Preliminary Characterization of a Ni2+-Activated and Mycothiol-Dependent Glyoxalase I Enzyme from Streptomyces coelicolor



Abstract
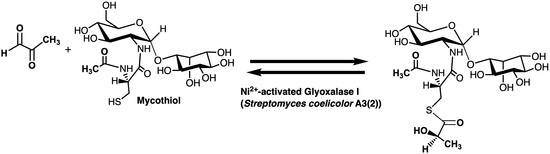
:
1. Introduction
2. Results and Discussion
2.1. Sequence Analysis
2.2. Overproduction, Isolation and Characterization of PDO and PLA
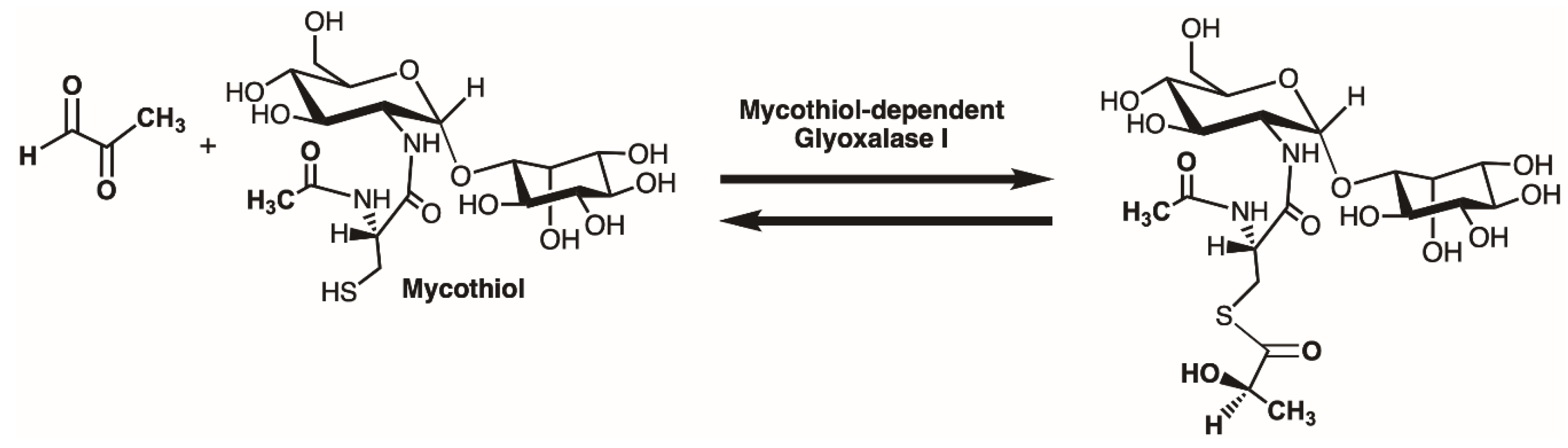
2.3. Enzyme Assay for Thiol Cofactors
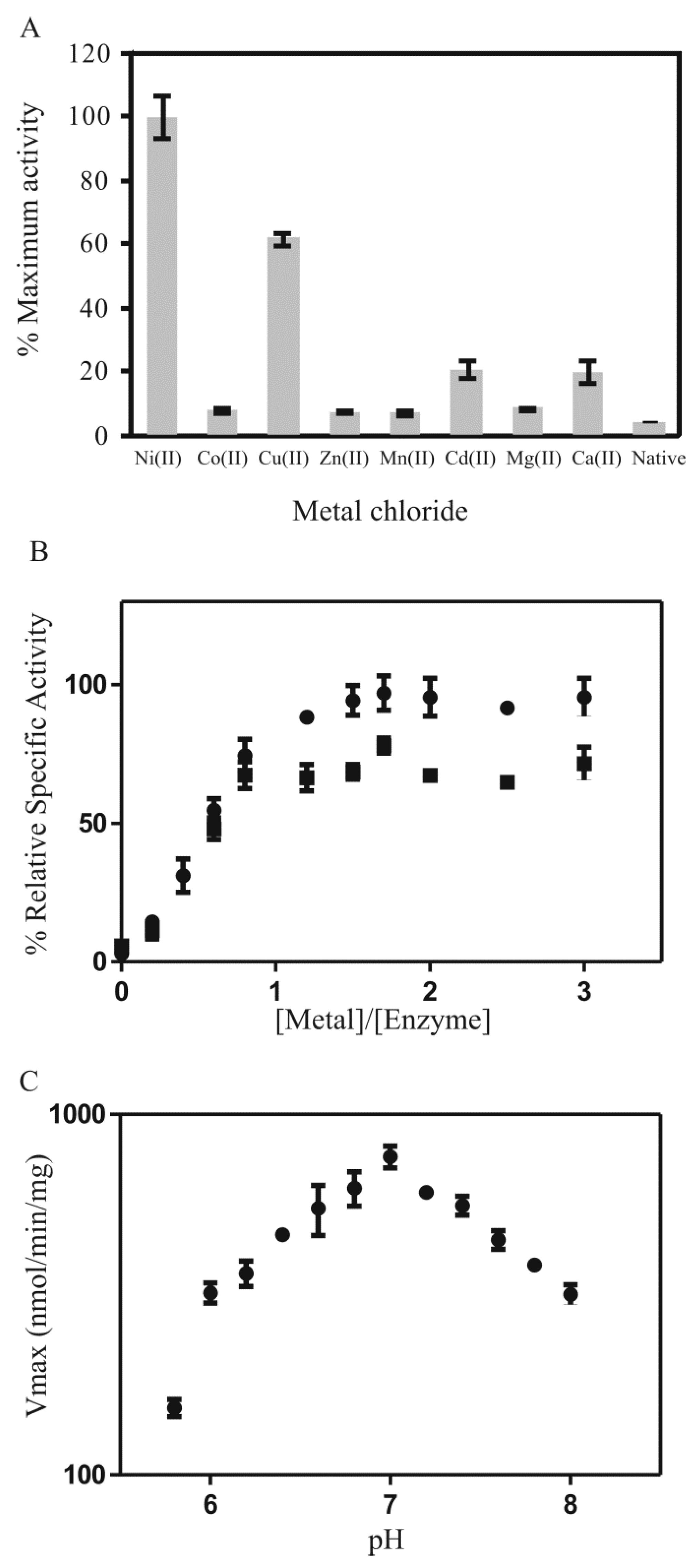
2.4. Metal Characterization and Kinetic Studies
3. Materials and Methods
3.1. DNA Cloning and Manipulation
3.2. Protein Induction, Expression, and Purification
3.3. Protein Secondary Structure and Stability Experiments
3.4. Preparation of Hemithioacetal Substrate
3.5. Enzyme Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thornalley, P.J. Dietary AGEs and ALEs and risk to human health by their interaction with the receptor for advanced glycation endproducts (RAGE)—An introduction. Mol. Nutr. Food Res. 2007, 51, 1107–1110. [Google Scholar] [CrossRef] [PubMed]
- Krautwald, M.; Munch, G. Advanced glycation end products as biomarkers and gerontotoxins—A basis to explore methylglyoxal-lowering agents for Alzheimer’s disease? Exp. Gerontol. 2010, 45, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem. Biophys. Res. Commun. 2015, 458, 221–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schalkwijk, C.G. Vascular AGE-ing by methylglyoxal: The past, the present and the future. Diabetologia 2015, 58, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Honek, J.F. Glyoxalase biochemistry. Biomol. Concepts 2015, 6, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Protein and nucleotide damage by glyoxal and methylglyoxal in physiological systems—Role in ageing and disease. Drug Metabol. Drug Interact. 2008, 23, 125–150. [Google Scholar] [CrossRef] [PubMed]
- Trellu, S.; Courties, A.; Jaisson, S.; Gorisse, L.; Gillery, P.; Kerdine-Romer, S.; Vaamonde-Garcia, C.; Houard, X.; Ekhirch, F.P.; Sautet, A.; et al. Impairment of glyoxalase-1, an advanced glycation end-product detoxifying enzyme, induced by inflammation in age-related osteoarthritis. Arthritis Res. Ther. 2019, 21, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bari, L.; Atlante, A.; Armeni, T.; Kalapos, M.P. Synthesis and metabolism of methylglyoxal, S-d-lactoylglutathione and d-lactate in cancer and Alzheimer’s disease. Exploring the crossroad of eternal youth and premature aging. Ageing Res. Rev. 2019, 53, 100915. [Google Scholar] [CrossRef] [PubMed]
- Sousa Silva, M.; Gomes, R.A.; Ferreira, A.E.; Ponces Freire, A.; Cordeiro, C. The glyoxalase pathway: The first hundred years... and beyond. Biochem. J. 2013, 453, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sukdeo, N.; Honek, J.F. Microbial glyoxalase enzymes: Metalloenzymes controlling cellular levels of methylglyoxal. Drug Metabol. Drug Interact. 2008, 23, 29–50. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Dicarbonyls and glyoxalase in disease mechanisms and clinical therapeutics. Glycoconj. J. 2016, 33, 513–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbani, N.; Thornalley, P.J. Glyoxalase Centennial conference: Introduction, history of research on the glyoxalase system and future prospects. Biochem. Soc. Trans. 2014, 42, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Glyoxalase I—Structure, function and a critical role in the enzymatic defence against glycation. Biochem. Soc. Trans. 2003, 31, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Ozyamak, E.; Black, S.S.; Walker, C.A.; Maclean, M.J.; Bartlett, W.; Miller, S.; Booth, I.R. The critical role of S-lactoylglutathione formation during methylglyoxal detoxification in Escherichia coli. Mol. Microbiol. 2010, 78, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Misra, K.; Banerjee, A.B.; Ray, S.; Ray, M. Glyoxalase III from Escherichia coli: A single novel enzyme for the conversion of methylglyoxal into d-lactate without reduced glutathione. Biochem. J. 1995, 305 Pt 3, 999–1003. [Google Scholar] [CrossRef]
- Choi, D.; Kim, J.; Ha, S.; Kwon, K.; Kim, E.H.; Lee, H.Y.; Ryu, K.S.; Park, C. Stereospecific mechanism of DJ-1 glyoxalases inferred from their hemithioacetal-containing crystal structures. FEBS J. 2014, 281, 5447–5462. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Su, Y.; Wang, Z.; Chen, C.; Wu, T.; Huang, Y. Identification of glutathione (GSH)-independent glyoxalase III from Schizosaccharomyces pombe. BMC Evol. Biol. 2014, 14, 86. [Google Scholar] [CrossRef] [PubMed]
- Subedi, K.P.; Choi, D.; Kim, I.; Min, B.; Park, C. Hsp31 of Escherichia coli K-12 is glyoxalase III. Mol. Microbiol. 2011, 81, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Sukdeo, N.; Clugston, S.L.; Daub, E.; Honek, J.F. Distinct classes of glyoxalase I: Metal specificity of the Yersinia pestis, Pseudomonas aeruginosa and Neisseria meningitidis enzymes. Biochem. J. 2004, 384, 111–117. [Google Scholar] [CrossRef]
- Aronsson, A.C.; Marmstal, E.; Mannervik, B. Glyoxalase I, a zinc metalloenzyme of mammals and yeast. Biochem. Biophys. Res. Commun. 1978, 81, 1235–1240. [Google Scholar] [CrossRef]
- Han, L.P.; Schimandle, C.M.; Davison, L.M.; Vander Jagt, D.L. Comparative kinetics of Mg2+-, Mn2+-, Co2+-, and Ni2+-activated glyoxalase I. Evaluation of the role of the metal ion. Biochemistry 1977, 16, 5478–5484. [Google Scholar] [CrossRef] [PubMed]
- Saint-Jean, A.P.; Phillips, K.R.; Creighton, D.J.; Stone, M.J. Active monomeric and dimeric forms of Pseudomonas putida glyoxalase I: Evidence for 3D domain swapping. Biochemistry 1998, 37, 10345–10353. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.D.; Olin, B.; Ridderstrom, M.; Mannervik, B.; Jones, T.A. Crystal structure of human glyoxalase I—Evidence for gene duplication and 3D domain swapping. EMBO J. 1997, 16, 3386–3395. [Google Scholar] [CrossRef] [PubMed]
- Clugston, S.L.; Barnard, J.F.; Kinach, R.; Miedema, D.; Ruman, R.; Daub, E.; Honek, J.F. Overproduction and characterization of a dimeric non-zinc glyoxalase I from Escherichia coli: Evidence for optimal activation by nickel ions. Biochemistry 1998, 37, 8754–8763. [Google Scholar] [CrossRef] [PubMed]
- Clugston, S.L.; Yajima, R.; Honek, J.F. Investigation of metal binding and activation of Escherichia coli glyoxalase I: Kinetic, thermodynamic and mutagenesis studies. Biochem. J. 2004, 377, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Davidson, G.; Clugston, S.L.; Honek, J.F.; Maroney, M.J. XAS investigation of the nickel active site structure in Escherichia coli glyoxalase I. Inorg. Chem. 2000, 39, 2962–2963. [Google Scholar] [CrossRef] [PubMed]
- He, M.M.; Clugston, S.L.; Honek, J.F.; Matthews, B.W. Determination of the structure of Escherichia coli glyoxalase I suggests a structural basis for differential metal activation. Biochemistry 2000, 39, 8719–8727. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.D.; Ridderstrom, M.; Olin, B.; Kavarana, M.J.; Creighton, D.J.; Mannervik, B. Reaction mechanism of glyoxalase I explored by an X-ray crystallographic analysis of the human enzyme in complex with a transition state analogue. Biochemistry 1999, 38, 13480–13490. [Google Scholar] [CrossRef]
- Sellin, S.; Eriksson, L.E.; Aronsson, A.C.; Mannervik, B. Octahedral metal coordination in the active site of glyoxalase I as evidenced by the properties of Co(II)-glyoxalase I. J. Biol. Chem. 1983, 258, 2091–2093. [Google Scholar]
- Himo, F.; Siegbahn, P.E. Catalytic mechanism of glyoxalase I: A theoretical study. J. Am. Chem. Soc. 2001, 123, 10280–10289. [Google Scholar] [CrossRef]
- Richter, U.; Krauss, M. Active site structure and mechanism of human glyoxalase I—An ab initio theoretical study. J. Am. Chem. Soc. 2001, 123, 6973–6982. [Google Scholar] [CrossRef] [PubMed]
- Davidson, G.; Clugston, S.L.; Honek, J.F.; Maroney, M.J. An XAS investigation of product and inhibitor complexes of Ni-containing GlxI from Escherichia coli: Mechanistic implications. Biochemistry 2001, 40, 4569–4582. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Suttisansanee, U.; Lau, K.; Lagishetty, S.; Rao, K.N.; Swaminathan, S.; Sauder, J.M.; Burley, S.K.; Honek, J.F. Structural variation in bacterial glyoxalase I enzymes: Investigation of the metalloenzyme glyoxalase I from Clostridium acetobutylicum. J. Biol. Chem. 2011, 286, 38367–38374. [Google Scholar] [CrossRef] [PubMed]
- Deponte, M.; Sturm, N.; Mittler, S.; Harner, M.; Mack, H.; Becker, K. Allosteric coupling of two different functional active sites in monomeric Plasmodium falciparum glyoxalase I. J. Biol. Chem. 2007, 282, 28419–28430. [Google Scholar] [CrossRef] [PubMed]
- Frickel, E.M.; Jemth, P.; Widersten, M.; Mannervik, B. Yeast glyoxalase I is a monomeric enzyme with two active sites. J. Biol. Chem. 2001, 276, 1845–1849. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Maeta, K.; Nomura, W. Glyoxalase system in yeasts: Structure, function, and physiology. Semin. Cell Dev. Biol. 2011, 22, 278–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iozef, R.; Rahlfs, S.; Chang, T.; Schirmer, H.; Becker, K. Glyoxalase I of the malarial parasite Plasmodium falciparum: Evidence for subunit fusion. FEBS Lett. 2003, 554, 284–288. [Google Scholar] [CrossRef]
- Turra, G.L.; Agostini, R.B.; Fauguel, C.M.; Presello, D.A.; Andreo, C.S.; Gonzalez, J.M.; Campos-Bermudez, V.A. Structure of the novel monomeric glyoxalase I from Zea mays. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 2009–2020. [Google Scholar] [CrossRef]
- Kaur, C.; Vishnoi, A.; Ariyadasa, T.U.; Bhattacharya, A.; Singla-Pareek, S.L.; Sopory, S.K. Episodes of horizontal gene-transfer and gene-fusion led to co-existence of different metal-ion specific glyoxalase I. Sci. Rep. 2013, 3, 3076. [Google Scholar] [CrossRef] [Green Version]
- Mustafiz, A.; Ghosh, A.; Tripathi, A.K.; Kaur, C.; Ganguly, A.K.; Bhavesh, N.S.; Tripathi, J.K.; Pareek, A.; Sopory, S.K.; Singla-Pareek, S.L. A unique Ni2+ -dependent and methylglyoxal-inducible rice glyoxalase I possesses a single active site and functions in abiotic stress response. Plant J. 2014, 78, 951–963. [Google Scholar] [CrossRef]
- Gonzalez, J.M.; Agostini, R.B.; Alvarez, C.E.; Klinke, S.; Andreo, C.S.; Campos-Bermudez, V.A. Deciphering the number and location of active sites in the monomeric glyoxalase I of Zea mays. FEBS J. 2019. [Google Scholar] [CrossRef]
- Su, Z.; Sukdeo, N.; Honek, J.F. 15N-1H HSQC NMR evidence for distinct specificity of two active sites in Escherichia coli glyoxalase I. Biochemistry 2008, 47, 13232–13241. [Google Scholar] [CrossRef]
- Bythell-Douglas, R.; Suttisansanee, U.; Flematti, G.R.; Challenor, M.; Lee, M.; Panjikar, S.; Honek, J.F.; Bond, C.S. The crystal structure of a homodimeric Pseudomonas glyoxalase I enzyme reveals asymmetric metallation commensurate with half-of-sites activity. Chemistry 2015, 21, 541–544. [Google Scholar] [CrossRef]
- Suttisansanee, U.; Ran, Y.; Mullings, K.Y.; Sukdeo, N.; Honek, J.F. Modulating glyoxalase I metal selectivity by deletional mutagenesis: Underlying structural factors contributing to nickel activation profiles. Metallomics 2015, 7, 605–612. [Google Scholar] [CrossRef]
- Hand, C.E.; Honek, J.F. Biological chemistry of naturally occurring thiols of microbial and marine origin. J. Nat. Prod. 2005, 68, 293–308. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, Q.; Liu, W. The versatile low-molecular-weight thiols: Beyond cell protection. Bioessays 2015, 37, 1262–1267. [Google Scholar] [CrossRef]
- Newton, G.L.; Arnold, K.; Price, M.S.; Sherrill, C.; Delcardayre, S.B.; Aharonowitz, Y.; Cohen, G.; Davies, J.; Fahey, R.C.; Davis, C. Distribution of thiols in microorganisms: Mycothiol is a major thiol in most actinomycetes. J. Bacteriol. 1996, 178, 1990–1995. [Google Scholar] [CrossRef]
- Gaballa, A.; Newton, G.L.; Antelmann, H.; Parsonage, D.; Upton, H.; Rawat, M.; Claiborne, A.; Fahey, R.C.; Helmann, J.D. Biosynthesis and functions of bacillithiol, a major low-molecular-weight thiol in Bacilli. Proc. Natl. Acad. Sci. USA 2010, 107, 6482–6486. [Google Scholar] [CrossRef] [Green Version]
- Jothivasan, V.K.; Hamilton, C.J. Mycothiol: Synthesis, biosynthesis and biological functions of the major low molecular weight thiol in actinomycetes. Nat. Prod. Rep. 2008, 25, 1091–1117. [Google Scholar] [CrossRef]
- Vickers, T.J.; Greig, N.; Fairlamb, A.H. A trypanothione-dependent glyoxalase I with a prokaryotic ancestry in Leishmania major. Proc. Natl. Acad. Sci. USA 2004, 101, 13186–13191. [Google Scholar] [CrossRef]
- Oza, S.L.; Shaw, M.P.; Wyllie, S.; Fairlamb, A.H. Trypanothione biosynthesis in Leishmania major. Mol. Biochem. Parasitol. 2005, 139, 107–116. [Google Scholar] [CrossRef]
- Greig, N.; Wyllie, S.; Patterson, S.; Fairlamb, A.H. A comparative study of methylglyoxal metabolism in trypanosomatids. FEBS J. 2009, 276, 376–386. [Google Scholar] [CrossRef]
- Ariza, A.; Vickers, T.J.; Greig, N.; Armour, K.A.; Dixon, M.J.; Eggleston, I.M.; Fairlamb, A.H.; Bond, C.S. Specificity of the trypanothione-dependent Leishmania major glyoxalase I: Structure and biochemical comparison with the human enzyme. Mol. Microbiol. 2006, 59, 1239–1248. [Google Scholar] [CrossRef]
- Greig, N.; Wyllie, S.; Vickers, T.J.; Fairlamb, A.H. Trypanothione-dependent glyoxalase I in Trypanosoma cruzi. Biochem. J. 2006, 400, 217–223. [Google Scholar] [CrossRef]
- Fahey, R.C. Glutathione analogs in prokaryotes. Biochim. Biophys. Acta 2013, 1830, 3182–3198. [Google Scholar] [CrossRef]
- Sharma, S.V.; Arbach, M.; Roberts, A.A.; Macdonald, C.J.; Groom, M.; Hamilton, C.J. Biophysical features of bacillithiol, the glutathione surrogate of Bacillus subtilis and other firmicutes. ChemBioChem 2013, 14, 2160–2168. [Google Scholar] [CrossRef]
- Perera, V.R.; Newton, G.L.; Pogliano, K. Bacillithiol: A key protective thiol in Staphylococcus aureus. Expert Rev. Anti Infect. Ther. 2015, 13, 1089–1107. [Google Scholar] [CrossRef]
- Chandrangsu, P.; Loi, V.V.; Antelmann, H.; Helmann, J.D. The Role of Bacillithiol in Gram-Positive Firmicutes. Antioxid. Redox Signal. 2018, 28, 445–462. [Google Scholar] [CrossRef]
- Chandrangsu, P.; Dusi, R.; Hamilton, C.J.; Helmann, J.D. Methylglyoxal resistance in Bacillus subtilis: Contributions of bacillithiol-dependent and independent pathways. Mol. Microbiol. 2014, 91, 706–715. [Google Scholar] [CrossRef]
- Sao Emani, C.; Williams, M.J.; Wiid, I.J.; Baker, B. The functional interplay of low molecular weight thiols in Mycobacterium tuberculosis. J. Biomed. Sci. 2018, 25, 55. [Google Scholar] [CrossRef]
- Sharma, S.V.; Van Laer, K.; Messens, J.; Hamilton, C.J. Thiol Redox and pKa Properties of Mycothiol, the Predominant Low-Molecular-Weight Thiol Cofactor in the Actinomycetes. ChemBioChem 2016, 17, 1689–1692. [Google Scholar] [CrossRef]
- Liu, Y.B.; Long, M.X.; Yin, Y.J.; Si, M.R.; Zhang, L.; Lu, Z.Q.; Wang, Y.; Shen, X.H. Physiological roles of mycothiol in detoxification and tolerance to multiple poisonous chemicals in Corynebacterium glutamicum. Arch. Microbiol. 2013, 195, 419–429. [Google Scholar] [CrossRef]
- Newton, G.L.; Buchmeier, N.; Fahey, R.C. Biosynthesis and functions of mycothiol, the unique protective thiol of Actinobacteria. Microbiol. Mol. Biol. Rev. 2008, 72, 471–494. [Google Scholar] [CrossRef]
- Bentley, S.D.; Chater, K.F.; Cerdeno-Tarraga, A.M.; Challis, G.L.; Thomson, N.R.; James, K.D.; Harris, D.E.; Quail, M.A.; Kieser, H.; Harper, D.; et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 2002, 417, 141–147. [Google Scholar] [CrossRef]
- Park, J.H.; Cha, C.J.; Roe, J.H. Identification of genes for mycothiol biosynthesis in Streptomyces coelicolor A3(2). J. Microbiol. 2006, 44, 121–125. [Google Scholar]
- Kizil, G.; Wilks, K.; Wells, D.; Ala’Aldeen, D.A. Detection and characterisation of the genes encoding glyoxalase I and II from Neisseria meningitidis. J. Med. Microbiol. 2000, 49, 669–673. [Google Scholar] [CrossRef]
- Sukdeo, N.; Honek, J.F. Pseudomonas aeruginosa contains multiple glyoxalase I-encoding genes from both metal activation classes. Biochim. Biophys. Acta 2007, 1774, 756–763. [Google Scholar] [CrossRef]
- Hamilton, C.J.; Finlay, R.M.; Stewart, M.J.; Bonner, A. Mycothiol disulfide reductase: A continuous assay for slow time-dependent inhibitors. Anal. Biochem. 2009, 388, 91–96. [Google Scholar] [CrossRef]
- Unson, M.D.; Newton, G.L.; Davis, C.; Fahey, R.C. An immunoassay for the detection and quantitative determination of mycothiol. J. Immunol. Methods 1998, 214, 29–39. [Google Scholar] [CrossRef]
- Patel, M.P.; Blanchard, J.S. Synthesis of des-myo-inositol mycothiol and demonstration of a mycobacterial specific reductase activity. J. Am. Chem. Soc. 1998, 120, 11538–11539. [Google Scholar] [CrossRef]
- Vander Jagt, D.L.; Han, L.P.; Lehman, C.H. Kinetic evaluation of substrate specificity in the glyoxalase-I-catalyzed disproportionation of α-ketoaldehydes. Biochemistry 1972, 11, 3735–3740. [Google Scholar] [CrossRef]
- Vince, R.; Daluge, S.; Wadd, W.B. Studies on the inhibition of glyoxalase I by S-substituted glutathiones. J. Med. Chem. 1971, 14, 402–404. [Google Scholar] [CrossRef]
- Cliffe, E.E.; Waley, S.G. The mechanism of the glyoxalase I reaction, and the effect of ophthalmic acid as an inhibitor. Biochem. J. 1961, 79, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Creighton, D.J.; Migliorini, M.; Pourmotabbed, T.; Guha, M.K. Optimization of efficiency in the glyoxalase pathway. Biochemistry 1988, 27, 7376–7384. [Google Scholar] [CrossRef]
- Griffis, C.E.; Ong, L.H.; Buettner, L.; Creighton, D.J. Nonstereospecific substrate usage by glyoxalase I. Biochemistry 1983, 22, 2945–2951. [Google Scholar] [CrossRef]
- Vander Jagt, D.L.; Daub, E.; Krohn, J.A.; Han, L.P. Effects of pH and thiols on the kinetics of yeast glyoxalase I. An evaluation of the random pathway mechanism. Biochemistry 1975, 14, 3669–3675. [Google Scholar] [CrossRef]
- Mullings, K.Y.; Sukdeo, N.; Suttisansanee, U.; Ran, Y.; Honek, J.F. Ni2+-activated glyoxalase I from Escherichia coli: Substrate specificity, kinetic isotope effects and evolution within the betaalphabetabetabeta superfamily. J. Inorg. Biochem. 2012, 108, 133–140. [Google Scholar] [CrossRef]
- Rae, C.; O’Donoghue, S.I.; Bubb, W.A.; Kuchel, P.W. Stereospecificity of substrate usage by glyoxalase 1: Nuclear magnetic resonance studies of kinetics and hemithioacetal substrate conformation. Biochemistry 1994, 33, 3548–3559. [Google Scholar] [CrossRef]
- Akoachere, M.; Iozef, R.; Rahlfs, S.; Deponte, M.; Mannervik, B.; Creighton, D.J.; Schirmer, H.; Becker, K. Characterization of the glyoxalases of the malarial parasite Plasmodium falciparum and comparison with their human counterparts. Biol. Chem. 2005, 386, 41–52. [Google Scholar] [CrossRef]
- Mannervik, B.; Ridderstrom, M. Catalytic and molecular properties of glyoxalase I. Biochem. Soc. Trans. 1993, 21, 515–517. [Google Scholar] [CrossRef] [Green Version]
- Bergdoll, M.; Eltis, L.D.; Cameron, A.D.; Dumas, P.; Bolin, J.T. All in the family: Structural and evolutionary relationships among three modular proteins with diverse functions and variable assembly. Protein Sci. 1998, 7, 1661–1670. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, R.N. Mechanistic diversity in a metalloenzyme superfamily. Biochemistry 2000, 39, 13625–13632. [Google Scholar] [CrossRef]
- Honek, J.F. Nickel Glyuoxalase I. In The Biological Chemistry of Nickel; Kozlowski, H., Zamble, D., Rowinska-Zyrek, M., Eds.; Royal Societ of Chemistry: London, UK, 2017; Volume 10. [Google Scholar]
- McCarthy, A.A.; Baker, H.M.; Shewry, S.C.; Patchett, M.L.; Baker, E.N. Crystal structure of methylmalonyl-coenzyme A epimerase from P. shermanii: A novel enzymatic function on an ancient metal binding scaffold. Structure 2001, 9, 637–646. [Google Scholar] [CrossRef]
- Dumas, P.; Bergdoll, M.; Cagnon, C.; Masson, J.M. Crystal structure and site-directed mutagenesis of a bleomycin resistance protein and their significance for drug sequestering. EMBO J. 1994, 13, 2483–2492. [Google Scholar] [CrossRef]
- Martin, T.W.; Dauter, Z.; Devedjiev, Y.; Sheffield, P.; Jelen, F.; He, M.; Sherman, D.H.; Otlewski, J.; Derewenda, Z.S.; Derewenda, U. Molecular basis of mitomycin C resistance in streptomyces: Structure and function of the MRD protein. Structure 2002, 10, 933–942. [Google Scholar] [CrossRef]
- Thompson, M.K.; Keithly, M.E.; Harp, J.; Cook, P.D.; Jagessar, K.L.; Sulikowski, G.A.; Armstrong, R.N. Structural and chemical aspects of resistance to the antibiotic fosfomycin conferred by FosB from Bacillus cereus. Biochemistry 2013, 52, 7350–7362. [Google Scholar] [CrossRef]
- Gonzalez-Quinonez, N.; Corte-Rodriguez, M.; Alvarez-Fernandez-Garcia, R.; Rioseras, B.; Lopez-Garcia, M.T.; Fernandez-Garcia, G.; Montes-Bayon, M.; Manteca, A.; Yague, P. Cytosolic copper is a major modulator of germination, development and secondary metabolism in Streptomyces coelicolor. Sci. Rep. 2019, 9, 4214. [Google Scholar] [CrossRef]
- Aronsson, A.C.; Mannervik, B. Characterization of glyoxalase I purified from pig erythrocytes by affinity chromatography. Biochem. J. 1977, 165, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Mannervik, B.; Lindstrom, L.; Bartfai, T. Partial purification and characterization of glyoxalase I from porcine erythrocytes. Eur. J. Biochem. 1972, 29, 276–281. [Google Scholar] [CrossRef]
- Uotila, L.; Koivusalo, M. Purification and properties of glyoxalase I from sheep liver. Eur. J. Biochem. 1975, 52, 493–503. [Google Scholar] [CrossRef]
- Takatsume, Y.; Izawa, S.; Inoue, Y. Identification of thermostable glyoxalase I in the fission yeast Schizosaccharomyces pombe. Arch. Microbiol. 2004, 181, 371–377. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- O’Young, J.; Sukdeo, N.; Honek, J.F. Escherichia coli glyoxalase II is a binuclear zinc-dependent metalloenzyme. Arch. Biochem. Biophys. 2007, 459, 20–26. [Google Scholar] [CrossRef]
- Hunt, J.B.; Neece, S.H.; Ginsburg, A. The use of 4-(2-pyridylazo)resorcinol in studies of zinc release from Escherichia coli aspartate transcarbamoylase. Anal. Biochem. 1985, 146, 150–157. [Google Scholar] [CrossRef]
- McCall, K.A.; Fierke, C.A. Colorimetric and fluorimetric assays to quantitate micromolar concentrations of transition metals. Anal. Biochem. 2000, 284, 307–315. [Google Scholar] [CrossRef]
- Louis-Jeune, C.; Andrade-Navarro, M.A.; Perez-Iratxeta, C. Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins 2011, 80, 374–381. [Google Scholar] [CrossRef]





 ) Ni2+ and (
) Ni2+ and (  ) Cu2+; (C) pH dependency of PDO (3.125 µg in 200 µL assay; 5 equivalents NiCl2) with the substrate MG-tMSH (0.5 mM, Kd = 3.3 mM) that was incubated for 30 min in potassium phosphate buffer at various pH (5.8–8) at 25 °C.
) Ni2+ and ( ) Cu2+; (C) pH dependency of PDO (3.125 µg in 200 µL assay; 5 equivalents NiCl2) with the substrate MG-tMSH (0.5 mM, Kd = 3.3 mM) that was incubated for 30 min in potassium phosphate buffer at various pH (5.8–8) at 25 °C.
) Cu2+; (C) pH dependency of PDO (3.125 µg in 200 µL assay; 5 equivalents NiCl2) with the substrate MG-tMSH (0.5 mM, Kd = 3.3 mM) that was incubated for 30 min in potassium phosphate buffer at various pH (5.8–8) at 25 °C.
) Ni2+ and ( ) Cu2+; (C) pH dependency of PDO (3.125 µg in 200 µL assay; 5 equivalents NiCl2) with the substrate MG-tMSH (0.5 mM, Kd = 3.3 mM) that was incubated for 30 min in potassium phosphate buffer at various pH (5.8–8) at 25 °C.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Thiol Cofactors | Vmax (μmol/min/mg) | Km (μM) | kcat (s−1) | kcat/Km (M−1s−1) |
|---|---|---|---|---|
| MSH | 11.5 ± 1.8 | 612 ± 56 | 6.4 | 10403 |
| tMSH | 5.8 ± 1.0 | 1247 ± 127 | 3.2 | 2545 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suttisansanee, U.; Honek, J.F. Preliminary Characterization of a Ni2+-Activated and Mycothiol-Dependent Glyoxalase I Enzyme from Streptomyces coelicolor. Inorganics 2019, 7, 99. https://doi.org/10.3390/inorganics7080099
Suttisansanee U, Honek JF. Preliminary Characterization of a Ni2+-Activated and Mycothiol-Dependent Glyoxalase I Enzyme from Streptomyces coelicolor. Inorganics. 2019; 7(8):99. https://doi.org/10.3390/inorganics7080099
Chicago/Turabian StyleSuttisansanee, Uthaiwan, and John F. Honek. 2019. "Preliminary Characterization of a Ni2+-Activated and Mycothiol-Dependent Glyoxalase I Enzyme from Streptomyces coelicolor" Inorganics 7, no. 8: 99. https://doi.org/10.3390/inorganics7080099
APA StyleSuttisansanee, U., & Honek, J. F. (2019). Preliminary Characterization of a Ni2+-Activated and Mycothiol-Dependent Glyoxalase I Enzyme from Streptomyces coelicolor. Inorganics, 7(8), 99. https://doi.org/10.3390/inorganics7080099