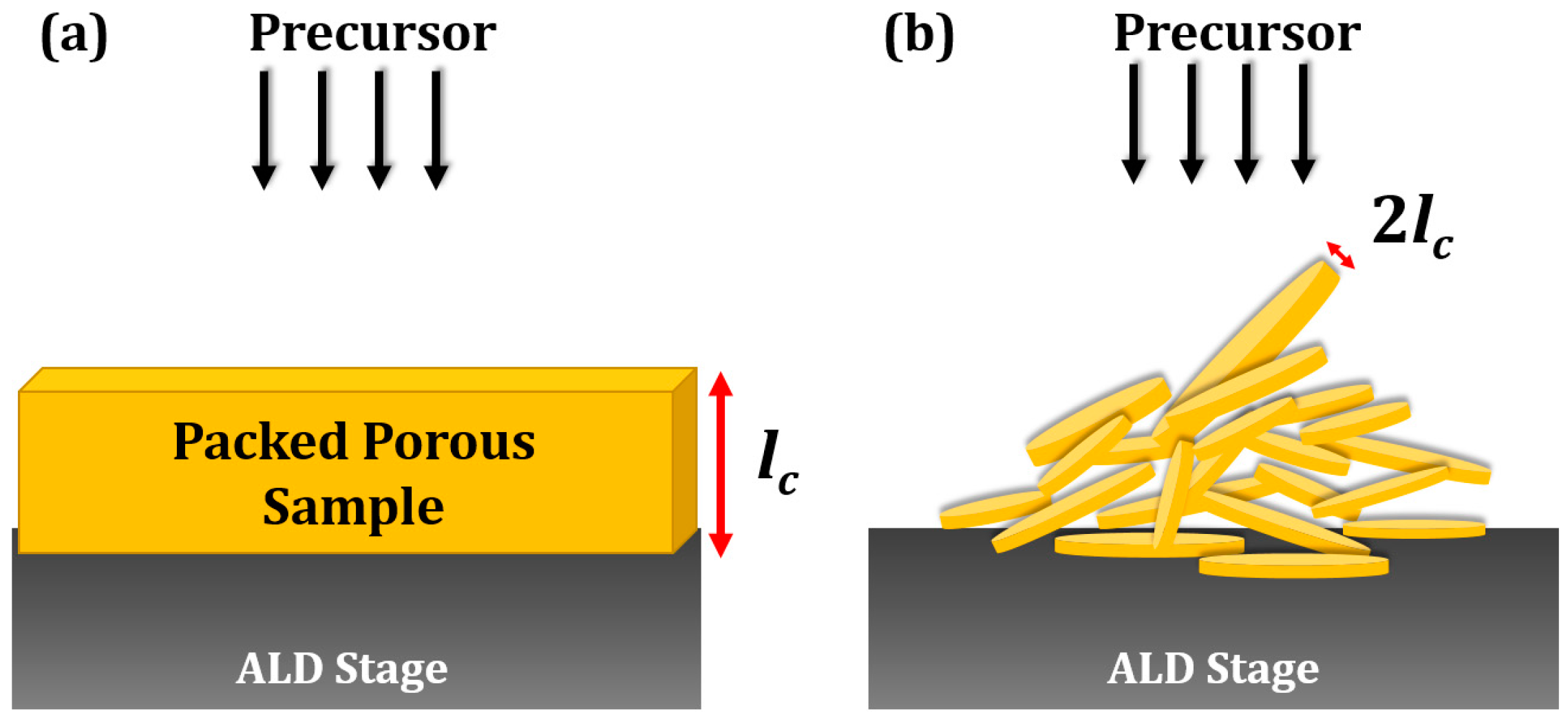
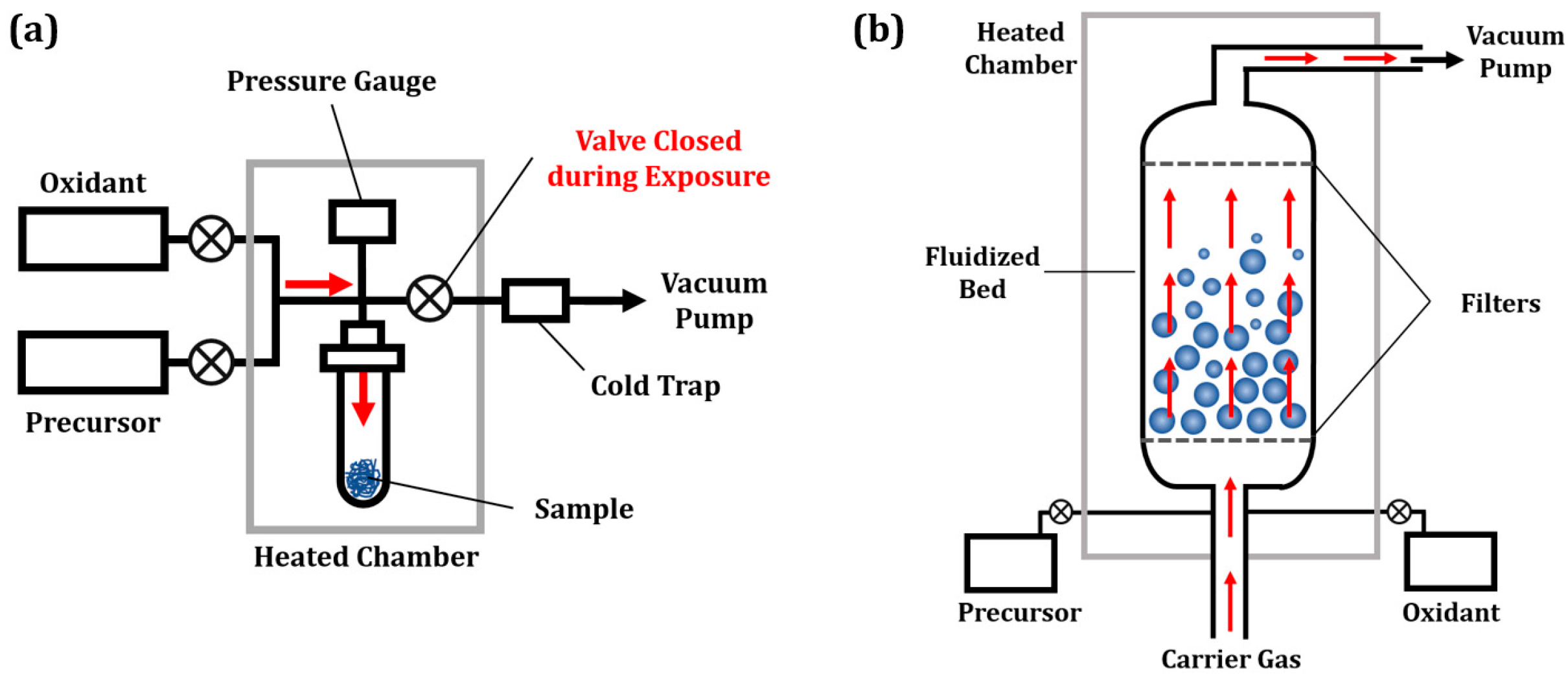
3. ALD Reactor Designs for Catalysts and SOFC Electrodes
4. Atomic Layer Deposition (ALD) in Catalyst Design and Synthesis
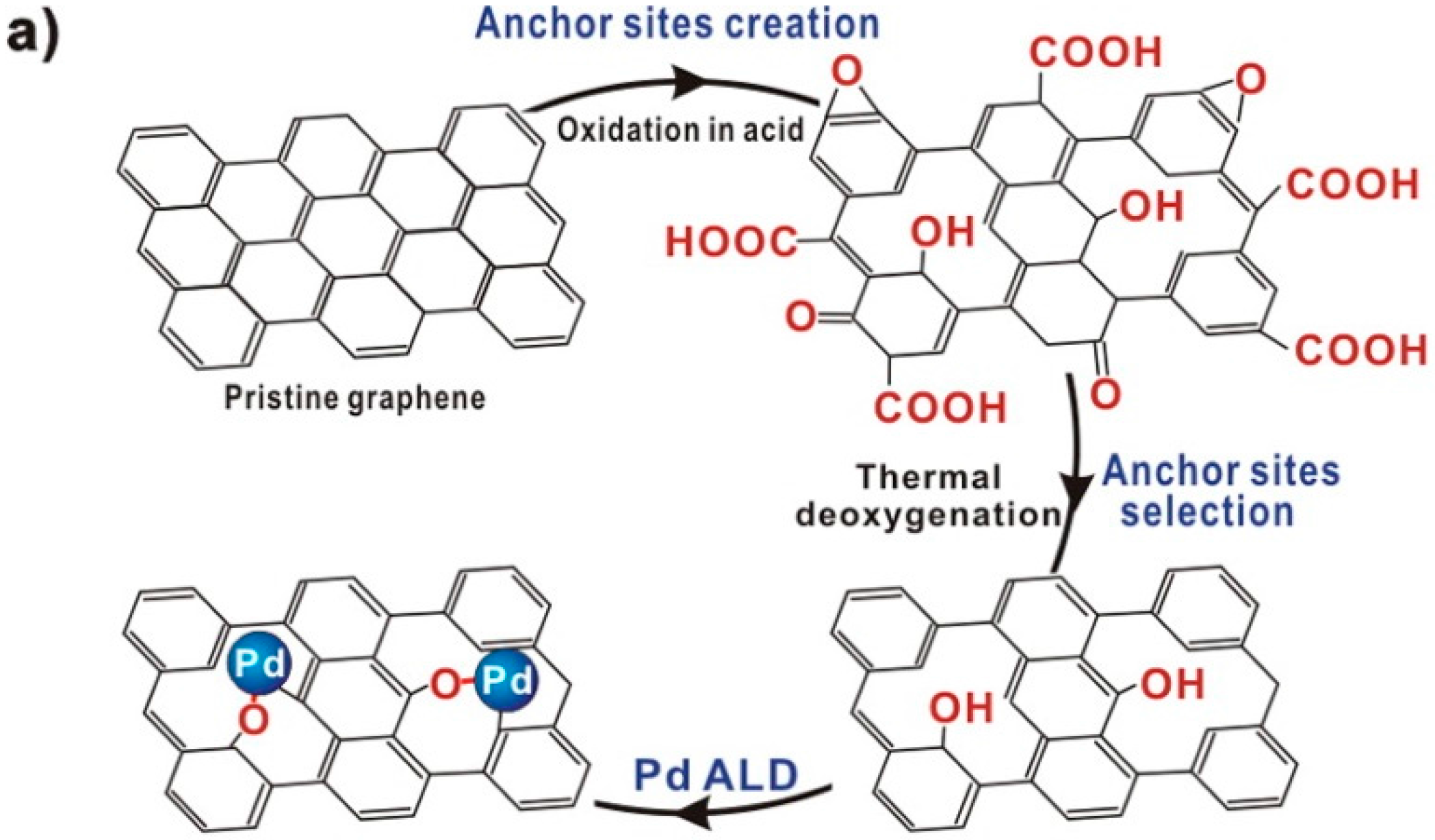
4.1. Formation of Isolated Sites
4.2. High-Surface-Area Functional Oxides
4.3. Coated Surfaces for Enhanced Stability
5. Applications to SOFC Electrodes
5.1. Modification of SOFC Cathodes
5.2. Modification of SOFC Anodes
6. Summary and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ma, Z.; Zaera, F. Heterogeneous catalysis by metals. In Encyclopedia of Inorganic and Bioinorganic Chemistry; Wiley: New York, NY, USA, 2014; pp. 1–16. [Google Scholar]
- Di Monte, R.; Kašpar, J. On the role of oxygen storage in three-way catalysis. Top. Catal. 2004, 28, 47–57. [Google Scholar] [CrossRef]
- Morikawa, A.; Suzuki, T.; Kanazawa, T.; Kikuta, K.; Suda, A.; Shinjo, H. A new concept in high performance ceria–zirconia oxygen storage capacity material with Al2O3 as a diffusion barrier. Appl. Catal. B Environ. 2008, 78, 210–221. [Google Scholar]
- Gorte, R.J. Ceria in catalysis: From automotive applications to the water–gas shift reaction. AIChE J. 2010, 56, 1126–1135. [Google Scholar]
- Montini, T.; Melchionna, M.; Monai, M.; Fornasiero, P. Fundamentals and catalytic applications of CeO2-based materials. Chem. Rev. 2016, 116, 5987–6041. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, M.; Wang, J.; Lee, W.-S.; Williams, W.D.; Kim, S.M.; Stach, E.A.; Miller, J.T.; Delgass, W.N.; Ribeiro, F.H. Size and support effects for the water–gas shift catalysis over gold nanoparticles supported on model Al2O3 and TiO2. J. Am. Chem. Soc. 2012, 134, 4700–4708. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Doan-Nguyen, V.V.; Gordon, T.R.; Diaz, R.E.; Stach, E.A.; Gorte, R.J.; Fornasiero, P.; Murray, C.B. Control of metal nanocrystal size reveals metal-support interface role for ceria catalysts. Science 2013, 341, 771–773. [Google Scholar] [PubMed]
- Zhu, Y.; Ramasse, Q.M.; Brorson, M.; Moses, P.G.; Hansen, L.P.; Kisielowski, C.F.; Helveg, S. Visualizing the stoichiometry of industrial-style Co–Mo–S catalysts with single-atom sensitivity. Angew. Chem. Int. Ed. 2014, 53, 10723–10727. [Google Scholar] [CrossRef] [PubMed]
- Kanno, D.; Shikazono, N.; Takagi, N.; Matsuzaki, K.; Kasagi, N. Evaluation of SOFC anode polarization simulation using three-dimensional microstructures reconstructed by FIB tomography. Electrochim. Acta 2011, 56, 4015–4021. [Google Scholar] [CrossRef]
- Bidrawn, F.; Kim, G.; Aramrueang, N.; Vohs, J.; Gorte, R. Dopants to enhance SOFC cathodes based on Sr-doped LaFeO3 and LaMnO3. J. Power Sources 2010, 195, 720–728. [Google Scholar] [CrossRef]
- Kiebach, R.; Knöfel, C.; Bozza, F.; Klemensø, T.; Chatzichristodoulou, C. Infiltration of ionic-, electronic- and mixed-conducting nano particles into La0.75Sr0.25MnO3-Y0.16Zr0.84O2 cathodes—A comparative study of performance enhancement and stability at different temperatures. J. Power Sources 2013, 228, 170–177. [Google Scholar] [CrossRef]
- Adijanto, L.; Sampath, A.; Yu, A.S.; Cargnello, M.; Fornasiero, P.; Gorte, R.J.; Vohs, J.M. Synthesis and stability of Pd@CeO2 core–shell catalyst films in solid oxide fuel cell anodes. ACS Catal. 2013, 3, 1801–1809. [Google Scholar] [CrossRef]
- Ding, D.; Li, X.; Lai, S.Y.; Gerdes, K.; Liu, M. Enhancing SOFC cathode performance by surface modification through infiltration. Energy Environ. Sci. 2014, 7, 552–575. [Google Scholar] [CrossRef]
- Yang, L.; Wang, S.; Blinn, K.; Liu, M.; Liu, Z.; Cheng, Z.; Liu, M. Enhanced sulfur and coking tolerance of a mixed ion conductor for SOFCs: BaZr0.1Ce0.7Y0.2−xYbxO3−δ. Science 2009, 326, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Choi, Y.; Qin, W.; Chen, H.; Blinn, K.; Liu, M.; Liu, P.; Bai, J.; Tyson, T.A.; Liu, M. Promotion of-mediated carbon removal by nanostructured/interfaces in solid oxide fuel cells. Nat. Commun. 2011, 2, 357. [Google Scholar] [CrossRef] [PubMed]
- Puurunen, R.L. Surface chemistry of atomic layer deposition: A case study for the trimethylaluminum/water process. J. Appl. Phys. 2005, 97, 121301. [Google Scholar] [CrossRef]
- Haukka, S.; Lakomaa, E.-L.; Suntola, T. Adsorption controlled preparation of heterogeneous catalysts. Stud. Surf. Sci. Catal. 1998, 120, 715–750. [Google Scholar]
- Ivers-Tiffée, E.; Weber, A.; Herbstritt, D. Materials and technologies for SOFC-components. J. Ceram. Soc. 2001, 21, 1805–1811. [Google Scholar] [CrossRef]
- Jacobson, A.J. Materials for solid oxide fuel cells. Chem. Mater. 2009, 22, 660–674. [Google Scholar] [CrossRef]
- Cassir, M.; Ringuedé, A.; Niinistö, L. Input of atomic layer deposition for solid oxide fuel cell applications. J. Mater. Chem. 2010, 20, 8987–8993. [Google Scholar] [CrossRef]
- George, S.M. Atomic layer deposition: An overview. Chem. Rev. 2010, 110, 111–131. [Google Scholar] [CrossRef] [PubMed]
- Detavernier, C.; Dendooven, J.; Sree, S.P.; Ludwig, K.F.; Martens, J.A. Tailoring nanoporous materials by atomic layer deposition. Chem. Soc. Rev. 2011, 40, 5242–5253. [Google Scholar] [CrossRef] [PubMed]
- Marichy, C.; Bechelany, M.; Pinna, N. Atomic layer deposition of nanostructured materials for energy and environmental applications. Adv. Mater. 2012, 24, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Van Delft, J.; Garcia-Alonso, D.; Kessels, W. Atomic layer deposition for photovoltaics: Applications and prospects for solar cell manufacturing. Semicond. Sci. Technol. 2012, 27, 074002. [Google Scholar] [CrossRef]
- Johnson, R.W.; Hultqvist, A.; Bent, S.F. A brief review of atomic layer deposition: From fundamentals to applications. Mater. Today 2014, 17, 236–246. [Google Scholar] [CrossRef]
- Singh, J.A.; Yang, N.; Bent, S.F. Nanoengineering heterogeneous catalysts by atomic layer deposition. Annu. Rev. Chem. Biomol. Eng. 2017, 8, 41–62. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, B.J.; Jackson, D.H.; Lee, J.; Canlas, C.; Stair, P.C.; Marshall, C.L.; Elam, J.W.; Kuech, T.F.; Dumesic, J.A.; Huber, G.W. Catalyst design with atomic layer deposition. ACS Catal. 2015, 5, 1804–1825. [Google Scholar] [CrossRef]
- Keuter, T.; Mauer, G.; Vondahlen, F.; Iskandar, R.; Menzler, N.H.; Vassen, R. Atomic-layer-controlled deposition of TEMAZ/O2–ZrO2 oxidation resistance inner surface coatings for solid oxide fuel cells. Surf. Coat. Technol. 2016, 288, 211–220. [Google Scholar] [CrossRef]
- Gong, Y.; Patel, R.L.; Liang, X.; Palacio, D.; Song, X.; Goodenough, J.B.; Huang, K. Atomic layer deposition functionalized composite SOFC cathode La0.6Sr0.4Fe0.8Co0.2O3−δ-Gd0.2Ce0.8O1.9: Enhanced long-term stability. Chem. Mater. 2013, 25, 4224–4231. [Google Scholar] [CrossRef]
- Chen, Y.; Gerdes, K.; Song, X. Nanoionics and nanocatalysts: Conformal mesoporous surface scaffold for cathode of solid oxide fuel cells. Sci. Rep. 2016, 6, 32997. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Goldstein, D.; McCormick, J.; Weimer, A.; George, S. Tungsten atomic layer deposition on cobalt nanoparticles. J. Vac. Sci. Technol. A 2008, 26, 430–437. [Google Scholar] [CrossRef]
- Yang, N.; Yoo, J.S.; Schumann, J.; Bothra, P.; Singh, J.A.; Valle, E.; Abild-Pedersen, F.; Nørskov, J.K.; Bent, S.F. Rh–MnO interface sites formed by atomic layer deposition promote syngas conversion to higher oxygenates. ACS Catal. 2017, 7, 5746–5757. [Google Scholar] [CrossRef]
- Pickrahn, K.L.; Park, S.W.; Gorlin, Y.; Lee, H.B.R.; Jaramillo, T.F.; Bent, S.F. Active MnOx electrocatalysts prepared by atomic layer deposition for oxygen evolution and oxygen reduction reactions. Adv. Energy Mater. 2012, 2, 1269–1277. [Google Scholar] [CrossRef]
- Martinson, A.B.; DeVries, M.J.; Libera, J.A.; Christensen, S.T.; Hupp, J.T.; Pellin, M.J.; Elam, J.W. Atomic layer deposition of Fe2O3 using ferrocene and ozone. J. Phys. Chem. C 2011, 115, 4333–4339. [Google Scholar] [CrossRef]
- Hu, Q.; Wang, S.; Gao, Z.; Li, Y.; Zhang, Q.; Xiang, Q.; Qin, Y. The precise decoration of Pt nanoparticles with Fe oxide by atomic layer deposition for the selective hydrogenation of cinnamaldehyde. Appl. Catal. B Environ. 2017, 218, 591–599. [Google Scholar] [CrossRef]
- Nieminen, M.; Putkonen, M.; Niinistö, L. Formation and stability of lanthanum oxide thin films deposited from β-diketonate precursor. Appl. Surf. Sci. 2001, 174, 155–166. [Google Scholar] [CrossRef]
- Rahmanipour, M.; Cheng, Y.; Onn, T.M.; Donazzi, A.; Vohs, J.M.; Gorte, R.J. Modification of LSF-YSZ composite cathodes by atomic layer deposition. J. Electrochem. Soc. 2017, 164, F879–F884. [Google Scholar] [CrossRef]
- Onn, T.M.; Monai, M.; Dai, S.; Fonda, E.; Montini, T.; Pan, X.; Graham, G.W.; Fornasiero, P.; Gorte, R.J. Smart Pd catalyst with improved thermal stability supported on high-surface-area LaFeO3 prepared by atomic layer deposition. J. Am. Chem. Soc. 2018. accepted. [Google Scholar] [CrossRef] [PubMed]
- Haukka, S. ALD technology-present and future challenges. ECS Trans. 2007, 3, 15–26. [Google Scholar]
- Raaijmakers, I.J. Current and future applications of ALD in micro-electronics. ECS Trans. 2011, 41, 3–17. [Google Scholar]
- Elam, J.; Groner, M.; George, S. Viscous flow reactor with quartz crystal microbalance for thin film growth by atomic layer deposition. Rev. Sci. Instrum. 2002, 73, 2981–2987. [Google Scholar] [CrossRef]
- Elers, K.E.; Blomberg, T.; Peussa, M.; Aitchison, B.; Haukka, S.; Marcus, S. Film uniformity in atomic layer deposition. Chem. Vap. Depos. 2006, 12, 13–24. [Google Scholar] [CrossRef]
- Elam, J.; Routkevitch, D.; Mardilovich, P.; George, S. Conformal coating on ultrahigh-aspect-ratio nanopores of anodic alumina by atomic layer deposition. Chem. Mater. 2003, 15, 3507–3517. [Google Scholar] [CrossRef]
- Longrie, D.; Deduytsche, D.; Detavernier, C. Reactor concepts for atomic layer deposition on agitated particles: A review. J. Vac. Sci. Technol. A 2014, 32, 010802. [Google Scholar] [CrossRef]
- Sun, S.; Zhang, G.; Gauquelin, N.; Chen, N.; Zhou, J.; Yang, S.; Chen, W.; Meng, X.; Geng, D.; Banis, M.N. Single-atom catalysis using Pt/graphene achieved through atomic layer deposition. Sci. Rep. 2013, 3, 1775. [Google Scholar] [CrossRef]
- Yan, H.; Cheng, H.; Yi, H.; Lin, Y.; Yao, T.; Wang, C.; Li, J.; Wei, S.; Lu, J. Single-atom Pd1/graphene catalyst achieved by atomic layer deposition: Remarkable performance in selective hydrogenation of 1, 3-butadiene. J. Am. Chem. Soc. 2015, 137, 10484–10487. [Google Scholar] [CrossRef] [PubMed]
- Piernavieja-Hermida, M.; Lu, Z.; White, A.; Low, K.-B.; Wu, T.; Elam, J.W.; Wu, Z.; Lei, Y. Towards ALD thin film stabilized single-atom Pd1 catalysts. Nanoscale 2016, 8, 15348–15356. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Y.; An, C.J.; Piao, S.J.; Ahn, D.Y.; Kim, M.-T.; Min, Y.-S. Shrinking core model for knudsen diffusion-limited atomic layer deposition on a nanoporous monolith with an ultrahigh aspect ratio. J. Phys. Chem. C 2010, 114, 18601–18606. [Google Scholar] [CrossRef]
- Grillo, F.; Kreutzer, M.T.; van Ommen, J.R. Modeling the precursor utilization in atomic layer deposition on nanostructured materials in fluidized bed reactors. Chem. Eng. J. 2015, 268, 384–398. [Google Scholar] [CrossRef]
- Duan, C.-L.; Zhu, P.-H.; Deng, Z.; Li, Y.; Shan, B.; Fang, H.-S.; Feng, G.; Chen, R. Mechanistic modeling study of atomic layer deposition process optimization in a fluidized bed reactor. J. Vac. Sci. Technol. A 2017, 35, 01B102. [Google Scholar] [CrossRef]
- Haukka, S.; Kytökivi, A.; Lakomaa, E.; Lehtovirta, U.; Lindblad, M.; Lujala, V.; Suntola, T. The utilization of saturated gas–solid reactions in the preparation of heterogeneous catalysts. Stud. Surf. Sci. Catal. 1995, 91, 957–966. [Google Scholar]
- Wind, R.; George, S. Quartz crystal microbalance studies of Al2O3 atomic layer deposition using trimethylaluminum and water at 125 °C. J. Phys. Chem. A 2010, 114, 1281–1289. [Google Scholar] [CrossRef] [PubMed]
- Miikkulainen, V.; Leskelä, M.; Ritala, M.; Puurunen, R.L. Crystallinity of inorganic films grown by atomic layer deposition: Overview and general trends. J. Appl. Phys. 2013, 113, 2. [Google Scholar] [CrossRef]
- Ylilammi, M. Monolayer thickness in atomic layer deposition. Thin Solid Films 1996, 279, 124–130. [Google Scholar] [CrossRef]
- Schindler, P.; Logar, M.; Provine, J.; Prinz, F.B. Enhanced step coverage of TiO2 deposited on high aspect ratio surfaces by plasma-enhanced atomic layer deposition. Langmuir 2015, 31, 5057–5062. [Google Scholar] [CrossRef] [PubMed]
- Strempel, V.; Naumann d’Alnoncourt, R.; Driess, M.; Rosowski, F. Atomic layer deposition on porous powders with in situ gravimetric monitoring in a modular fixed bed reactor setup. Rev. Sci. Instrum. 2017, 88, 074102. [Google Scholar] [CrossRef] [PubMed]
- Kucheyev, S.; Biener, J.; Baumann, T.; Wang, Y.; Hamza, A.; Li, Z.; Lee, D.; Gordon, R. Mechanisms of atomic layer deposition on substrates with ultrahigh aspect ratios. Langmuir 2008, 24, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Mao, X.; Onn, T.M.; Jang, J.; Gorte, R.J. Stabilization of ZrO2 Powders via ALD of CeO2 and ZrO2. Inorganics 2017, 5, 65. [Google Scholar] [CrossRef]
- Demmin, R.; Gorte, R. Design parameters for temperature-programmed desorption from a packed bed. J. Catal. 1984, 90, 32–39. [Google Scholar] [CrossRef]
- Hutchings, G.; Copperthwaite, R.; Themistocleous, T.; Foulds, G.; Bielovitch, A.; Loots, B.; Nowitz, G.; Van Eck, P. A comparative study of reactivation of zeolite Y using oxygen and ozone/oxygen mixtures. Appl. Catal. 1987, 34, 153–161. [Google Scholar] [CrossRef]
- Mackus, A.J.; MacIsaac, C.; Kim, W.-H.; Bent, S.F. Incomplete elimination of precursor ligands during atomic layer deposition of Zinc-Oxide, Tin-Oxide, and Zinc-Tin-Oxide. J. Chem. Phys. 2017, 146, 052802. [Google Scholar] [CrossRef] [PubMed]
- HafezKhiabani, N.; Fathi, S.; Shokri, B.; Hosseini, S.I. A novel method for decoking of Pt–Sn/Al2O3 in the naphtha reforming process using RF and pin-to-plate DBD plasma systems. Appl. Catal. A Gen. 2015, 493, 8–16. [Google Scholar] [CrossRef]
- Jia, L.; Farouha, A.; Pinard, L.; Hedan, S.; Comparot, J.-D.; Dufour, A.; Tayeb, K.B.; Vezin, H.; Batiot-Dupeyrat, C. New routes for complete regeneration of coked zeolite. Appl. Catal. B Environ. 2017, 219, 82–91. [Google Scholar] [CrossRef]
- Stanmore, B.R.; Brilhac, J.-F.; Gilot, P. The oxidation of soot: A review of experiments, mechanisms and models. Carbon 2001, 39, 2247–2268. [Google Scholar] [CrossRef]
- Bibby, D.; Milestone, N.; Patterson, J.; Aldridge, L. Coke formation in zeolite ZSM-5. J. Catal. 1986, 97, 493–502. [Google Scholar] [CrossRef]
- McCormick, J.; Cloutier, B.; Weimer, A.; George, S. Rotary reactor for atomic layer deposition on large quantities of nanoparticles. J. Vac. Sci. Technol. A 2007, 25, 67–74. [Google Scholar] [CrossRef]
- Cavanagh, A.S.; Wilson, C.A.; Weimer, A.W.; George, S.M. Atomic layer deposition on gram quantities of multi-walled carbon nanotubes. Nanotechnology 2009, 20, 255602. [Google Scholar] [CrossRef] [PubMed]
- Lindblad, M.; Lindfors, L.P.; Suntola, T. Preparation of Ni/Al2O3 catalysts from vapor phase by atomic layer epitaxy. Catal. Lett. 1994, 27, 323–336. [Google Scholar] [CrossRef]
- Anthony, S.Y.; Küngas, R.; Vohs, J.M.; Gorte, R.J. Modification of SOFC cathodes by atomic layer deposition. J. Electrochem. Soc. 2013, 160, F1225–F1231. [Google Scholar]
- Onn, T.M.; Zhang, S.; Arroyo-Ramirez, L.; Chung, Y.-C.; Graham, G.W.; Pan, X.; Gorte, R.J. Improved thermal stability and methane-oxidation activity of Pd/Al2O3 catalysts by atomic layer deposition of ZrO2. ACS Catal. 2015, 5, 5696–5701. [Google Scholar] [CrossRef]
- Onn, T.M.; Monai, M.; Dai, S.; Arroyo-Ramirez, L.; Zhang, S.; Pan, X.; Graham, G.W.; Fornasiero, P.; Gorte, R.J. High-surface-area, iron-oxide films prepared by atomic layer deposition on γ-Al2O3. Appl. Catal. A Gen. 2017, 534, 70–77. [Google Scholar] [CrossRef]
- Onn, T.M.; Zhang, S.; Arroyo-Ramirez, L.; Xia, Y.; Wang, C.; Pan, X.; Graham, G.W.; Gorte, R.J. High-surface-area ceria prepared by ALD on Al2O3 support. Appl. Catal. B Environ. 2017, 201, 430–437. [Google Scholar] [CrossRef]
- Onn, T.M.; Dai, S.; Chen, J.; Pan, X.; Graham, G.W.; Gorte, R.J. High-surface area ceria–zirconia films prepared by atomic layer deposition. Catal. Lett. 2017, 147, 1464–1470. [Google Scholar] [CrossRef]
- Wang, C.; Hu, L.; Lin, Y.; Poeppelmeier, K.; Stair, P.; Marks, L. Controllable ALD synthesis of platinum nanoparticles by tuning different synthesis parameters. J. Phys. D Appl. Phys. 2017, 50, 415301. [Google Scholar] [CrossRef]
- Ramachandran, R.K.; Filez, M.; Dendooven, J.; Galvita, V.V.; Poelman, H.; Solano, E.; Fonda, E.; Marin, G.B.; Detavernier, C. Size- and composition-controlled Pt–Sn bimetallic nanoparticles prepared by atomic layer deposition. RSC Adv. 2017, 7, 20201–20205. [Google Scholar] [CrossRef]
- Berland, B.; Gartland, I.; Ott, A.; George, S.M. In situ monitoring of atomic layer controlled pore reduction in alumina tubular membranes using sequential surface reactions. Chem. Mater. 1998, 10, 3941–3950. [Google Scholar] [CrossRef]
- King, D.M.; Spencer, J.A.; Liang, X.; Hakim, L.F.; Weimer, A.W. Atomic layer deposition on particles using a fluidized bed reactor with in situ mass spectrometry. Surf. Coat. Technol. 2007, 201, 9163–9171. [Google Scholar] [CrossRef]
- De Martin, L.; Bouwman, W.G.; van Ommen, J.R. Multidimensional nature of fluidized nanoparticle agglomerates. Langmuir 2014, 30, 12696–12702. [Google Scholar] [CrossRef] [PubMed]
- De Martín, L.; Fabre, A.; van Ommen, J.R. The fractal scaling of fluidized nanoparticle agglomerates. Chem. Eng. Sci. 2014, 112, 79–86. [Google Scholar] [CrossRef]
- Feng, H.; Libera, J.A.; Stair, P.C.; Miller, J.T.; Elam, J.W. Subnanometer palladium particles synthesized by atomic layer deposition. ACS Catal. 2011, 1, 665–673. [Google Scholar] [CrossRef]
- Najafabadi, A.T.; Khodadadi, A.A.; Parnian, M.J.; Mortazavi, Y. Atomic layer deposited Co/γ-Al2O3 catalyst with enhanced cobalt dispersion and Fischer–Tropsch synthesis activity and selectivity. Appl. Catal. A Gen. 2016, 511, 31–46. [Google Scholar] [CrossRef]
- Cheng, N.; Sun, X. Single atom catalyst by atomic layer deposition technique. Chin. J. Catal. 2017, 38, 1508–1514. [Google Scholar] [CrossRef]
- Gould, T.D.; Lubers, A.M.; Neltner, B.T.; Carrier, J.V.; Weimer, A.W.; Falconer, J.L.; Medlin, J.W. Synthesis of supported Ni catalysts by atomic layer deposition. J. Catal. 2013, 303, 9–15. [Google Scholar] [CrossRef]
- He, B.J.-J.; Wang, C.-X.; Zheng, T.-T.; Zhao, Y.-K. Thermally induced deactivation and the corresponding strategies for improving durability in automotive three-way catalysts. Johns. Matthey Technol. Rev. 2016, 60, 196–203. [Google Scholar] [CrossRef]
- Onn, T.M.; Mao, X.; Lin, C.; Wang, C.; Gorte, R.J. Investigation of the thermodynamic properties of surface ceria and ceria–zirconia solid solution films prepared by atomic layer deposition on Al2O3. Inorganics 2017, 5, 69. [Google Scholar]
- Senthilkumar, B.; Selvan, R.K.; Meyrick, D.; Minakshi, M. Synthesis and Characterization of Manganese Molybdate for Symmetric Capacitor Applications. Int. J. Electrochem. Sci. 2015, 10, 185–193. [Google Scholar]
- Barmi, M.J.; Minakshi, M. Tuning the Redox Properties of the Nanostructured CoMoO4Electrode: Effects of Surfactant Content and Synthesis Temperature. ChemCatChem 2016, 81, 964–977. [Google Scholar]
- Héroguel, F.; Le Monnier, B.P.; Brown, K.S.; Siu, J.C.; Luterbacher, J.S. Catalyst stabilization by stoichiometrically limited layer-by-layer overcoating in liquid media. Appl. Catal. B Environ. 2017, 218, 643–649. [Google Scholar] [CrossRef]
- Lu, J.; Liu, B.; Greeley, J.P.; Feng, Z.; Libera, J.A.; Lei, Y.; Bedzyk, M.J.; Stair, P.C.; Elam, J.W. Porous alumina protective coatings on palladium nanoparticles by self-poisoned atomic layer deposition. Chem. Mater. 2012, 24, 2047–2055. [Google Scholar] [CrossRef]
- Lu, J.; Elam, J.W.; Stair, P.C. Synthesis and stabilization of supported metal catalysts by atomic layer deposition. Acc. Chem. Res. 2013, 46, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jackson, D.H.; Li, T.; Winans, R.E.; Dumesic, J.A.; Kuech, T.F.; Huber, G.W. Enhanced stability of cobalt catalysts by atomic layer deposition for aqueous-phase reactions. Energy Environ. Sci. 2014, 7, 1657–1660. [Google Scholar] [CrossRef]
- Lu, J.; Fu, B.; Kung, M.C.; Xiao, G.; Elam, J.W.; Kung, H.H.; Stair, P.C. Coking- and sintering-resistant palladium catalysts achieved through atomic layer deposition. Science 2012, 335, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.B.; Holme, T.P.; Gür, T.M.; Prinz, F.B. Surface-modified low-temperature solid oxide fuel cell. Adv. Funct. Mater. 2011, 21, 4684–4690. [Google Scholar] [CrossRef]
- Nieminen, M.; Lehto, S.; Niinistö, L. Atomic layer epitaxy growth of LaGaO3 thin films. J. Mater. Chem. 2001, 11, 3148–3153. [Google Scholar] [CrossRef]
- Gourba, E.; Ringuede, A.; Cassir, M.; Billard, A.; Päiväsaari, J.; Niinistö, J.; Putkonen, M.; Niinistö, L. Characterisation of thin films of ceria-based electrolytes for intermediatetemperature—Solid oxide fuel cells (IT-SOFC). Ionics 2003, 9, 15–20. [Google Scholar] [CrossRef]
- Fan, Z.; Chao, C.-C.; Hossein-Babaei, F.; Prinz, F.B. Improving solid oxide fuel cells with yttria-doped ceria interlayers by atomic layer deposition. J. Mater. Chem. 2011, 21, 10903–10906. [Google Scholar] [CrossRef]
- Chang, I.; Ji, S.; Park, J.; Lee, M.H.; Cha, S.W. Ultrathin YSZ coating on Pt cathode for high thermal stability and enhanced oxygen reduction reaction activity. Adv. Energy Mater. 2015, 5, 1402251. [Google Scholar] [CrossRef]
- Jiang, X.; Huang, H.; Prinz, F.B.; Bent, S.F. Application of atomic layer deposition of platinum to solid oxide fuel cells. Chem. Mater. 2008, 20, 3897–3905. [Google Scholar] [CrossRef]
- Holme, T.P.; Lee, C.; Prinz, F.B. Atomic layer deposition of LSM cathodes for solid oxide fuel cells. Solid State Ion. 2008, 179, 1540–1544. [Google Scholar] [CrossRef]
- Lie, M.; Nilsen, O.; Fjellvåg, H.; Kjekshus, A. Growth of La1−xSrxFeO3 thin films by atomic layer deposition. Dalton Trans. 2009, 3, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Kenjo, T.; Osawa, S.; Fujikawa, K. High temperature air cathodes containing ion conductive oxides. J. Electrochem. Soc. 1991, 138, 349–355. [Google Scholar] [CrossRef]
- Østergård, M.; Clausen, C.; Bagger, C.; Mogensen, M. Manganite-zirconia composite cathodes for SOFC: Influence of structure and composition, Electrochim. Acta 1995, 40, 1971–1981. [Google Scholar] [CrossRef]
- Gong, Y.; Palacio, D.; Song, X.; Patel, R.L.; Liang, X.; Zhao, X.; Goodenough, J.B.; Huang, K. Stabilizing nanostructured solid oxide fuel cell cathode with atomic layer deposition. Nanoletters 2013, 13, 4340–4345. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Bae, K.; Jang, D.Y.; Kim, J.W.; Shim, J.H. Performance degradation of lanthanum strontium cobaltite after surface modification. J. Electrochem. Soc. 2015, 162, F622–F626. [Google Scholar] [CrossRef]
- Sbrockey, N.; Aindow, M.; Deljoo, B.; Ghezel-Ayagh, H.; Torabi, A.; Tompa, G. Fluidized bed production of surface functionalized powders for solid oxide fuel cell cathodes. ECS Trans. 2017, 78, 817–825. [Google Scholar] [CrossRef]
- Karimaghaloo, A.; Andrade, A.M.; Grewal, S.; Shim, J.H.; Lee, M.H. Mechanism of cathodic performance enhancement by a few-nanometer-thick oxide overcoat on porous Pt cathodes of solid oxide fuel cells. ACS Omega 2017, 2, 806–813. [Google Scholar] [CrossRef]
- Liu, K.-Y.; Fan, L.; Yu, C.-C.; Su, P.-C. Thermal stability and performance enhancement of nano-porous platinum cathode in solid oxide fuel cells by nanoscale ZrO2 capping. Electrochem. Commun. 2015, 56, 65–69. [Google Scholar] [CrossRef]
- Koide, H.; Someya, Y.; Yoshida, T.; Maruyama, T. Properties of Ni/YSZ cermet as anode for SOFC. Solid State Ion. 2000, 132, 253–260. [Google Scholar] [CrossRef]
- Malzbender, J.; Wessel, E.; Steinbrech, R.W. Reduction and re-oxidation of anodes for solid oxide fuel cells. Solid State Ion. 2005, 176, 2201–2203. [Google Scholar] [CrossRef]
- Klemensø, T.; Chung, C.; Larsen, P.H.; Mogensen, M. The mechanism behind redox instability of anodes in high-temperature SOFCs. J. Electrochem. Soc. 2005, 152, A2186–A2192. [Google Scholar] [CrossRef] [Green Version]
- Noh, H.-S.; Son, J.-W.; Lee, H.; Ji, H.-I.; Lee, J.-H.; Lee, H.-W. Suppression of Ni agglomeration in PLD fabricated Ni-YSZ composite for surface modification of SOFC anode. J. Eur. Ceram. Soc. 2010, 30, 3415–3423. [Google Scholar] [CrossRef]
- Matsui, T.; Eguchi, K.; Shirai, K.; Ozeki, T.; Okanishi, T.; Muroyama, H.; Eguchi, K. Redox-induced self-modification of cermet anodes of Ni–CeO2-based oxide for solid oxide fuel cells. J. Electrochem. Soc. 2017, 164, F1368–F1374. [Google Scholar] [CrossRef]










© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onn, T.M.; Küngas, R.; Fornasiero, P.; Huang, K.; Gorte, R.J. Atomic Layer Deposition on Porous Materials: Problems with Conventional Approaches to Catalyst and Fuel Cell Electrode Preparation. Inorganics 2018, 6, 34. https://doi.org/10.3390/inorganics6010034
Onn TM, Küngas R, Fornasiero P, Huang K, Gorte RJ. Atomic Layer Deposition on Porous Materials: Problems with Conventional Approaches to Catalyst and Fuel Cell Electrode Preparation. Inorganics. 2018; 6(1):34. https://doi.org/10.3390/inorganics6010034
Chicago/Turabian StyleOnn, Tzia Ming, Rainer Küngas, Paolo Fornasiero, Kevin Huang, and Raymond J. Gorte. 2018. "Atomic Layer Deposition on Porous Materials: Problems with Conventional Approaches to Catalyst and Fuel Cell Electrode Preparation" Inorganics 6, no. 1: 34. https://doi.org/10.3390/inorganics6010034
APA StyleOnn, T. M., Küngas, R., Fornasiero, P., Huang, K., & Gorte, R. J. (2018). Atomic Layer Deposition on Porous Materials: Problems with Conventional Approaches to Catalyst and Fuel Cell Electrode Preparation. Inorganics, 6(1), 34. https://doi.org/10.3390/inorganics6010034