
Density Functional Theory Study on the Adsorption Mechanism of Sulphide Gas Molecules on α-Fe2O3(001) Surface


Abstract
:1. Introduction
2. Computational Methods and Models
2.1. Theoretical Calculation Methods
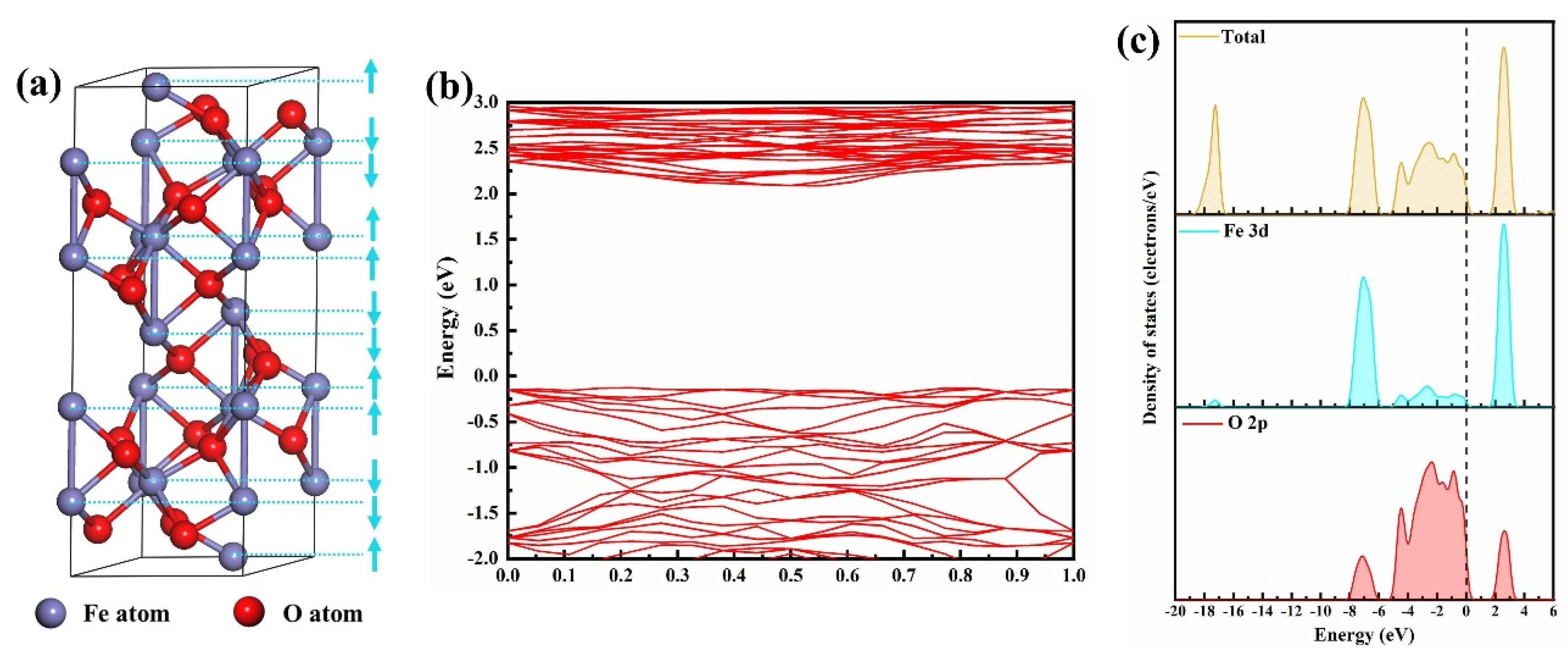
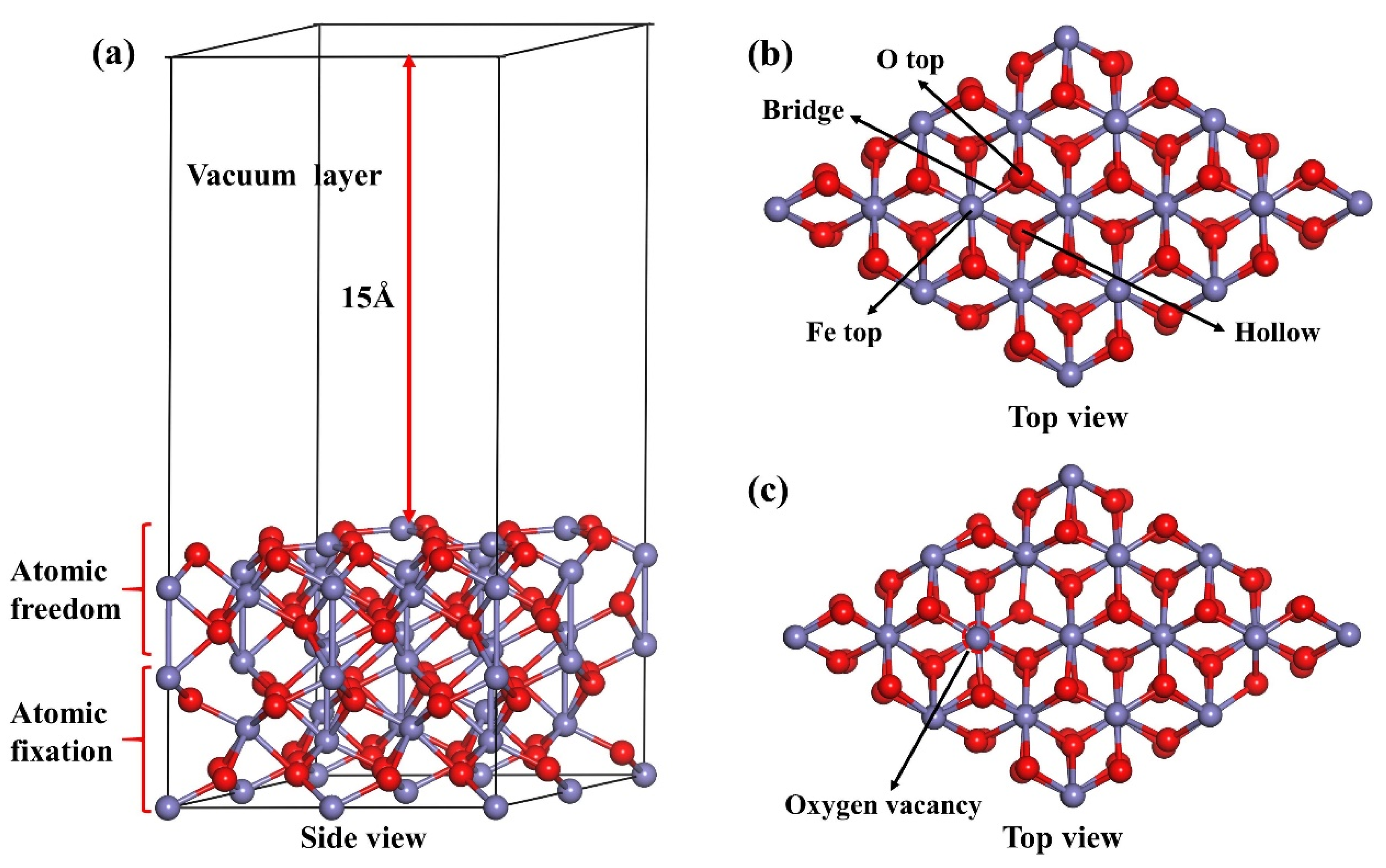
2.2. Models
3. Results and Discussion
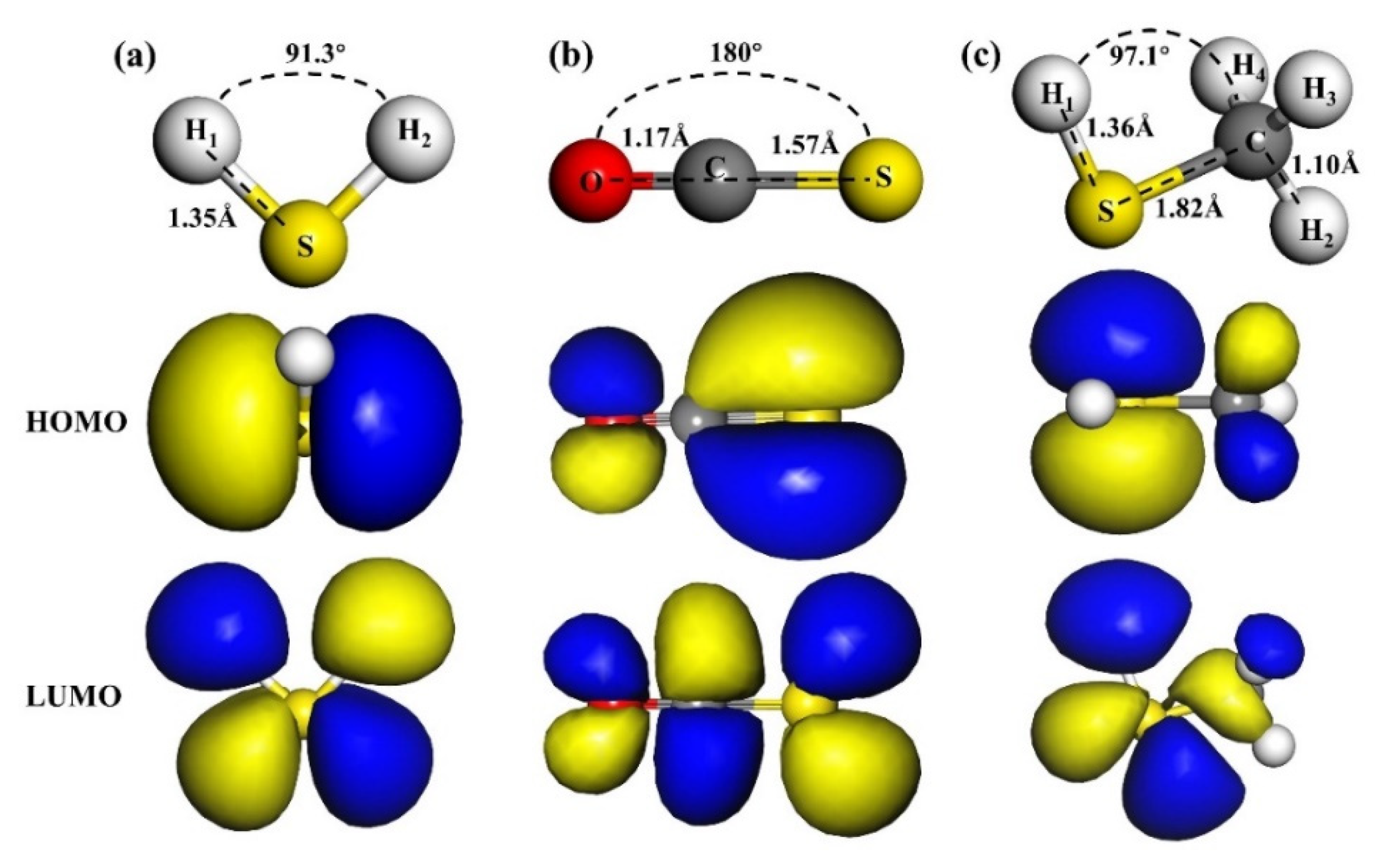
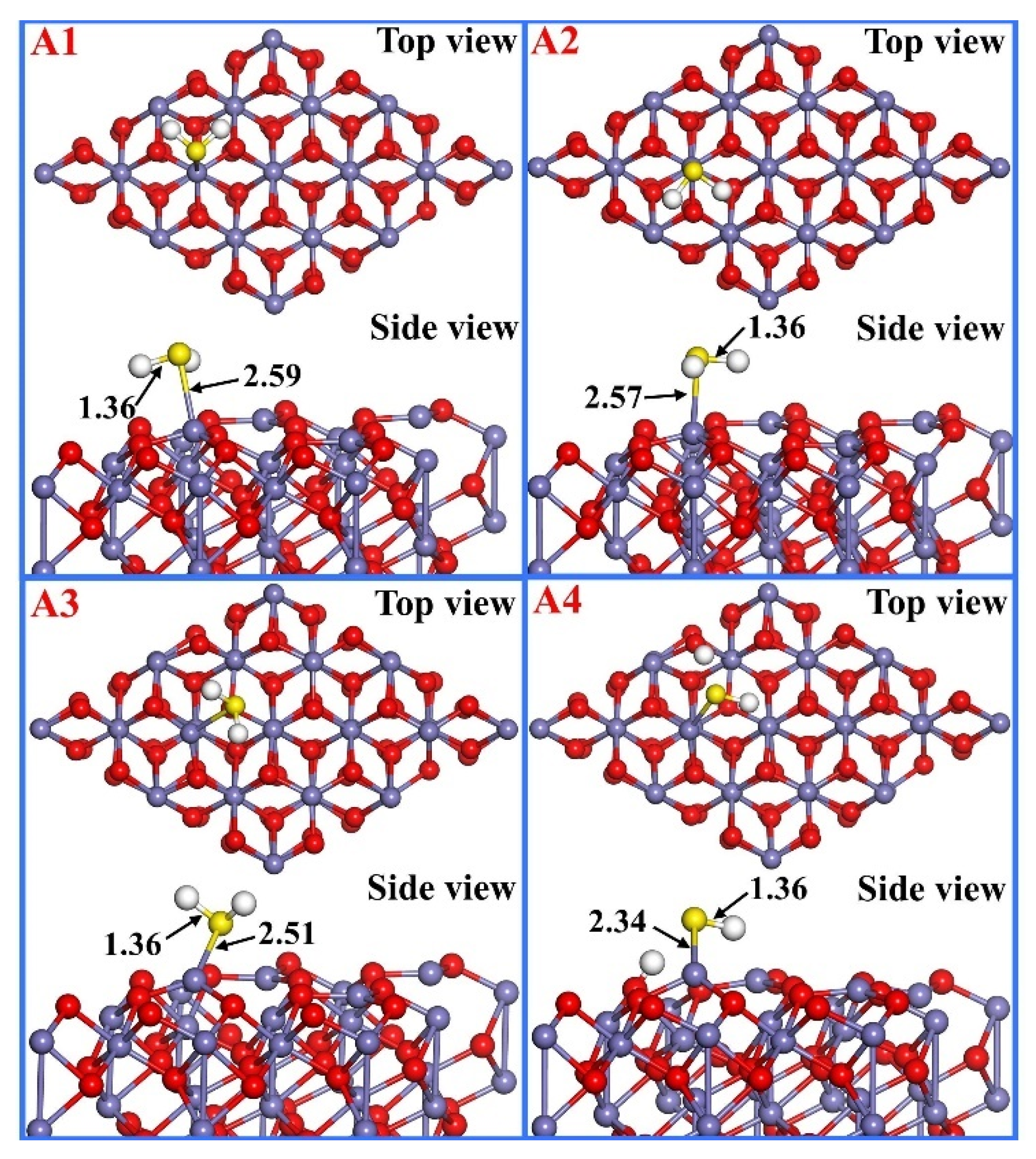
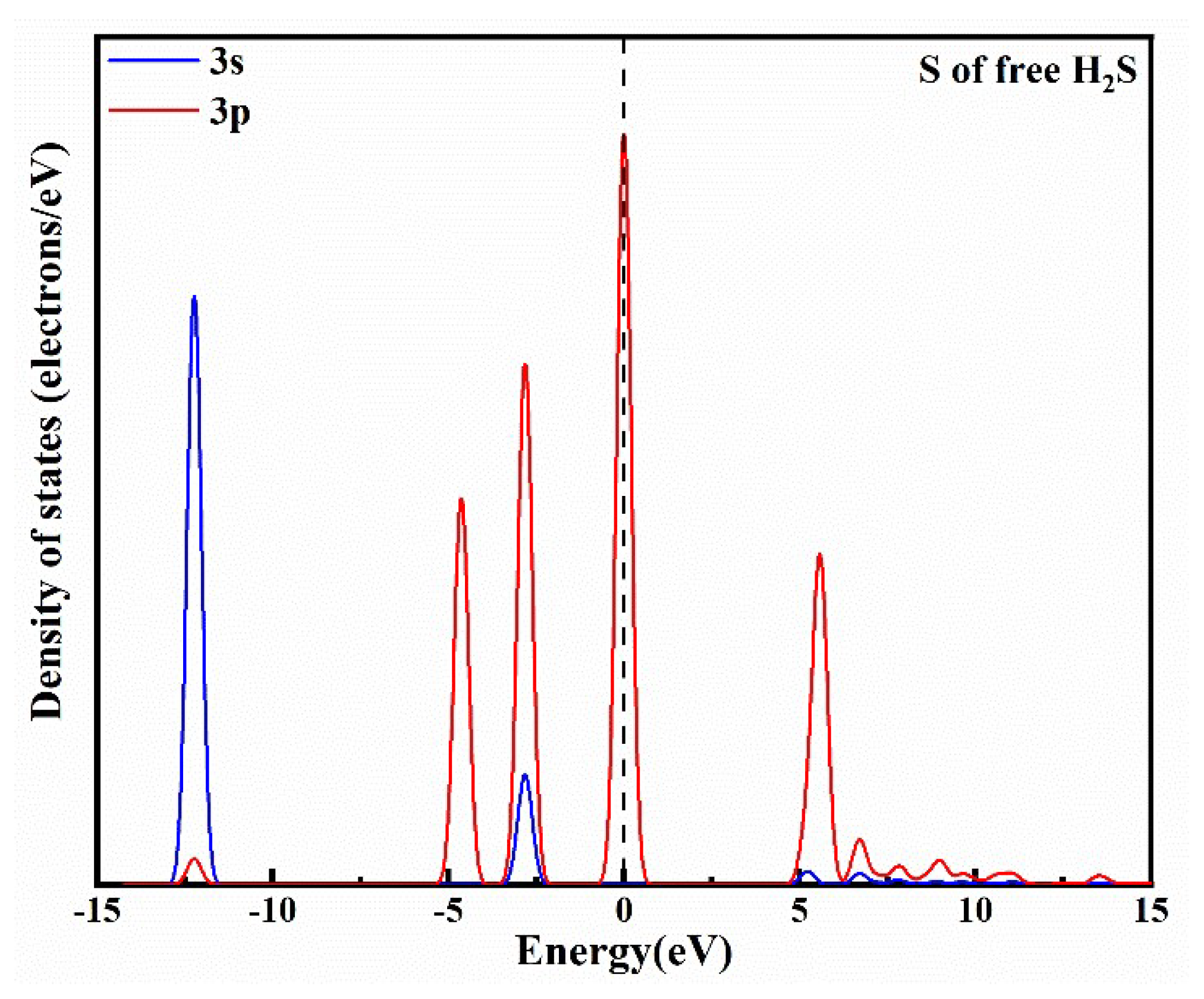
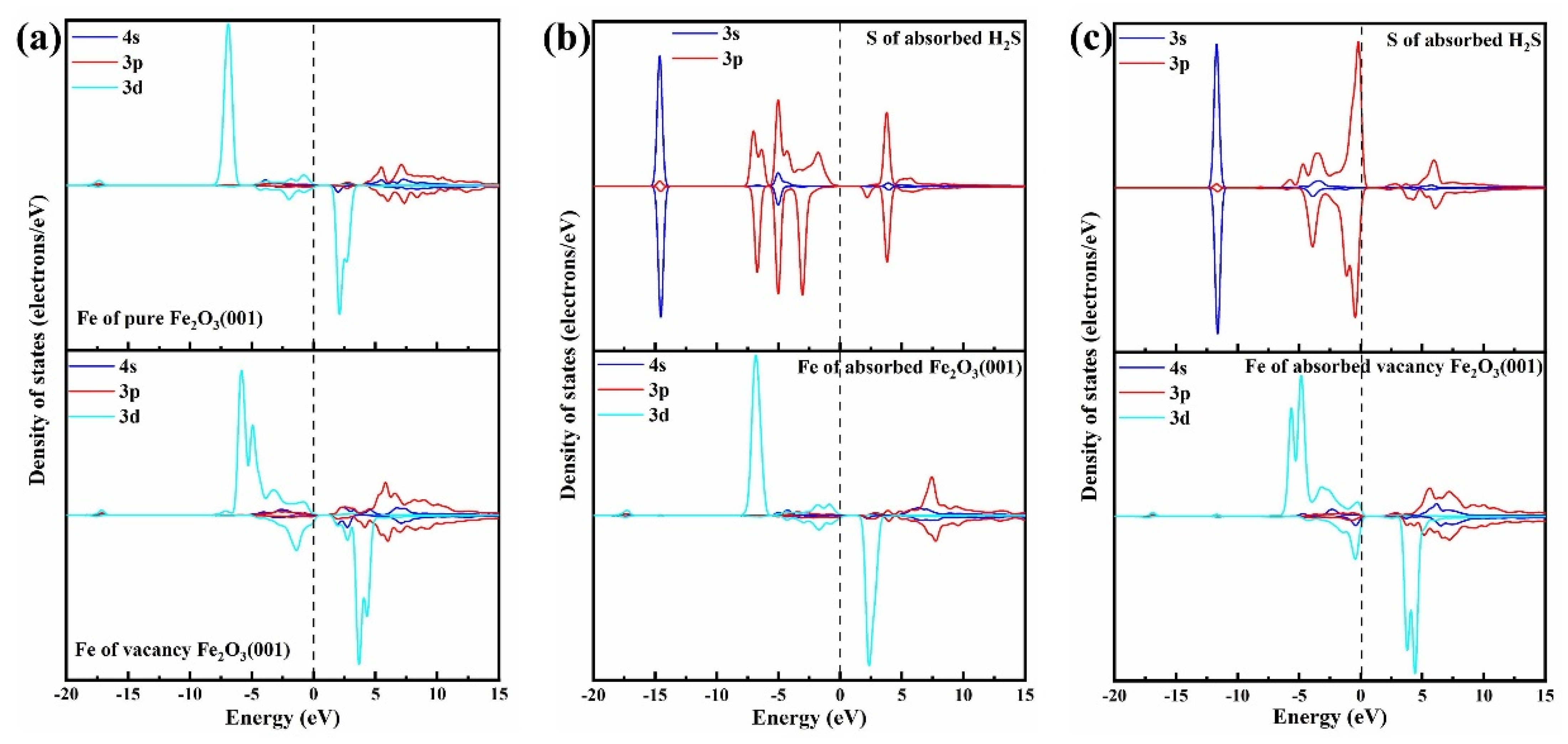
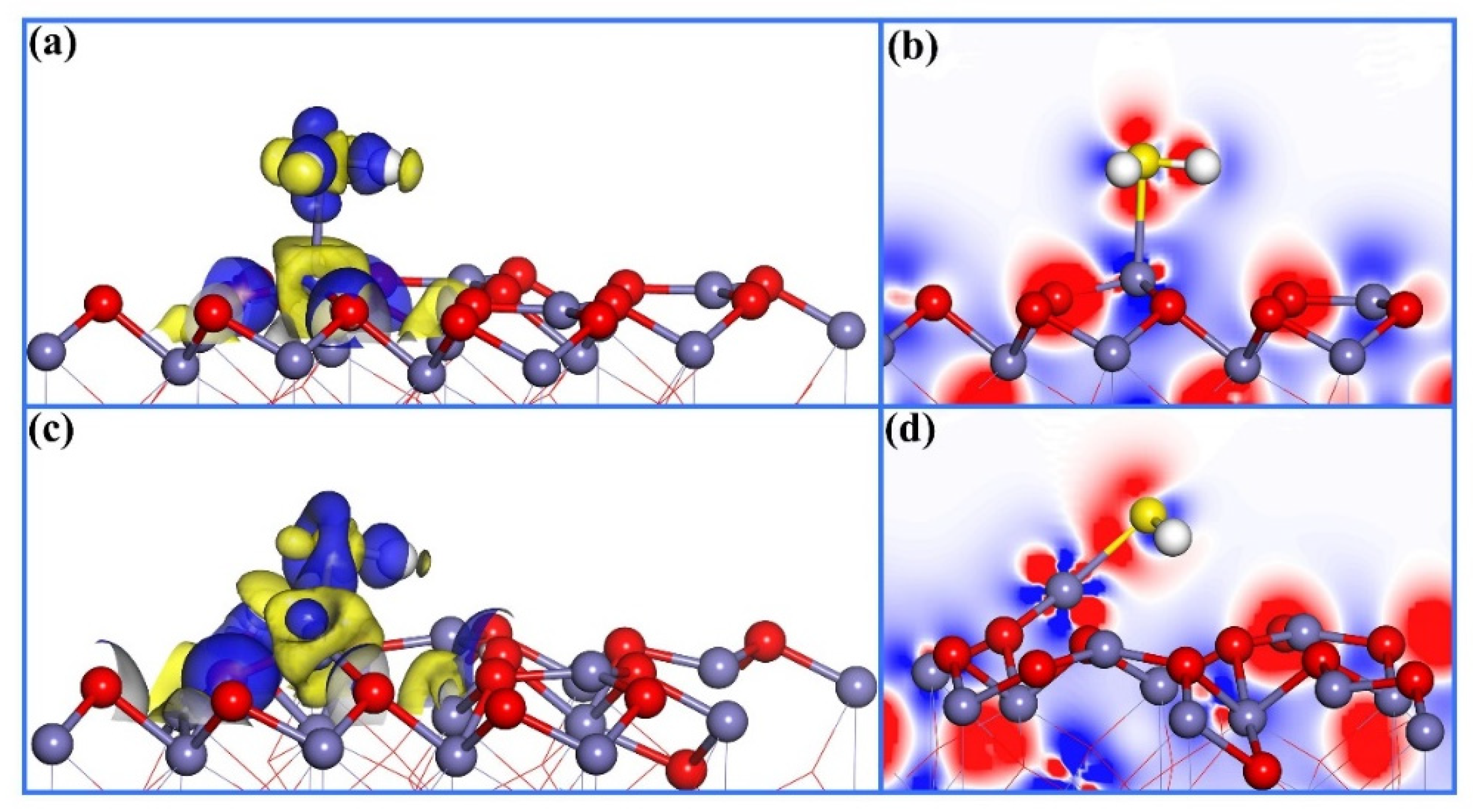
3.1. H2S Adsorption
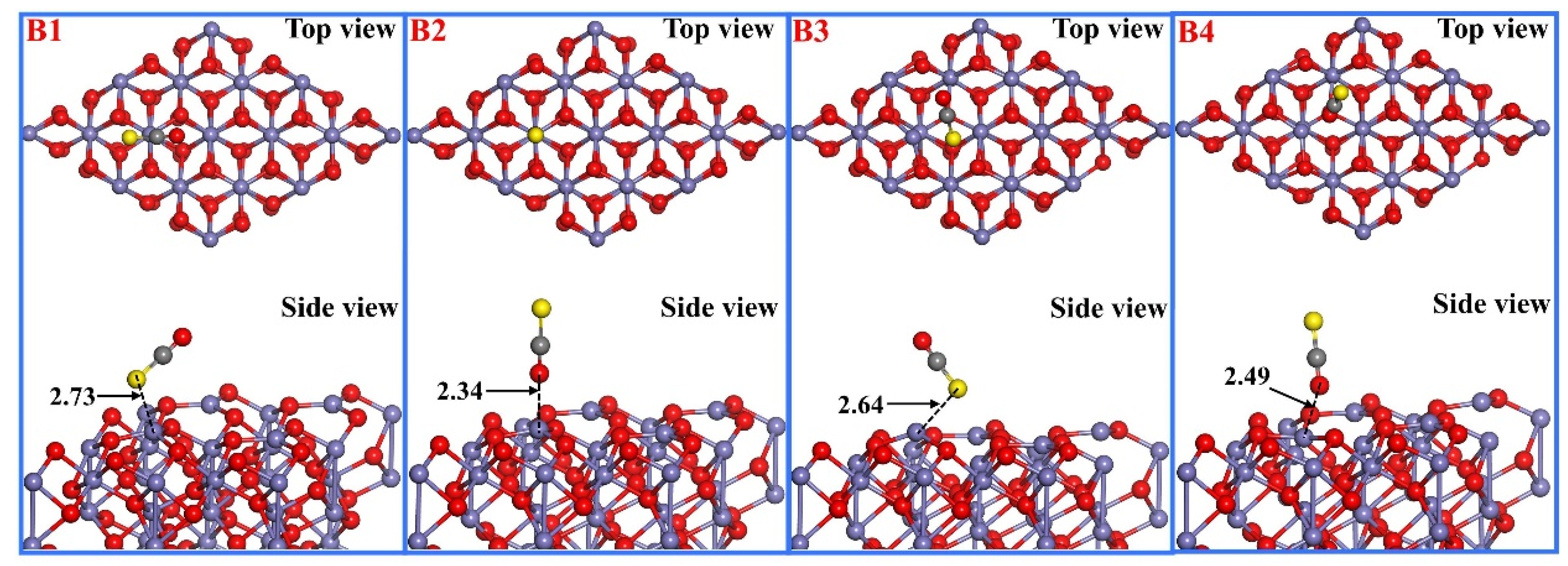
3.2. COS Adsorption
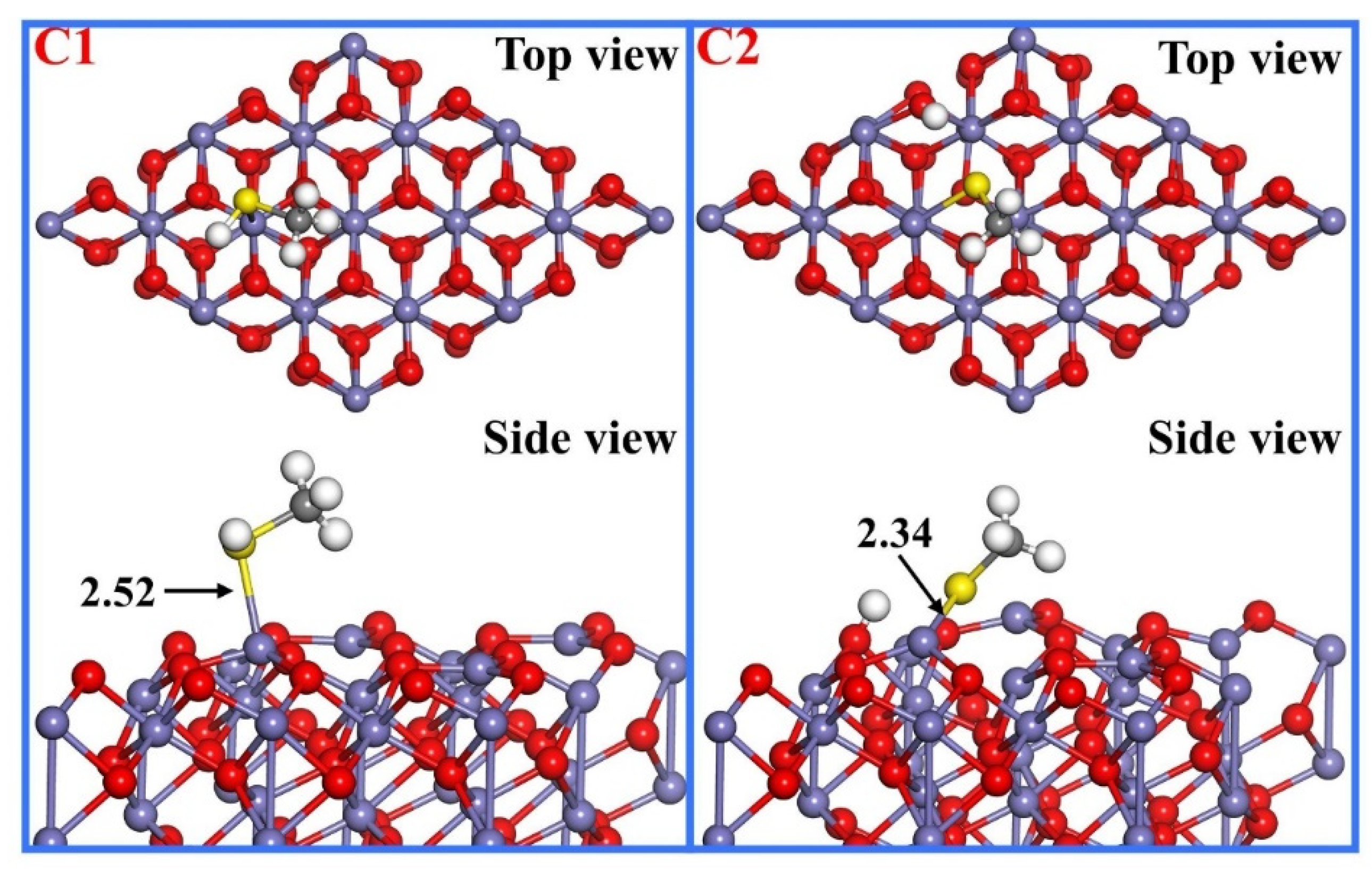
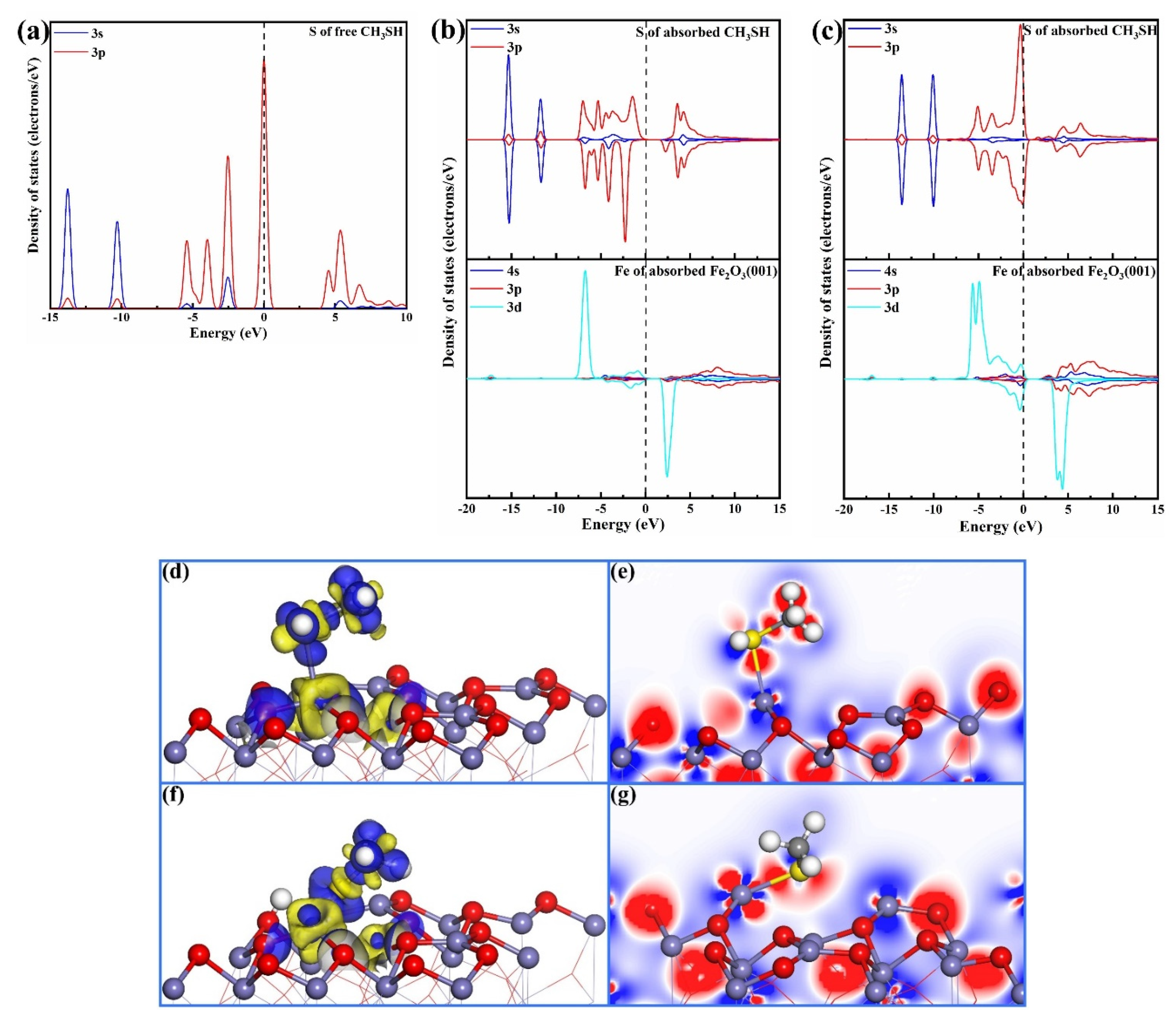
3.3. CH3SH Adsorption
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, J.; Song, Z.; Cheng, H.; Chen, L.; Deng, L.; Qi, Z. Multilevel screening of ionic liquid absorbents for simultaneous removal of CO2 and H2S from natural gas. Sep. Purif. Technol. 2020, 248, 117053. [Google Scholar] [CrossRef]
- Faramawy, S.; Zaki, T.; Sakr, A.-E. Natural gas origin, composition, and processing: A review. J. Nat. Gas Sci. Eng. 2016, 34, 34–54. [Google Scholar] [CrossRef]
- Watanabe, S. Chemistry of H2S over the surface of Common solid sorbents in industrial natural gas desulfurization. Catal. Today 2021, 371, 204–220. [Google Scholar] [CrossRef]
- Zeng, Z.; Dlugogorski, B.Z.; Oluwoye, I.; Altarawneh, M. Combustion chemistry of COS and occurrence of intersystem cross-ing. Fuel 2021, 283, 119257. [Google Scholar] [CrossRef]
- Alzueta, M.U.; Pernía, R.; Abián, M.; Millera, Á.; Bilbao, R. CH3SH conversion in a tubular flow reactor. Experiments and ki-netic modelling. Combust. Flame 2019, 203, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Kahyarian, A.; Nesic, S. H2S corrosion of mild steel: A quantitative analysis of the mechanism of the cathodic reaction. Electrochim. Acta 2019, 297, 676–684. [Google Scholar] [CrossRef]
- Latosov, E.; Loorits, M.; Maaten, B.; Volkova, A.; Soosaar, S. Corrosive effects of H2S and NH3 on natural gas piping systems manufactured of carbon steel. Energy Procedia 2017, 128, 316–323. [Google Scholar] [CrossRef]
- Yi, H.; Tao, T.; Zhao, S.; Yu, Q.; Gao, F.; Zhou, Y.; Tang, X. Promoted adsorption of methyl mercaptan by γ-Al2O3 catalyst loaded with Cu/Mn. Environ. Technol. Innov. 2021, 21, 101349. [Google Scholar] [CrossRef]
- Fu, Y.; Chen, J.; Zhang, H. Synthesis of Fe2O3 nanowires by oxidation of iron. Chem. Phys. Lett. 2001, 350, 491–494. [Google Scholar] [CrossRef]
- Shi, C.; Chen, Y.; Liu, H.; Cui, G.; Ju, L.; Chen, L. Adsorption and gas-sensing characteristics of a stoichiometric α-Fe2O3 (001) nano thin film for carbon dioxide and carbon monoxide with and without pre-adsorbed O2. RSC Adv. 2016, 6, 3514–3525. [Google Scholar] [CrossRef]
- Saritas, S.; Kundakci, M.; Coban, O.; Tuzemen, S.; Yildirim, M. Ni:Fe2O3, Mg:Fe2O3 and Fe2O3 thin films gas sensor application. Phys. B Condens. Matter 2018, 541, 14–18. [Google Scholar] [CrossRef]
- Hitam, C.; Jalil, A.; Izan, S.; Azami, M.; Hassim, M.; Chanlek, N. The unforeseen relationship of Fe2O3 and ZnO on fibrous silica KCC-1 catalyst for fabricated Z-scheme extractive-photooxidative desulphurization. Powder Technol. 2020, 375, 397–408. [Google Scholar] [CrossRef]
- Song, Z.; Wang, B.; Yu, J.; Ma, C.; Zhou, C.; Chen, T.; Yan, Q.; Wang, K.; Sun, L. Density functional study on the heterogeneous oxidation of NO over α-Fe2O3 catalyst by H2O2: Effect of oxygen vacancy. Appl. Surf. Sci. 2017, 413, 292–301. [Google Scholar] [CrossRef]
- Ling, L.; Song, J.; Zhao, S.; Zhang, R.; Wang, B. DFT study on the effects of defect and metal-doping on the decomposition of H2S on the α-Fe2O3(0001) surface. RSC Adv. 2014, 4, 22411–22418. [Google Scholar] [CrossRef]
- Li, F.; Shi, C.; Wang, X.; Cui, G.; Wang, D.; Chen, L. The important role of oxygen defect for NO gas-sensing behavior of α-Fe2O3 (001) surface: Predicted by density functional theory. Comput. Mater. Sci. 2018, 146, 1–8. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, C.; Zhou, X.; Chen, J.; Chen, L.; Li, F. DFT study on the interaction of H2O and O2 with α-Fe2O3 (001) surface. Vacuum 2021, 188, 110164. [Google Scholar] [CrossRef]
- Hao, M.; Zeng, W.; Li, Y.Q. Adsorption mechanism of H2S and CH3SH on Fe(110) surface: A density functional theory study. Physica E 2022, 135, 114938. [Google Scholar] [CrossRef]
- Li, B.; Zhou, Q.; Peng, R.; Liao, Y.; Zeng, W. Adsorption of SF6 decomposition gases (H2S, SO2, SOF2 and SO2F2) on Sc-doped MoS2 surface: A DFT study. Appl. Surf. Sci. 2021, 549, 149271. [Google Scholar] [CrossRef]
- Shang, J.; Li, C.; Tang, X.; Du, A.; Liao, T.; Gu, Y.; Ma, Y.; Kou, L.; Chen, C. Multiferroic decorated Fe2O3 monolayer predicted from first principles. Nanoscale 2020, 12, 14847–14852. [Google Scholar] [CrossRef] [PubMed]
- Ta, H.T.T.; Tieu, A.; Zhu, H.; Yu, H.; Tran, N.V. A First-Principles Study of Impurity-Enhanced Adhesion and Lubricity of Gra-phene on Iron Oxide Surface. J. Phys. Chem. C 2021, 125, 4310–4321. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First Principles Methods Using CASTEP. Z. Krist. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Meunier, M. Introduction to materials studio. In EPJ Web of Conferences; EDP Sciences: Les Ulis, France, 2012; Volume 10, p. 04001. [Google Scholar]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993. [Google Scholar] [CrossRef]
- Ziesche, P.; Kurth, S.; Perdew, J.P. Density functionals from LDA to GGA. Comput. Mater. Sci. 1998, 11, 122–127. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865, Erratum in 1997, 78, 1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head, J.D.; Zerner, M.C. A Broyden—Fletcher—Goldfarb—Shanno optimization procedure for molecular geometries. Chem. Phys. Lett. 1985, 122, 264–270. [Google Scholar] [CrossRef]
- Froyen, S. Brillouin-zone integration by Fourier quadrature: Special points for superlattice and supercell calculations. Phys. Rev. B 1989, 39, 3168–3172. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, A.; Hafner, J.; Kresse, G. Molecular adsorption on the surface of strongly correlated transition-metal oxides: A case study for CO/NiO(100). Phys. Rev. B 2004, 69, 075413. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.; Velev, J.; Butler, W.; Sarker, S.K.; Bengone, O. Effect of electron correlations on the electronic and magnet-ic structure of Ti-doped α-hematite. Phys. Rev. B 2004, 69, 174429. [Google Scholar] [CrossRef]
- Sandratskii, L.M.; Uhl, M.; Kübler, J. Band theory for electronic and magnetic properties of. J. Phys. Condens. Matter 1996, 8, 983–989. [Google Scholar] [CrossRef]
- Wong, K.; Zeng, Q.H.; Yu, A.B. Electronic Structure of Metal (M = Au, Pt, Pd, or Ru) Bilayer Modified α-Fe2O3(0001) Surfaces. J. Phys. Chem. C 2011, 115, 4656–4663. [Google Scholar] [CrossRef]
- Rohrbach, A.; Hafner, J.; Kresse, G. Ab initio study of the (0001) surfaces of hematite and chromia: Influence of strong electronic correlations. Phys. Rev. B 2004, 70, 125426. [Google Scholar] [CrossRef]
- Glasscock, J.; Barnes, P.; Plumb, I.; Bendavid, A.; Martin, P. Structural, optical and electrical properties of undoped polycrystal-line hematite thin films produced using filtered arc deposition. Thin Solid Films 2008, 516, 1716–1724. [Google Scholar] [CrossRef]
- Merchant, P.; Collins, R.; Kershaw, R.; Dwight, K.; Wold, A. The electrical, optical and photoconducting properties of Fe2−xCrxO3 (0 ≤ x ≤ 0.47). J. Solid State Chem. 1979, 27, 307–315. [Google Scholar] [CrossRef]
- Zhang, Y.; Zeng, W.; Li, Y. NO2 and H2 sensing properties for urchin-like hexagonal WO3 based on experimental and first-principle investigations. Ceram. Int. 2019, 45, 6043–6050. [Google Scholar] [CrossRef]
- Zhang, Y.; Zeng, W.; Li, Y. Computational study of surface orientation effect of rutile TiO2 on H2S and CO sensing mechanism. Appl. Surf. Sci. 2019, 495, 143619. [Google Scholar] [CrossRef]
- Guerra, C.F.; Handgraaf, J.W.; Baerends, E.J.; Bickelhaupt, F.M. Voronoi deformation density (VDD) charges: Assessment of the Mulliken, Bader, Hirshfeld, Weinhold, and VDD methods for charge analysis. J. Comput. Chem. 2004, 25, 189–210. [Google Scholar] [CrossRef]
- Finger, L.W.; Hazen, R.M. Crystal structure and isothermal compression of Fe2O3, Cr2O3, and V2O3 to 50 kbars. J. Appl. Phys. 1980, 51, 5362. [Google Scholar] [CrossRef]
- Wang, X.-G.; Weiss, W.; Shaikhutdinov, S.K.; Ritter, M.; Petersen, M.; Wagner, F.; Schlögl, R.; Scheffler, M. The hematite (α-Fe2O3) (0001) surface: Evidence for domains of distinct chemistry. Phys. Rev. Lett. 1998, 81, 1038. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liu, J. Density Functional Theory Study of Arsenic Adsorption on the Fe2O3 (001) Surface. Energy Fuels 2019, 33, 1414–1421. [Google Scholar] [CrossRef]
- Alvarez-Ramırez, F.; Martınez-Magadán, J.; Gomes, J.; Illas, F. On the geometric structure of the (0001) hematite surface. Surf. Sci. 2004, 558, 4–14. [Google Scholar] [CrossRef]
- Perepichka, D.F.; Bryce, M.R. Molecules with Exceptionally Small HOMO-LUMO Gaps. Angew. Chem. Int. Ed. 2005, 44, 5370–5373. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.C.; Zeng, W.; Gu, L.; Saito, M.; Tsukimoto, S.; Ikuhara, Y. Atomic scale structure and electronic property of the LaA-lO3/TiO2 interface. J. Appl. Phys. 2010, 108, 113701. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorption Configuration | Bond Length (Å) | Bond Angle (°) | Eads (eV) | d (Å) | ||
|---|---|---|---|---|---|---|
| A1 | S-H1 | 1.36 | H1-S-H2 | 91.3 | −0.56 | 2.59 |
| S-H2 | 1.36 | |||||
| A2 | S-H1 | 1.36 | H1-S-H2 | 91.9 | −0.53 | 2.57 |
| S-H2 | 1.36 | |||||
| A3 | S-H1 | 1.36 | H1-S-H2 | 93.0 | −0.64 | 2.51 |
| S-H2 | 1.36 | |||||
| A4 | - | - | - | - | −1.53 | - |
| Adsorption Configuration | Mulliken Charge (e) | Q (e) | |
|---|---|---|---|
| A2 | S | −0.25 | 0.09 |
| H1 | 0.17 | ||
| H2 | 0.17 | ||
| A4 | S | −0.54 | −0.47 |
| H1 | 0.07 | ||
| Adsorption System | Bond Length (Å) | Bond Angle (°) | Eads (eV) | d (Å) | ||
|---|---|---|---|---|---|---|
| B1 | C-O | 1.17 | S-C-O | 179.6 | 0.36 | 2.73 |
| C-S | 1.57 | |||||
| B2 | C-O | 1.17 | S-C-O | 179.9 | 0.44 | 2.34 |
| C-S | 1.56 | |||||
| B3 | C-O | 1.17 | S-C-O | 179.7 | 0.22 | 2.64 |
| C-S | 1.59 | |||||
| B4 | C-O | 1.17 | S-C-O | 179.3 | 0.42 | 2.49 |
| C-S | 1.56 | |||||
| Adsorption Configuration | Bond Length (Å) | Bond Angle (°) | Eads (eV) | d (Å) | ||
|---|---|---|---|---|---|---|
| C1 | S-H1 S-C C-H2 C-H3 C-H4 | 1.36 1.83 1.10 1.10 1.10 | H1-S-C S-C-H2 S-C-H3 S-C-H4 | 98.2 108.7 109.9 108.0 | −0.50 | 2.52 |
| C2 | S-H1 S-C C-H2 C-H3 C-H4 | - 1.84 1.10 1.10 1.10 | H1-S-C S-C-H2 S-C-H3 S-C-H4 | - 112.1 107.3 108.9 | −1.40 | 2.34 |
| Adsorption Configuration | Mulliken Charge (e) | Q (e) | |
|---|---|---|---|
| C1 | S C H1 H2 H3 H4 | −0.04 −0.88 0.15 0.31 0.30 0.27 | 0.11 |
| C2 | S C H1 H2 H3 H4 | −0.31 −0.83 0.42 0.28 0.24 0.22 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.; Zhu, H.; Zeng, W. Density Functional Theory Study on the Adsorption Mechanism of Sulphide Gas Molecules on α-Fe2O3(001) Surface. Inorganics 2021, 9, 80. https://doi.org/10.3390/inorganics9110080
Zhou L, Zhu H, Zeng W. Density Functional Theory Study on the Adsorption Mechanism of Sulphide Gas Molecules on α-Fe2O3(001) Surface. Inorganics. 2021; 9(11):80. https://doi.org/10.3390/inorganics9110080
Chicago/Turabian StyleZhou, Li, Huadong Zhu, and Wen Zeng. 2021. "Density Functional Theory Study on the Adsorption Mechanism of Sulphide Gas Molecules on α-Fe2O3(001) Surface" Inorganics 9, no. 11: 80. https://doi.org/10.3390/inorganics9110080
APA StyleZhou, L., Zhu, H., & Zeng, W. (2021). Density Functional Theory Study on the Adsorption Mechanism of Sulphide Gas Molecules on α-Fe2O3(001) Surface. Inorganics, 9(11), 80. https://doi.org/10.3390/inorganics9110080