NHI- and NHC-Supported Al(III) Hydrides for Amine–Borane Dehydrocoupling Catalysis




Abstract
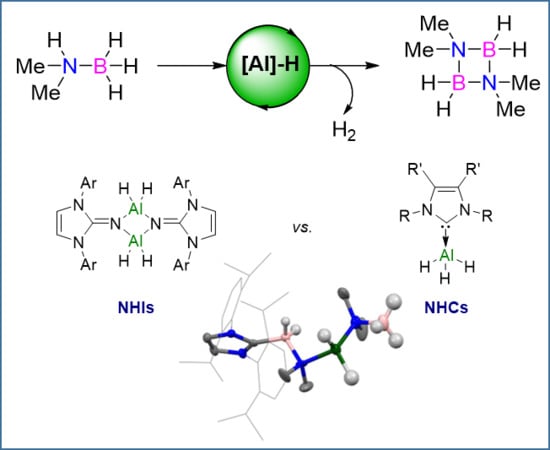
:
1. Introduction
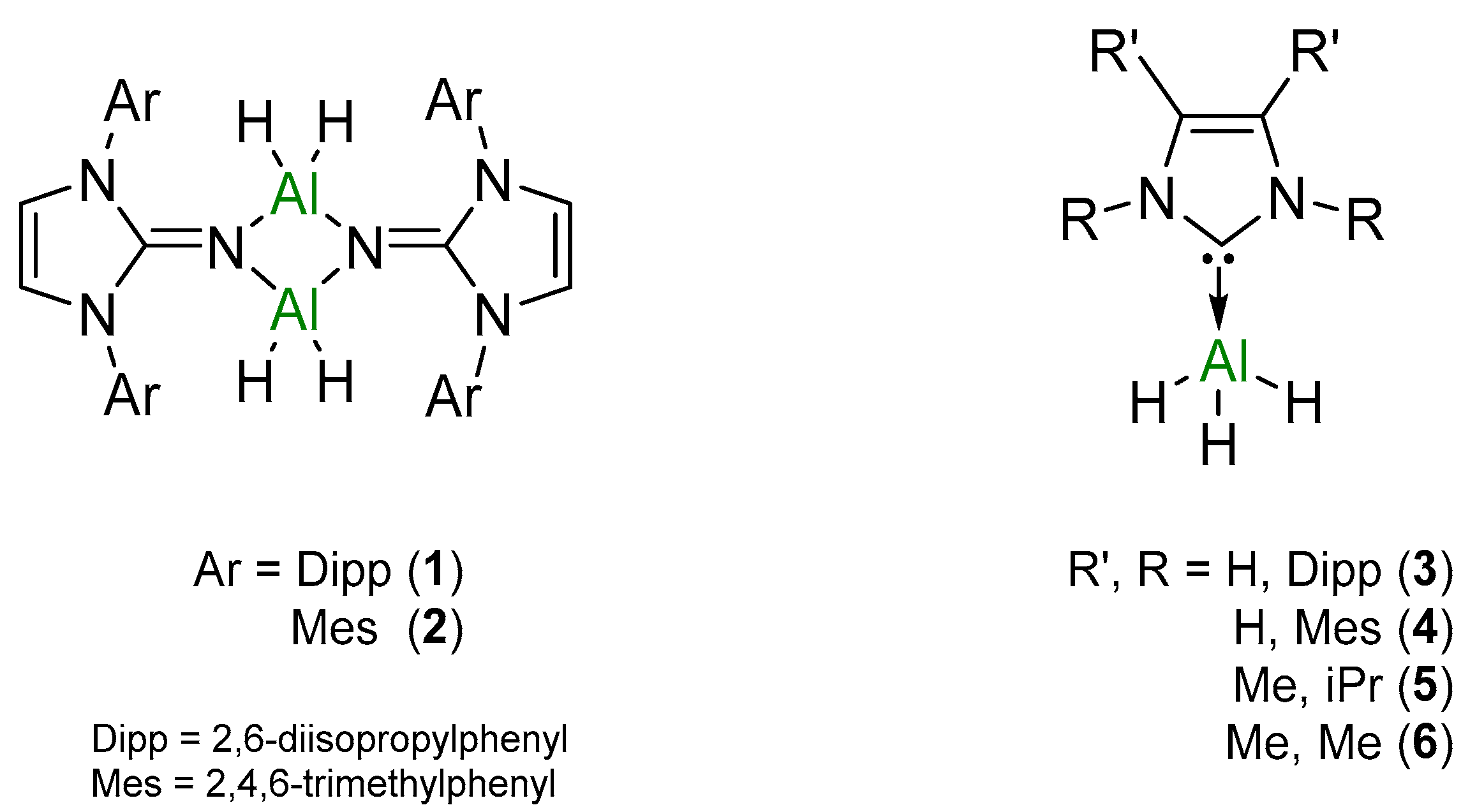
2. Results and Discussion
2.1. Catalysis
2.2. Mechanistic Studies
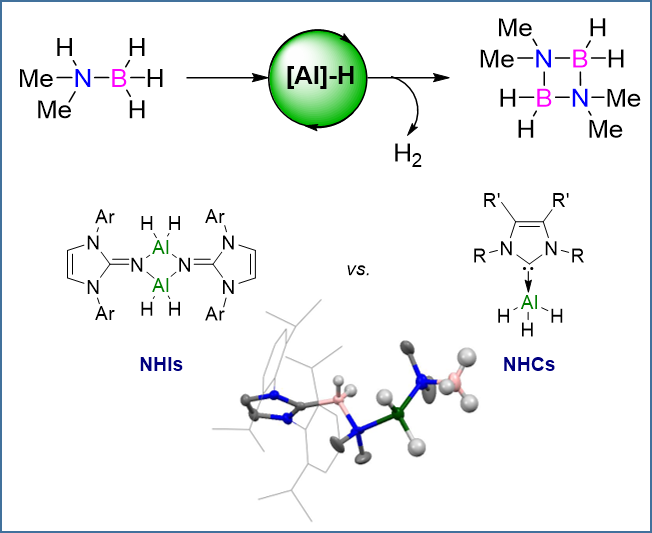
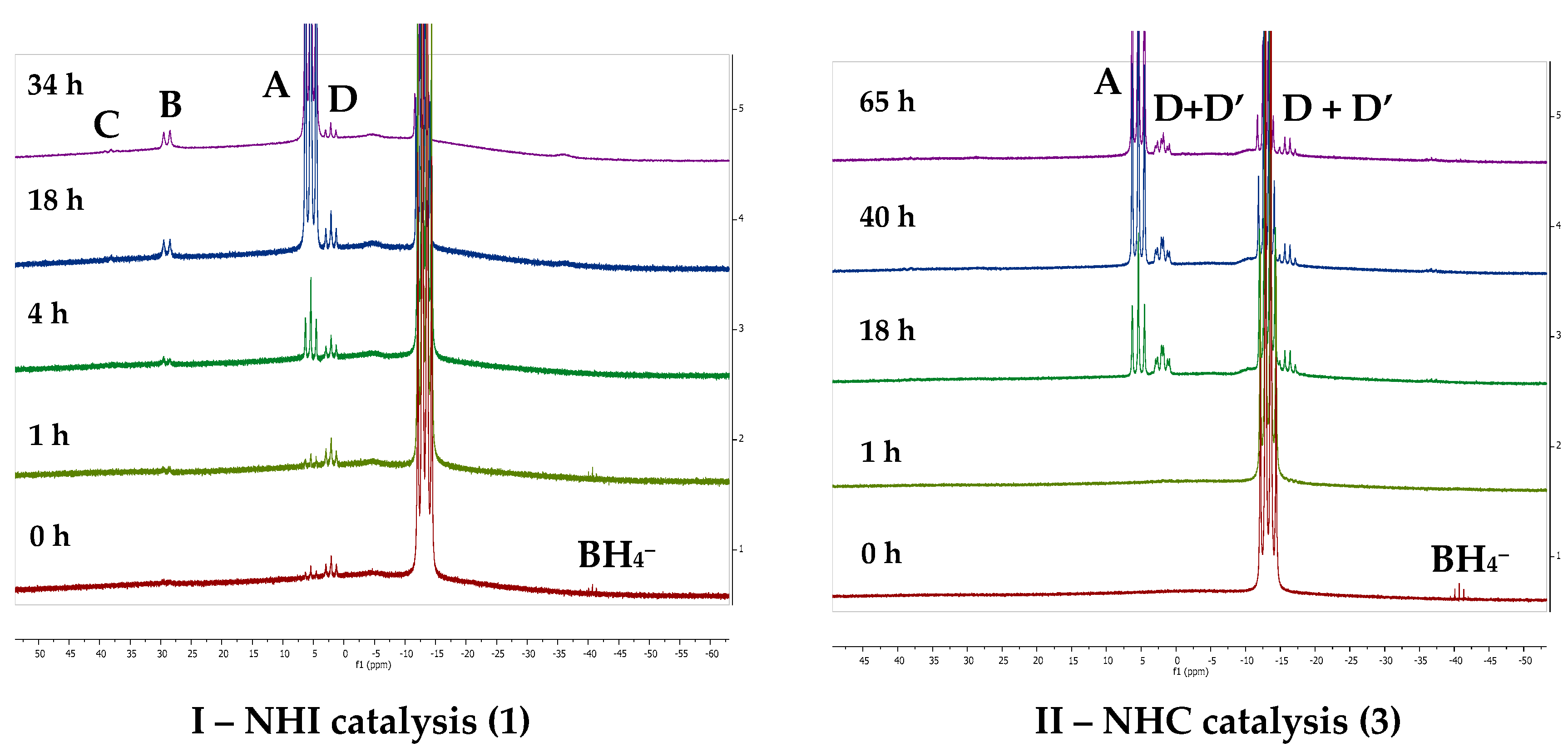
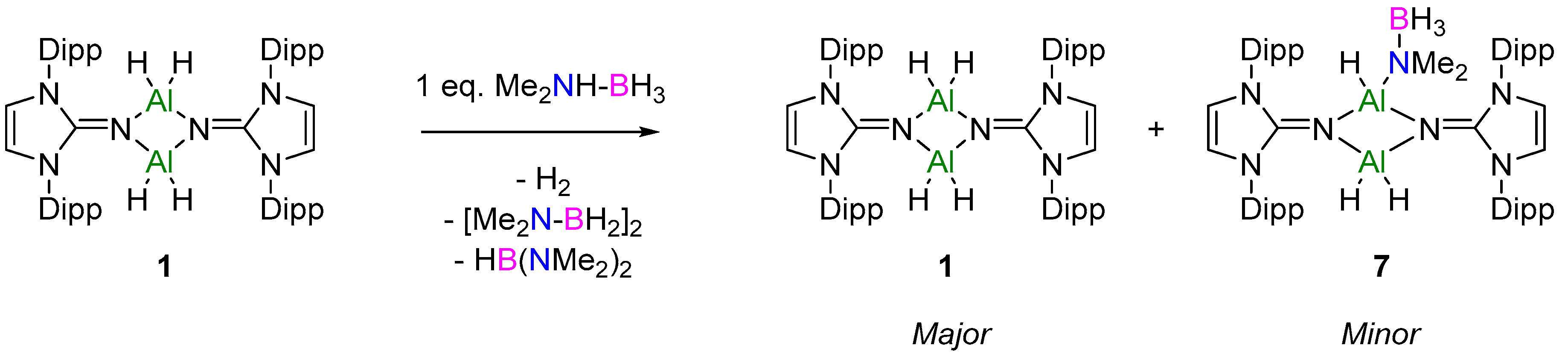
2.2.1. NHI Mechanism, Catalysts 1 and 2
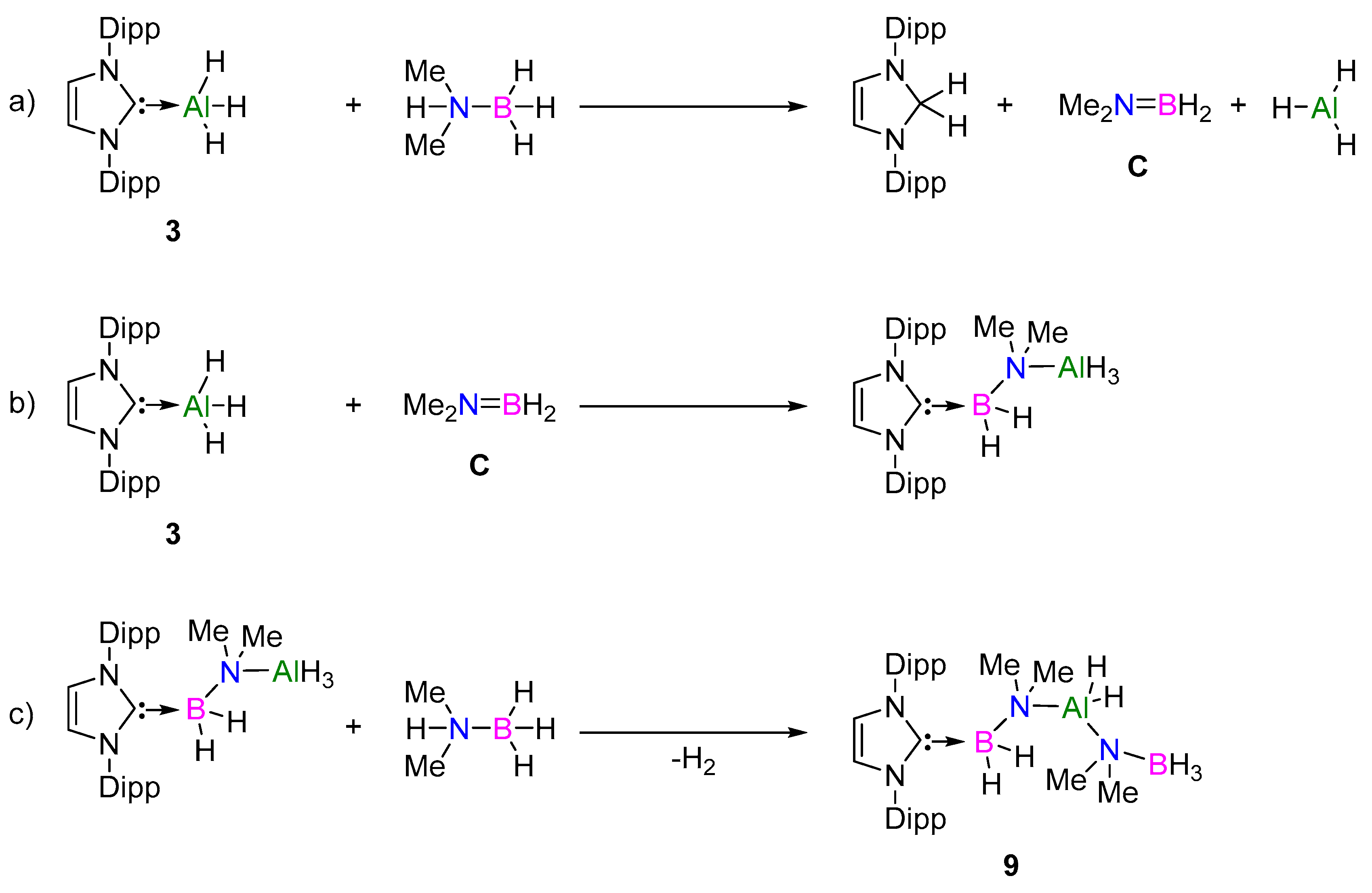
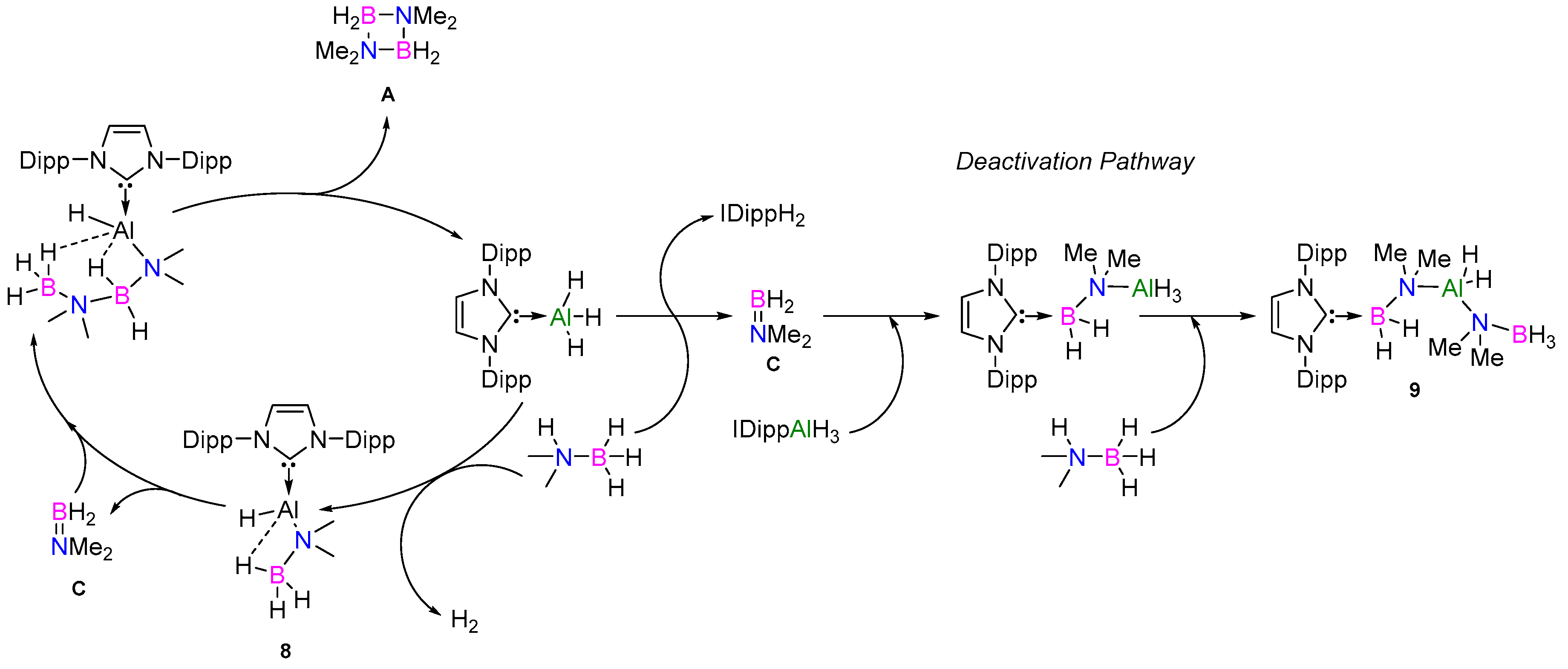
2.2.2. NHC Mechanism
3. Conclusions
4. Materials and Methods
4.1. General Methods and Instruments
4.2. General Catalytic Procedure for Catalysis of Me2NHBH3
4.3. General Procedure of Stoichiometric Reactions
4.4. Synthesis of Diisopropylamineborane, iPr2NHBH3
4.5. Synthesis of Compound 8iPr
4.6. Synthesis of Compound 9
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Boor, J., Jr. Zieglar-Natta Catalysts and Polymerisations; Academic Press Inc.: New York, NY, USA, 1979. [Google Scholar]
- Olah, G.A. Friedel-Crafts and Related Reactions; Wiley: New York, NY, USA, 1963. [Google Scholar]
- Power, P.P. Main-group elements as transition metals. Nature 2010, 463, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Weetman, C.; Inoue, S. The Road Travelled: After Main-Group Elements as Transition Metals. ChemCatChem 2018, 10, 4213–4228. [Google Scholar] [CrossRef]
- Melen, R.L. Frontiers in molecular p-block chemistry: From structure to reactivity. Science 2019, 363, 479. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-C.; Reed, C.A.; Long, G.S.; Sen, A. Et2Al+ Alumenium Ion-like Chemistry. Synthesis and Reactivity toward Alkenes and Alkene Oxides. J. Am. Chem. Soc. 2002, 124, 7662–7663. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, M.; Wehmschulte, R.J. Deoxygenative Reduction of Carbon Dioxide to Methane, Toluene, and Diphenylmethane with [Et2Al]+ as Catalyst. Angew. Chem. Int. Ed. 2012, 51, 7323–7326. [Google Scholar] [CrossRef] [PubMed]
- Weetman, C.; Bag, P.; Silvási, T.; Jandl, C.; Inoue, S. CO2 Fixation and Catalytic Reduction by a Neutral Aluminium Double bond. Angew. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W.; Erker, G. Frustrated Lewis Pairs: Metal-free Hydrogen Activation and More. Angew. Chem. Int. Ed. 2009, 49, 46–76. [Google Scholar] [CrossRef]
- Stephan, D.W.; Erker, G. Frustrated Lewis Pair Chemistry: Development and Perspectives. Angew. Chem. Int. Ed. 2015, 54, 6400–6441. [Google Scholar] [CrossRef]
- Stephan, D.W. The broadening reach of frustrated Lewis pair chemistry. Science 2016, 354, aff7229. [Google Scholar] [CrossRef]
- Hill, M.S.; Liptrot, D.J.; Weetman, C. Alkaline earths as main group reagents in molecular catalysis. Chem. Soc. Rev. 2016, 45, 972–988. [Google Scholar] [CrossRef]
- Hadlington, T.J.; Driess, M.; Jones, C. Low-valent group 14 element hydride chemistry: Towards catalysis. Chem. Soc. Rev. 2018, 47, 4176–4197. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhong, M.; Ma, X.; De, S.; Anusha, C.; Parameswaran, P.; Roesky, H.W. An Aluminum Hydride That Functions like a Transition-Metal Catalyst. Angew. Chem. Int. Ed. 2015, 54, 10225–10229. [Google Scholar] [CrossRef] [PubMed]
- Elsen, H.; Färber, C.; Ballmann, G.; Harder, S. LiAlH4: From Stoichiometric Reduction to Imine Hydrogenation Catalysis. Angew. Chem. Int. Ed. 2018, 57, 7156–7160. [Google Scholar] [CrossRef] [PubMed]
- Bismuto, A.; Thomas Stephen, P.; Cowley Michael, J. Aluminum Hydride Catalyzed Hydroboration of Alkynes. Angew. Chem. Int. Ed. 2016, 55, 15356–15359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitao, E.M.; Jurca, T.; Manners, I. Catalysis in service of main group chemistry offers a versatile approach to p-block molecules and materials. Nat. Chem. 2013, 5, 817. [Google Scholar] [CrossRef] [PubMed]
- Melen, R.L. Dehydrocoupling routes to element-element bonds catalysed by main group compounds. Chem. Soc. Rev. 2016, 45, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Slootweg, C.; Jupp, A.; Boom, D. Dehydrogenation of Amine–Boranes using p-block Compounds. Chem. Eur. J. 2019. [Google Scholar] [CrossRef]
- Keaton, R.J.; Blacquiere, J.M.; Baker, R.T. Base Metal Catalyzed Dehydrogenation of Ammonia−Borane for Chemical Hydrogen Storage. J. Am. Chem. Soc. 2007, 129, 1844–1845. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, C.W.; Baker, R.T.; Staubitz, A.; Manners, I. B–N compounds for chemical hydrogen storage. Chem. Soc. Rev. 2009, 38, 279–293. [Google Scholar] [CrossRef]
- Rossin, A.; Peruzzini, M. Ammonia–Borane and Amine–Borane Dehydrogenation Mediated by Complex Metal Hydrides. Chem. Rev. 2016, 116, 8848–8872. [Google Scholar] [CrossRef]
- Liptrot, D.J.; Hill, M.S.; Mahon, M.F.; MacDougall, D.J. Group 2 Promoted Hydrogen Release from NMe2H⋅BH3: Intermediates and Catalysis. Chem. Eur. J. 2010, 16, 8508–8515. [Google Scholar] [CrossRef] [PubMed]
- McLellan, R.; Kennedy, A.R.; Orr, S.A.; Robertson, S.D.; Mulvey, R.E. Lithium Dihydropyridine Dehydrogenation Catalysis: A Group 1 Approach to the Cyclization of Diamine Boranes. Angew. Chem. Int. Ed. 2017, 56, 1036–1041. [Google Scholar] [CrossRef] [PubMed]
- Bellham, P.; Hill, M.S.; Kociok-Köhn, G. Alkali metal-mediated dehydrocoupling of Me2NH·BH3. Dalton Trans. 2015, 44, 12078–12081. [Google Scholar] [CrossRef] [PubMed]
- Bellham, P.; Anker, M.D.; Hill, M.S.; Kociok-Köhn, G.; Mahon, M.F. The significance of secondary interactions during alkaline earth-promoted dehydrogenation of dialkylamine–boranes. Dalton Trans. 2016, 45, 13969–13978. [Google Scholar] [CrossRef] [PubMed]
- Erickson, K.A.; Kiplinger, J.L. Catalytic Dehydrogenation of Dimethylamine Borane by Highly Active Thorium and Uranium Metallocene Complexes. ACS Catal. 2017, 7, 4276–4280. [Google Scholar] [CrossRef]
- Cui, P.; Spaniol, T.P.; Maron, L.; Okuda, J. Dehydrogenation of Amine-Borane Me2NH⋅BH3 Catalyzed by a Lanthanum-Hydride Complex. Chem. Eur. J. 2013, 19, 13437–13444. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, N.E.; Robertson, A.P.M.; Leitao, E.M.; Manners, I. Amine–borane dehydrogenation chemistry: Metal-free hydrogen transfer, new catalysts and mechanisms, and the synthesis of polyaminoboranes. J. Organomet. Chem. 2013, 730, 84–89. [Google Scholar] [CrossRef]
- Sabourin, K.J.; Malcolm, A.C.; McDonald, R.; Ferguson, M.J.; Rivard, E. Metal-free dehydrogenation of amine–boranes by an N-heterocyclic carbene. Dalton Trans. 2013, 42, 4625–4632. [Google Scholar] [CrossRef]
- Lui Melanie, W.; Paisley Nathan, R.; McDonald, R.; Ferguson Michael, J.; Rivard, E. Metal-Free Dehydrogenation of Amine-Boranes by Tunable N-Heterocyclic Iminoboranes. Chem. Eur. J. 2016, 22, 2134–2145. [Google Scholar] [CrossRef]
- Hansmann, M.M.; Melen, R.L.; Wright, D.S. Group 13 BN dehydrocoupling reagents, similar to transition metal catalysts but with unique reactivity. Chem. Sci. 2011, 2, 1554–1559. [Google Scholar] [CrossRef]
- Cowley, H.J.; Holt, M.S.; Melen, R.L.; Rawson, J.M.; Wright, D.S. Catalytic dehydrocoupling of Me2NHBH3 with Al(NMe2)3. Chem. Commun. 2011, 47, 2682–2684. [Google Scholar] [CrossRef] [PubMed]
- Less, R.J.; Simmonds, H.R.; Dane, S.B.J.; Wright, D.S. Stoichiometric and catalytic reactions of LiAlH4 with Me2NHBH3. Dalton Trans. 2013, 42, 6337–6343. [Google Scholar] [CrossRef] [PubMed]
- Less, R.J.; Simmonds, H.R.; Wright, D.S. Reactivity and catalytic activity of tert-butoxy-aluminium hydride reagents. Dalton Trans. 2014, 43, 5785–5792. [Google Scholar] [CrossRef] [PubMed]
- Less, R.J.; García-Rodríguez, R.; Simmonds, H.R.; Allen, L.K.; Bond, A.D.; Wright, D.S. Use of crown ethers to isolate intermediates in ammonia–borane dehydrocoupling reactions. Chem. Commun. 2016, 52, 3650–3652. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.K.; García-Rodríguez, R.; Wright, D.S. Stoichiometric and catalytic Si–N bond formation using the p-block base Al(NMe2)3. Dalton Trans. 2015, 44, 12112–12118. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, T.; Franz, D.; Inoue, S. Applications of N-heterocyclic imines in main group chemistry. Chem. Soc. Rev. 2016, 45, 6327–6344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesterov, V.; Reiter, D.; Bag, P.; Frisch, P.; Holzner, R.; Porzelt, A.; Inoue, S. NHCs in Main Group Chemistry. Chem. Rev. 2018. [Google Scholar] [CrossRef]
- Franz, D.; Inoue, S. Activation of Elemental Sulfur by Aluminum Dihydride: Isolation of Mono-and Bis(hydrogensulfide) Complexes of Aluminum. Chem. Eur. J. 2014, 20, 10645–10649. [Google Scholar] [CrossRef]
- Franz, D.; Szilvasi, T.; Irran, E.; Inoue, S. A monotopic aluminum telluride with an Al=Te double bond stabilized by N-heterocyclic carbenes. Nat. Commun. 2015, 6, 10037–10042. [Google Scholar] [CrossRef]
- Franz, D.; Sirtl, L.; Pothig, A.; Inoue, S. Aluminum Hydrides Stabilized by N-Heterocyclic Imines as Catalysts for Hydroborations with Pinacolborane. Z. Anorg. Allg. Chem. 2016, 642, 1245–1250. [Google Scholar] [CrossRef]
- Spielmann, J.; Bolte, M.; Harder, S. Synthesis and structure of a magnesium–amidoborane complex and its role in catalytic formation of a new bis-aminoborane ligand. Chem. Commun. 2009, 6934–6936. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.S.; Kociok-Köhn, G.; Robinson, T.P. Group 3-centred dehydrocoupling of Me2NH·BH3. Chem. Comm. 2010, 46, 7587–7589. [Google Scholar] [CrossRef] [PubMed]
- Malcolm, A.C.; Sabourin, K.J.; McDonald, R.; Ferguson, M.J.; Rivard, E. Donor–Acceptor Complexation and Dehydrogenation Chemistry of Aminoboranes. Inorg. Chem. 2012, 51, 12905–12916. [Google Scholar] [CrossRef] [PubMed]
- Dübek, G.; Franz, D.; Eisenhut, C.; Altmann, P.J.; Inoue, S. Reactivity of an NHC-stabilized pyramidal hydrosilylene with electrophilic boron sources. Dalton Trans. 2019, 48, 5756–5765. [Google Scholar] [CrossRef] [PubMed]
- Francis, M.D.; Hibbs, D.E.; Hursthouse, M.B.; Jones, C.; Smithies, N.A. Carbene complexes of Group 13 trihydrides: Synthesis and characterisation of MH3{CN(Pri)C2Me2N(Pri)}, M = Al, Ga or In. J. Chem. Soc. Dalton Trans. 1998, 3249–3254. [Google Scholar] [CrossRef]
- Coles, N.T.; Mahon, M.F.; Webster, R.L. Phosphine– and Amine–Borane Dehydrocoupling Using a Three-Coordinate Iron(II) β-Diketiminate Precatalyst. Organometallics 2017, 36, 2262–2268. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Catalyst | Temp/°C | Time/h | Conversion a/% | TOF b/h−1 |
|---|---|---|---|---|
| 1 | 80 | 34 | 90 | 0.53 |
| 2 | 80 | 34 | 84 | 0.49 |
| 3 | 50 | 65 | 80 | 0.25 |
| 4 | 50 | 26 | 92 | 0.74 |
| 5 | 50 | 65 | 93 | 0.29 |
| 6 | 50 | 40 | 92 | 0.46 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weetman, C.; Ito, N.; Unno, M.; Hanusch, F.; Inoue, S. NHI- and NHC-Supported Al(III) Hydrides for Amine–Borane Dehydrocoupling Catalysis. Inorganics 2019, 7, 92. https://doi.org/10.3390/inorganics7080092
Weetman C, Ito N, Unno M, Hanusch F, Inoue S. NHI- and NHC-Supported Al(III) Hydrides for Amine–Borane Dehydrocoupling Catalysis. Inorganics. 2019; 7(8):92. https://doi.org/10.3390/inorganics7080092
Chicago/Turabian StyleWeetman, Catherine, Nozomi Ito, Masafumi Unno, Franziska Hanusch, and Shigeyoshi Inoue. 2019. "NHI- and NHC-Supported Al(III) Hydrides for Amine–Borane Dehydrocoupling Catalysis" Inorganics 7, no. 8: 92. https://doi.org/10.3390/inorganics7080092
APA StyleWeetman, C., Ito, N., Unno, M., Hanusch, F., & Inoue, S. (2019). NHI- and NHC-Supported Al(III) Hydrides for Amine–Borane Dehydrocoupling Catalysis. Inorganics, 7(8), 92. https://doi.org/10.3390/inorganics7080092