
Synthesis of N,O-Chelating Hydrazidopalladium Complexes from 1,2-Bis(trifluoroacetyl)hydrazine



Abstract
:1. Introduction
2. Results and Discussion
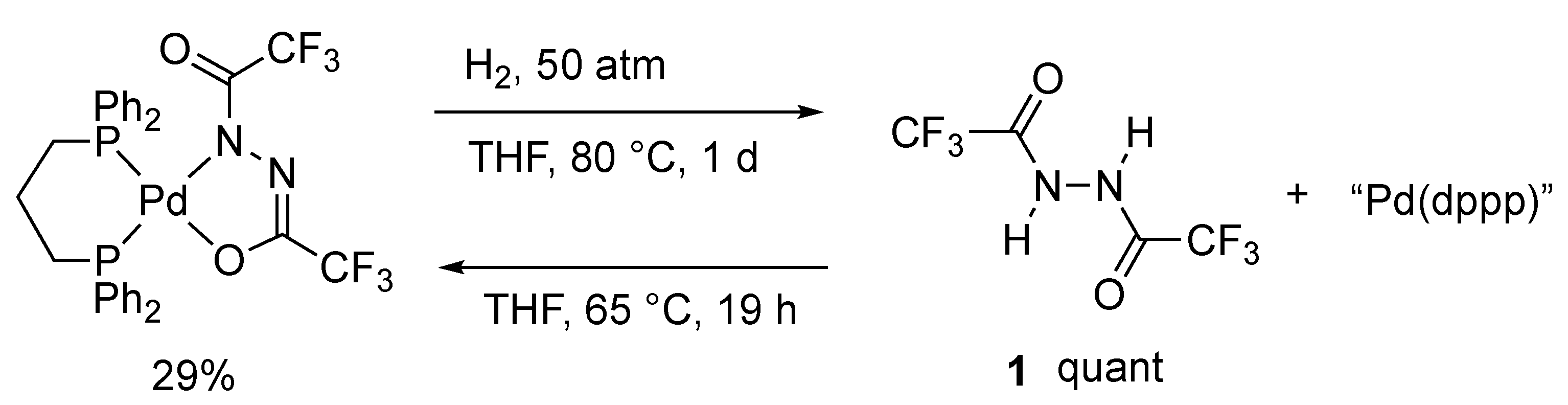
2.1. Reaction of 1,2-Bis(trifluoroacetyl)hydrazine in the Presence of Pd(0)
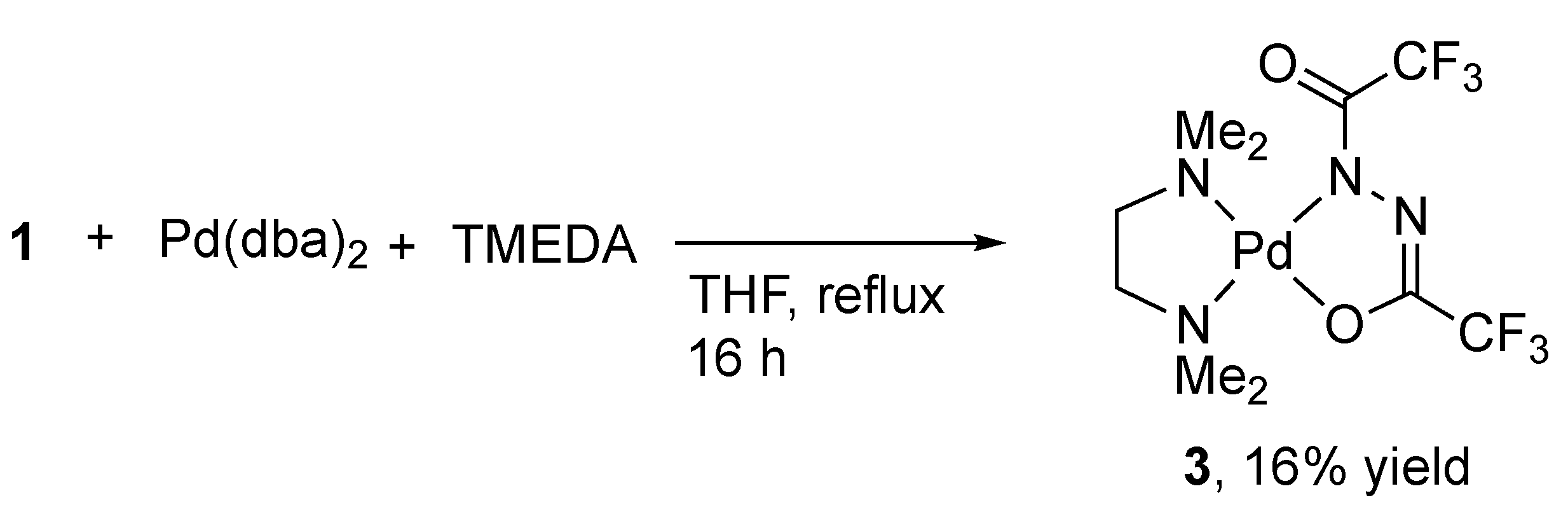
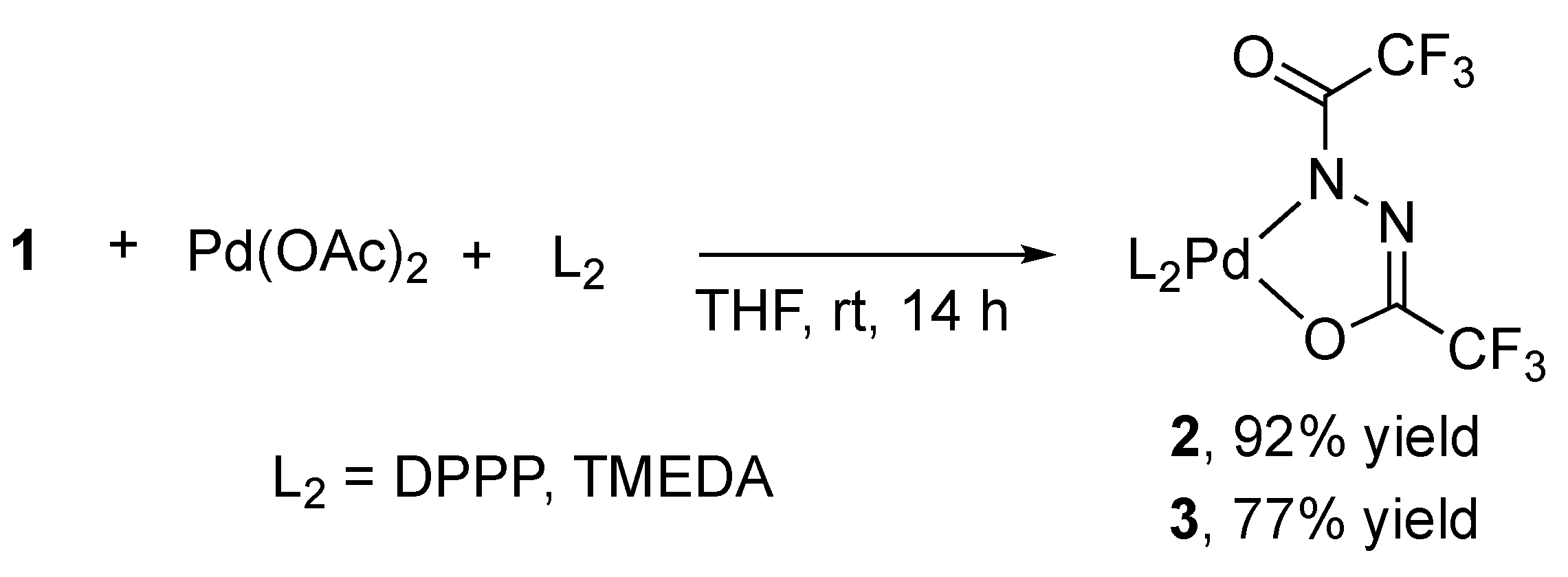
2.2. Alternative Synthesis of Diacylhydrazido Complexes from 1 and Pd(OAc)2
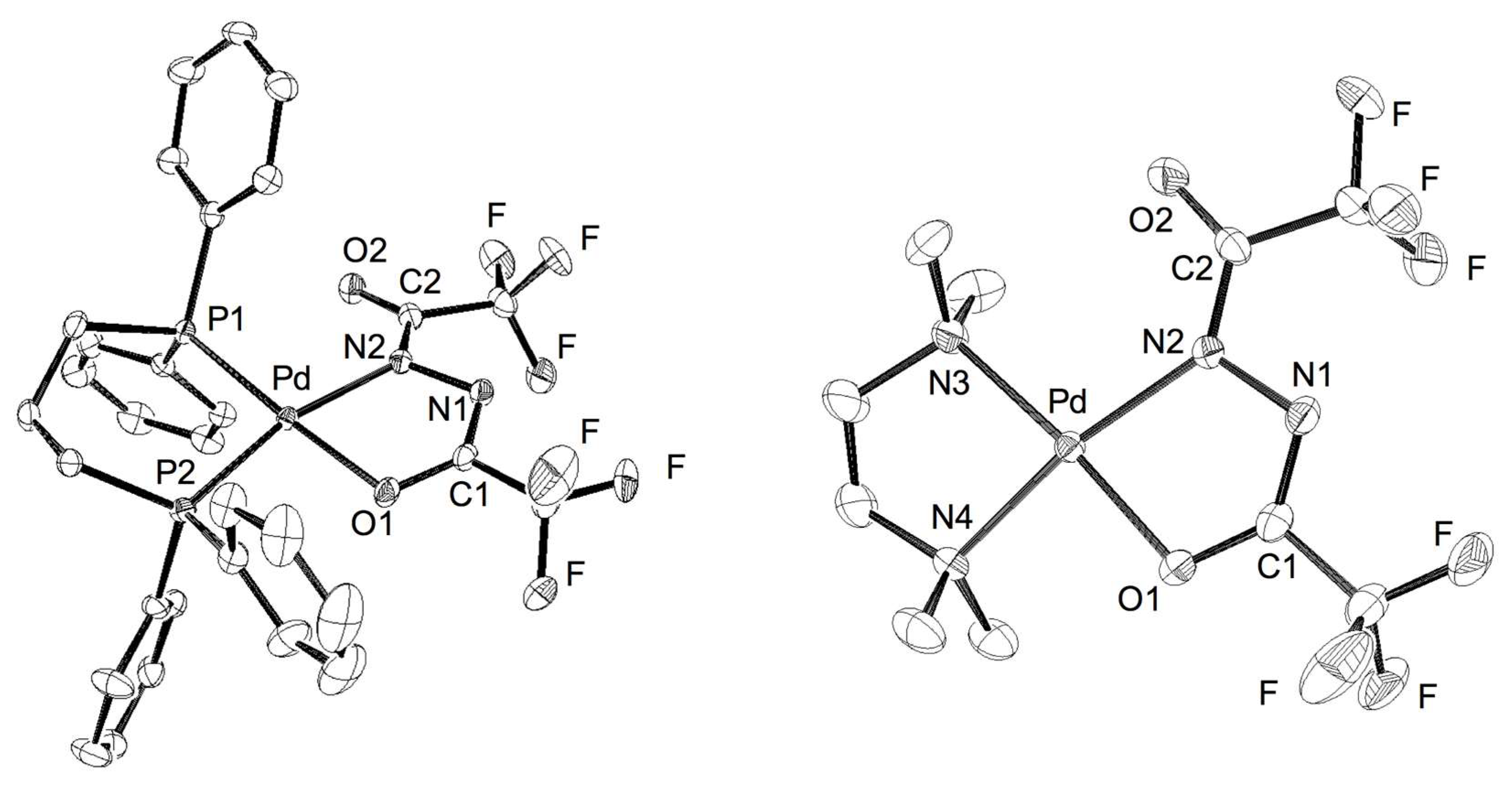
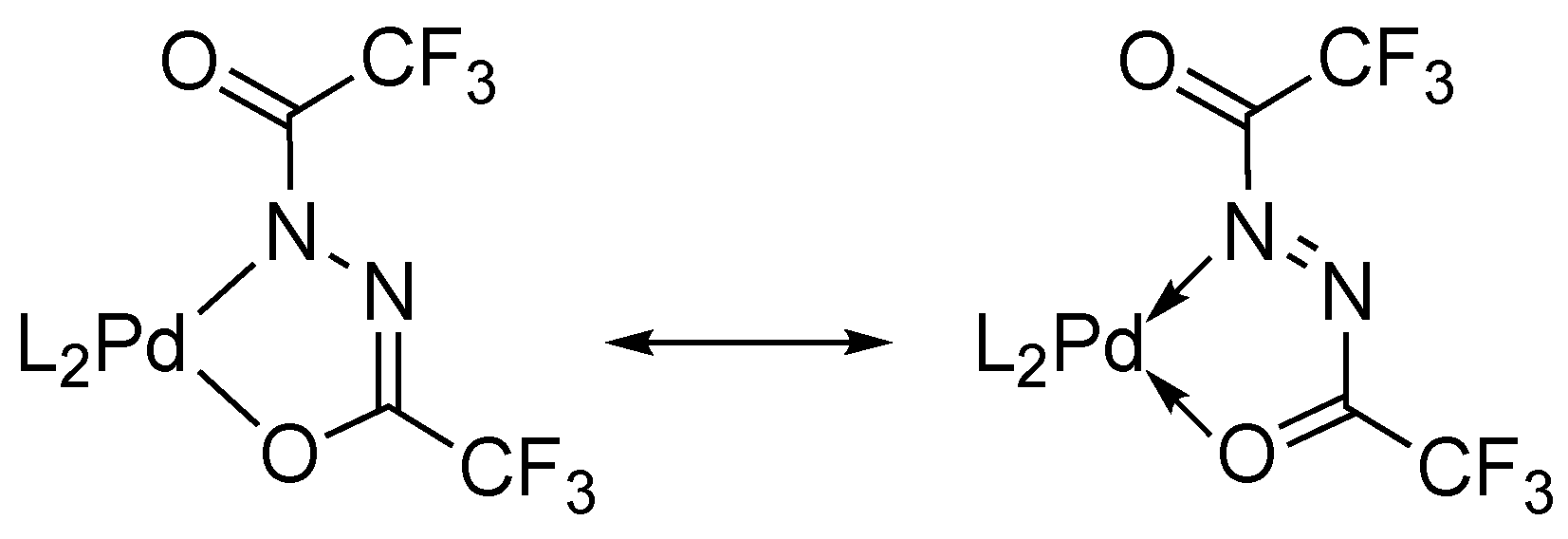
2.3. Crystal Structure of Diacylhydrazido Complexes 2 and 3
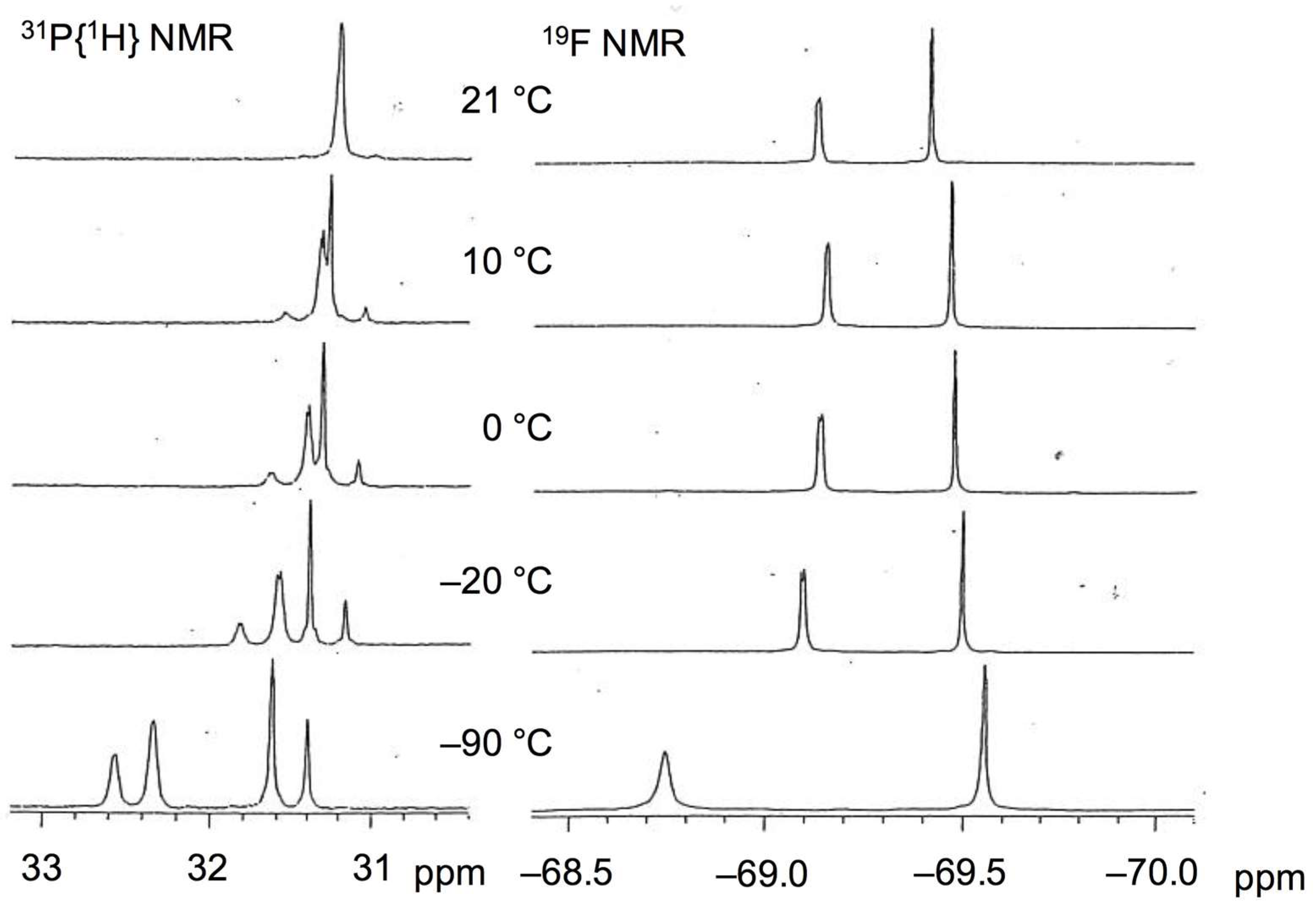
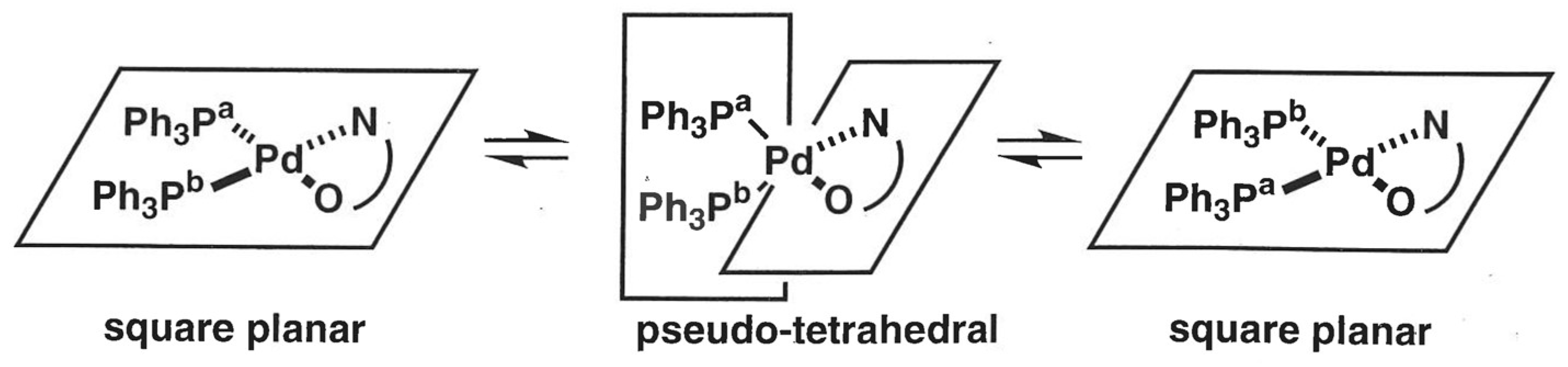
2.4. Synthesis and NMR Analyses of PPh3-Coordinated Diacylhydrazido Complex 4
3. Materials and Methods
3.1. General Information
3.2. Reaction of 1 with Pd(0) Species
3.2.1. Formation of 2 by Treatment of 1 with [Pd(dba)2] and DPPP
3.2.2. Formation of 3 by Treatment of 1 with [Pd(dba)2] and TMEDA
3.2.3. Formation of 4 by Treatment of 1 with [Pd(dba)2] and PPh3
3.3. Alternative Synthesis of 2 and 3 Derived from a Pd(II) Precursor
3.4. NMR Monitoring on the Formation of 2
3.5. Reaction of 2 with Pressurized H2 and Subsequent Dehydrogenation
3.6. X-ray Crystal Structure Determination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lundgren, R.J.; Stradiotto, M. Palladium-Catalyzed Cross-Coupling of Aryl Chlorides and Tosylates with Hydrazine. Angew. Chem. Int. Ed. 2010, 49, 8686–8690. [Google Scholar] [CrossRef]
- Kinjo, R.; Donnadieu, B.; Bertrand, G. Gold-Catalyzed Hydroamination of Alkynes and Allenes with Parent Hydrazine. Angew. Chem. Int. Ed. 2011, 50, 5560–5563. [Google Scholar] [CrossRef] [PubMed]
- Umehara, K.; Kuwata, S.; Ikariya, T. N–N Bond Cleavage of Hydrazines with a Multi-Proton Responsive Pincer-Type Iron Complex. J. Am. Chem. Soc. 2010, 135, 6754–6757. [Google Scholar] [CrossRef] [PubMed]
- Kiernicki, J.J.; Zeller, M.; Szymczak, N.K. Hydrazine Capture and N–N Bond Cleavage at Iron Enabled by Flexible Appended Lewis Acids. J. Am. Chem. Soc. 2017, 139, 18194–18197. [Google Scholar] [CrossRef]
- Heaton, B.T.; Jacob, C.; Page, P. Transition metal complexes containing hydrazine and substituted hydrazines. Coord. Chem. Rev. 1996, 154, 193–229. [Google Scholar] [CrossRef]
- Mellor, J.M.; Smith, N.M. Reductive cleavage of the nitrogen-nitrogen bond in hydrazine derivatives. J. Chem. Soc. Perkin Trans. 1984, 1, 2927–2931. [Google Scholar] [CrossRef]
- Ding, H.; Friestad, G.K. Trifluoroacetyl-Activated Nitrogen–Nitrogen Bond Cleavage of Hydrazines by Samarium(II) Iodide. Org. Lett. 2004, 6, 637–640. [Google Scholar] [CrossRef]
- Dey, S.; Gadakh, S.K.; Ahuja, B.B.; Kamble, S.P.; Sudalai, A. Pd-catalyzed reductive cleavage of N–N bond in dibenzyl-1-alkylhydrazine-1,2-dicarboxylates with PMHS: Application to a formal enantioselective synthesis of (R)-sitagliptin. Tetrahedron Lett. 2016, 57, 684–687. [Google Scholar] [CrossRef]
- Hoover, J.M.; Freudenthal, J.; Michael, F.E.; Mayer, J.M. Reactivity of Low-Valent Iridium, Rhodium, and Platinum Complexes with Di- and Tetrasubstituted Hydrazines. Organometallics 2008, 27, 2238–2245. [Google Scholar] [CrossRef]
- Zhao, B.; Du, H.; Cui, S.; Shi, Y. Synthetic and Mechanistic Studies on Pd(0)-Catalyzed Diamination of Conjugated Dienes. J. Am. Chem. Soc. 2010, 132, 3523–3532. [Google Scholar] [CrossRef] [Green Version]
- Ligandro, E.; Perdicchia, D. N-Acylhydrazines: Future Perspectives Offered by New Syntheses and Chemistry. Eur. J. Org. Chem. 2004, 2004, 665–675. [Google Scholar] [CrossRef]
- Hoover, J.M.; DiPasquale, A.; Mayer, J.M.; Michael, F.E. Platinum-Catalyzed Intramolecular Hydrohydrazination: Evidence for Alkene Insertion into a Pt–N Bond. J. Am. Chem. Soc. 2010, 132, 5043–5053. [Google Scholar] [CrossRef] [PubMed]
- Dilworth, J.R.; Kasenally, A.S. Dibenzoyl- and diacetylhydrazido(2–) complexes of platinum. J. Organomet. Chem. 1973, 60, 203–207. [Google Scholar] [CrossRef]
- Ittel, S.D.; Ibers, J.A. The Structure of Bis(triphenylphosphine)(dibenzoylhydrazido)platinum, Pt[P(C6H5)3]2[C6H5CONNCOC6H5]·C2H5OH. Inorg. Chem. 1973, 12, 2290–2295. [Google Scholar] [CrossRef]
- Chan, D.; Cronin, L.; Duckett, S.B.; Hupfield, P.; Perutz, R.N. Synthesis, structure and reactivity of N,O-metallacyclic (dicarbonyldiazene) platinum complexes. New. J. Chem. 1998, 22, 511–516. [Google Scholar] [CrossRef]
- Kasenally, A.S.; Hussein, F.M. Hydrazido complexes of the transition metals. Synthesis and reactions of dibenzoyl- and diacetylhydrazido(2–) (N,N′,O,O′)-bis(carbonyltriphenylphosphine)-rhodium(I) and –iridium(I). J. Organomet. Chem. 1976, 111, 355–359. [Google Scholar] [CrossRef]
- Zambrano, C.H.; Sharp, P.R.; Barnes, C.L. Synthesis and X-ray crystal structure of an iridium N,O-chelating hydrazido complex. Polyhedron 1996, 15, 3653–3657. [Google Scholar] [CrossRef]
- Fujisawa, K.; Sugiyama, M.; Miyashita, Y.; Okamoto, K.-i. Structure and chemical properties of a copper(II) hydrazido complex: [{Cu(HB(3,5-iPr2pz)3)}2(μ-NCOOEt)2]. Inorg. Chem. Commun. 2009, 12, 246–248. [Google Scholar] [CrossRef]
- Mondal, S.; Filippou, V.; Bubrin, M.; Schwederski, B.; Fiedler, J.; Kaim, W. Metallo with 1,2-Dipivaloylhydrazido Ligands: Electron Transfer and Alkylation/Protonation Effects. Eur. J. Inorg. Chem. 2019, 2019, 2639–2647. [Google Scholar] [CrossRef]
- Jana, R.; Sarkar, B.; Bubrin, D.; Fiedler, J.; Kaim, W. Structure electrochemistry and spectroscopy of a new diacylhydrazido-bridged diruthenium complex with a strongly near-infrared absorbing RuIIIRuII intermediate. Inorg. Chem. Commun. 2010, 13, 1160–1162. [Google Scholar] [CrossRef]
- Congrave, D.G.; Hsu, Y.-T.; Batsanov, A.S.; Beeby, A.; Bryce, M.R. Sky-blue emitting bridged diiridium complexes: Beneficial effects of intramolecular π–π stacking. Dalton Trans. 2018, 47, 2086–2098. [Google Scholar] [CrossRef] [Green Version]
- Congrave, D.G.; Batsanov, A.S.; Bryce, M.R. Highly luminescent 2-phenylpyridine-free diiridium complexes with bulky 1,2-diarylimidazole cyclometalating ligands. Dalton Trans. 2018, 47, 16524–16533. [Google Scholar] [CrossRef] [Green Version]
- Nagashima, K.; Kihara, N.; Iino, Y. Oxidative Coupling Polymerization of Bishydrazide for the Synthesis of Poly(diacylhydrazine): Oxidative Preparation of Oxidatively Degradable Polymer. J. Polym. Sci. Part A: Polym. Chem. 2012, 50, 4230–4238. [Google Scholar] [CrossRef]
- De Oliveira, C.S.; Lira, B.F.; Barbosa-Filho, J.M.; Lorenzo, J.G.F.; De Athayde-Filho, P.F. Synthetic Approaches and Pharmacological Activity of 1,3,4-Oxadiazoles: A Review of the Literature from 2000–2012. Molecules 2012, 17, 10192–10231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrero, M.A.; Wannberg, J.; Larhed, M. Direct Microwave Synthesis of N,N′-Diacylhydrazines and Boc-protected Hydrazines by in situ Carbonylations under Air. Synlett 2004, 2004, 2335–2338. [Google Scholar]
- Andersen, T.L.; Caneschi, W.; Ayoub, A.; Lindhardt, A.T.; Couri, M.R.C.; Skrydstrup, T. 1,2,4- and 1,3,4-Oxadiazole Synthesis by Palladium-Catalyzed Carbonylative Assembly of Aryl Bromides with Amidoximes or Hydrazides. Adv. Synth. Catal. 2014, 356, 3074–3082. [Google Scholar] [CrossRef]
- Mloston, G.; Obijalska, E.; Zurawik, A.; Heimgartner, H. Efficient synthesis of tri- and difluoroacetyl hydrazides as useful building blocks for non-symmetrically substituted, fluoroalkylated 1,3,4-oxadiazoles. Chem. Heterocycl. Compd. 2016, 52, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Tšupova, S.; Mäeorg, U. Hydrazines and azo-compounds in the synthesis of heterocycles comprising N-N bond. Heterocycles 2014, 88, 129–173. [Google Scholar]
- Bredikhin, A.; Tšubrik, O.; Sillard, R.; Mäeorg, U. Increasing the N–H Acidity: Introduction of Highly Electronegative Groups into the Hydrazine Molecule. Synlett 2005, 2005, 1939–1941. [Google Scholar]
- Takahashi, Y.; Ito, T.; Sakai, S.; Ishii, Y.; Bonnet, J.J.; Ibers, J.A. A novel palladium(0) complex; bis(dibenzylideneacetone)palladium(0). J. Chem. Soc. D Chem. Commun. 1970, 17, 1065–1066. [Google Scholar] [CrossRef]
- Unlike BzOOBz: Bird, C.; Booth, B.L.; Haszeldine, R.N.; Neuss, G.R.H.; Smith, M.A.; Flood, A. Reactions Involving Transition Metals. Part 18. Reactions of Transition-metal Complexes with Peroxycarboxylic Acids, Diacyl Peroxides, and t-Butyl Peroxybenzoate. J. Chem. Soc. Dalton Trans. 1982, 6, 1109–1114. [Google Scholar] [CrossRef]
- He, J.; Shigenari, T.; Yu, J.-Q. Palladium(0)/PAr3-Catalyzed Intermolecular Amination of C(sp3)–H Bonds: Synthesis of β-Amino Acids. Angew. Chem. Int. Ed. 2015, 54, 6545–6549. [Google Scholar] [CrossRef]
- Azizian, H.; Dixon, K.R.; Eaborn, C.; Pidcock, A.; Shuaib, N.M.; Vinaixa, J. Dynamic Stereochemistry of cis-[PtH(SiR3)(PPh3)2]. Spontaneous Ligand Interchange with Retention of Nuclear Spin-Spin Correlation. J. Chem. Soc. Chem. Commun. 1982, 17, 1020–1022. [Google Scholar] [CrossRef]
- Wouters, J.M.A.; Klein, R.A.; Elsevier, C.J.; Häming, L.; Stam, C.H. Synthesis of trans-(σ-Allenyl)platinum(II) and -palladium(II) Compounds. X-ray Crystal Structure of trans-[PtBr{C(H)=C=Me2}(PPh3)2] and Highly Diastereoselective trans-cis Isomerization of (σ-Allenyl)palladium(II) Bromides. Organometallics 1994, 13, 4586–4593. [Google Scholar] [CrossRef] [Green Version]
- Obora, Y.; Tsuji, Y.; Nishiyama, K.; Ebihara, M.; Kawamura, T. Structure and Fluxional Behavior of cis-Bis(stannyl)bis(phosphine)platinum: Oxidative Addition of Organodistannane to Platinum(0) Complex. J. Am. Chem. Soc. 1996, 118, 10922–10923. [Google Scholar] [CrossRef]
- Tsuji, Y.; Nishiyama, K.; Hori, S.-i.; Ebihara, M.; Kawamura, T. Structure and Facile Unimolecular Twist Rotation of cis-Bis(silyl)bis(phosphine)platinum and cis-Bis(stannyl)bis(phosphine)palladium Complexes. Organometallics 1998, 17, 507–512. [Google Scholar] [CrossRef]
- Mori, Y.; Shirase, H.; Fukuda, Y. Substituent and Solvent Effects on Square-Planar–Tetrahedral Equilibria of Bis[4-(arylimino)pentan-2-onato]nickel(II) and Bis[1-aryl-3-(phenylimino)butan-1-onato]nickel(II) Complexes. Bull. Chem. Soc. Jpn. 2008, 81, 1108–1115. [Google Scholar] [CrossRef]
- Maganas, D.; Grigoropoulos, A.; Staniland, S.S.; Chatziefthimiou, S.D.; Harrison, A.; Robertson, N.; Kyritsis, P.; Neese, F. Tetrahedral and Aquare Planar Ni[(SPR2)2N]2 Complexes, R = Ph & iPr Revisited: Experimetal and Theoretical Analysis of Interconversion Pathways, Structural Preferences, and Spin Delocalization. Inorg. Chem. 2010, 49, 5079–5093. [Google Scholar]
- Sherbo, R.S.; Bindra, G.S.; Budzelaar, P.H.M. Square-Planar–Tetrahedral Interconversion without Spin Flip in (β-diiminate)Rh(1,3-diene) Complexes. Organometallics 2016, 35, 2039–2048. [Google Scholar] [CrossRef]
- Zhou, X.; Lau, K.-C.; Petro, B.J.; Jordan, R.F. cis/trans Isomerization of o-Phosphino-Arenesulfonate Palladium Methyl Complexes. Organometallics 2014, 33, 7209–7214. [Google Scholar] [CrossRef]
- CrystalStructure 4.1: Crystal Structure Analysis Package; Rigaku Coorporation: Tokyo, Japan, 2015.
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M.; Polidori, G.; Camalli, M. SIR92—A program for automatic solution of crystal structures by direct methods. J. Appl. Cryst. 1994, 27, 435. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pd–O1 | 2.046 (4) | Pd–P1 | 2.2522 (13) |
| Pd–N2 | 2.063 (4) | Pd–P2 | 2.2692 (14) |
| N1–N2 | 1.425 (6) | ||
| N1–C1 | 1.281 (7) | N2–C2 | 1.327 (6) |
| C1–O1 | 1.297 (6) | C2–O2 | 1.228 (6) |
| O1–Pd–N2 | 78.15 (15) | Pd–N2–N1 | 113.5 (3) |
| N2–Pd–P1 | 100.63 (12) | N2–N1–C1 | 109.5 (4) |
| P1–Pd–P2 | 93.60 (5) | N1–C1–O1 | 128.8 (5) |
| P2–Pd–O1 | 87.78 (11) | C1–O1–Pd | 109.5 (3) |
| Pd–O1 | 1.997 (3) | Pd–N3 | 2.059 (4) |
| Pd–N2 | 2.034 (3) | Pd–N4 | 2.058 (3) |
| N1–N2 | 1.418 (5) | ||
| N1–C1 | 1.289 (5) | N2–C2 | 1.322 (5) |
| C1–O1 | 1.295 (5) | C2–O2 | 1.227 (5) |
| O1–Pd–N2 | 80.20 (12) | Pd–N2–N1 | 112.3 (2) |
| N2–Pd–N3 | 104.62 (13) | N2–N1–C1 | 109.8 (3) |
| N3–Pd–N4 | 85.16 (13) | N1–C1–O1 | 128.6 (4) |
| N4–Pd–O1 | 90.11 (12) | C1–O1–Pd | 109.0 (2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kayaki, Y.; Hayakawa, T.; Ikariya, T. Synthesis of N,O-Chelating Hydrazidopalladium Complexes from 1,2-Bis(trifluoroacetyl)hydrazine. Inorganics 2021, 9, 76. https://doi.org/10.3390/inorganics9100076
Kayaki Y, Hayakawa T, Ikariya T. Synthesis of N,O-Chelating Hydrazidopalladium Complexes from 1,2-Bis(trifluoroacetyl)hydrazine. Inorganics. 2021; 9(10):76. https://doi.org/10.3390/inorganics9100076
Chicago/Turabian StyleKayaki, Yoshihito, Tomohiro Hayakawa, and Takao Ikariya. 2021. "Synthesis of N,O-Chelating Hydrazidopalladium Complexes from 1,2-Bis(trifluoroacetyl)hydrazine" Inorganics 9, no. 10: 76. https://doi.org/10.3390/inorganics9100076
APA StyleKayaki, Y., Hayakawa, T., & Ikariya, T. (2021). Synthesis of N,O-Chelating Hydrazidopalladium Complexes from 1,2-Bis(trifluoroacetyl)hydrazine. Inorganics, 9(10), 76. https://doi.org/10.3390/inorganics9100076