
Utilizing Big Data to Identify Tiny Toxic Components: Digitalis


Abstract
: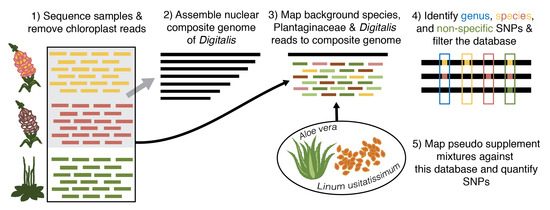
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sample Acquisition
2.1.1. DNA Extraction and Sequencing
2.1.2. Companion Data from Public Archives
2.2. Nuclear Enrichment of Digitalis WGS Data
2.3. Composite Genome Assembly
2.4. Nuclear Digitalis Read Mapping and SNP Dataset Development
2.5. Background Species Mapping and Data Filtration
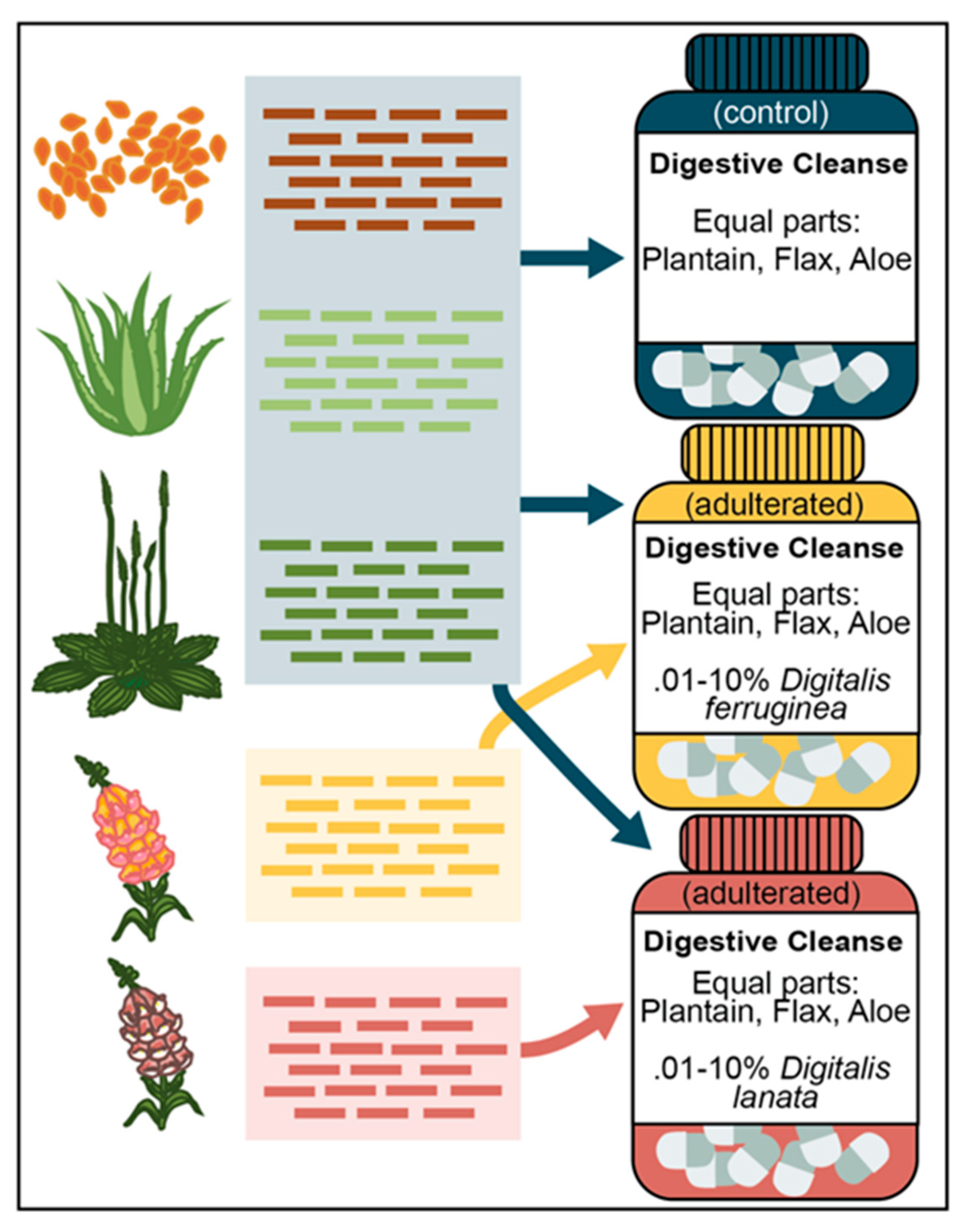
2.6. Generating Mixed Samples
2.7. Screening Mixed Samples for Digitalis SNPs
2.7.1. Genus-Level Digitalis Detection
2.7.2. Species-Level Digitalis Detection
3. Results
3.1. Assembly of the Nuclear Digitalis Composite Genome
3.2. Nuclear Digitalis Mapping and SNP Dataset Generation
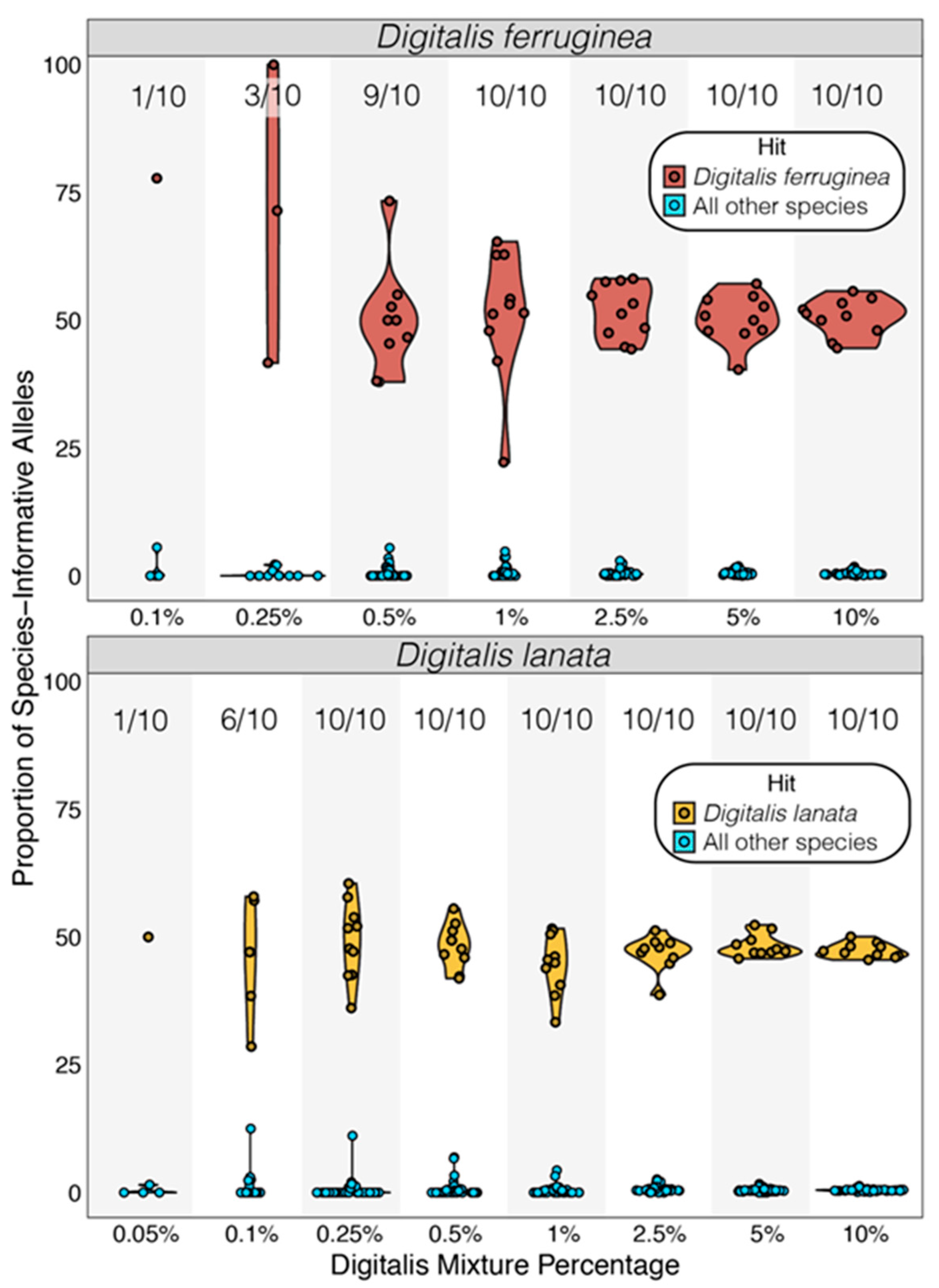
3.3. Screening Mixed Samples
3.3.1. Genus-Level Screening
3.3.2. Species-Level Screening
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Withering, W. An Account of the Foxglove and Some of Its Medical Uses; Cambridge University Press: Cambridge, UK, 1785. [Google Scholar]
- Beller, G.A.; Smith, T.W.; Abelmann, W.H.; Haber, E.; Hood, W.B.J. Digitalis Intoxication—A Prospective Clinical Study with Serum Level Correlations. N. Engl. J. Med. 1971, 284, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Lapostolle, F.; Borron, S.W. Chapter 58—Digitalis. In Haddad and Winchester’s Clinical Management of Poisoning and Drug Overdose, 4th ed.; Shannon, M.W., Borron, S.W., Burns, M.J., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2007; pp. 949–962. ISBN 978-0-7216-0693-4. [Google Scholar]
- Haruna, Y.; Kawasaki, T.; Kikkawa, Y.; Mizuno, R.; Matoba, S. Xanthopsia Due to Digoxin Toxicity as a Cause of Traffic Accidents: A Case Report. Am. J. Case Rep. 2020, 21, e924025-1–e924025-4. [Google Scholar] [CrossRef] [PubMed]
- Somberg, J.C. Van Gogh and Digitalis. Am. J. Cardiol. 2020, 136, 164–165. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Why Is the Use of Digitalis Withering? Another Reason That We Need Medical Heart Failure Specialists. Eur. J. Heart Fail. 2018, 20, 851–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slifman, N.R.; Obermeyer, W.R.; Aloi, B.K.; Musser, S.M.; Correll, W.A.; Cichowicz, S.M.; Betz, J.M.; Love, L.A. Contamination of Botanical Dietary Supplements by Digitalis Lanata. N. Engl. J. Med. 1998, 339, 806–811. [Google Scholar] [CrossRef]
- Harbaugh Reynaud, D.T.; Mishler, B.D.; Neal-Kababick, J.; Brown, P.N. The Capabilities and Limitations of DNA Barcoding of Dietary Supplements 2015. Available online: https://www.ahpa.org/portals/0/pdfs/the-capabilities-and-limitations-of-dna-testing-final_ahpa.pdf (accessed on 17 May 2021).
- Parveen, I.; Gafner, S.; Techen, N.; Murch, S.; Khan, I. DNA Barcoding for the Identification of Botanicals in Herbal Medicine and Dietary Supplements: Strengths and Limitations. Planta Med. 2016, 82, 1225–1235. [Google Scholar] [CrossRef] [Green Version]
- Pawar, R.; Handy, S.; Cheng, R.; Shyong, N.; Grundel, E. Assessment of the Authenticity of Herbal Dietary Supplements: Comparison of Chemical and DNA Barcoding Methods. Planta Med. 2017, 83, 921–936. [Google Scholar] [CrossRef] [Green Version]
- Mueller, S.; Handy, S.M.; Deeds, J.R.; George, G.O.; Broadhead, W.J.; Pugh, S.E.; Garrett, S.D. Development of a COX1 Based PCR-RFLP Method for Fish Species Identification. Food Control 2015, 55, 39–42. [Google Scholar] [CrossRef]
- Handy, S.M.; Chizhikov, V.; Yakes, B.J.; Paul, S.Z.; Deeds, J.R.; Mossoba, M.M. Microarray Chip Development Using Infrared Imaging for the Identification of Catfish Species. Appl. Spectrosc. 2014, 68, 1365–1373. [Google Scholar] [CrossRef]
- Rasmussen Hellberg, R.S.; Naaum, A.M.; Handy, S.M.; Hanner, R.H.; Deeds, J.R.; Yancy, H.F.; Morrissey, M.T. Interlaboratory Evaluation of a Real-Time Multiplex Polymerase Chain Reaction Method for Identification of Salmon and Trout Species in Commercial Products. J. Agric. Food Chem. 2011, 59, 876–884. [Google Scholar] [CrossRef]
- Handy, S.M.; Timme, R.E.; Jacob, S.M.; Deeds, J.R. Development of a Locked Nucleic Acid Real-Time Polymerase Chain Reaction Assay for the Detection of Pinus Armandii in Mixed Species Pine Nut Samples Associated with Dysgeusia. J. Agric. Food Chem. 2013, 61, 1060–1066. [Google Scholar] [CrossRef]
- Shokralla, S.; Hellberg, R.S.; Handy, S.M.; King, I.; Hajibabaei, M. A DNA Mini-Barcoding System for Authentication of Processed Fish Products. Sci. Rep. 2015, 5, 15894. [Google Scholar] [CrossRef]
- Puente-Lelievre, C.; Eischeid, A.C. Development and Validation of a Duplex Real-Time PCR Assay with Locked Nucleic Acid (LNA) Probes for the Specific Detection of Allergenic Walnut in Complex Food Matrices. Food Control 2021, 121, 107644. [Google Scholar] [CrossRef]
- Shanmughanandhan, J.; Shanmughanandhan, D.; Ragupathy, S.; Henry, T.A.; Newmaster, S.G. Quantification of Actaea racemosa L. (Black Cohosh) from Some of Its Potential Adulterants Using QPCR and DPCR Methods. Sci. Rep. 2021, 11, 4331. [Google Scholar] [CrossRef] [PubMed]
- Handy, S.M.; Deeds, J.R.; Ivanova, N.V.; Hebert, P.D.N.; Hanner, R.H.; Ormos, A.; Weigt, L.A.; Moore, M.M.; Yancy, H.F. A Single-Laboratory Validated Method for the Generation of DNA Barcodes for the Identification of Fish for Regulatory Compliance. J. AOAC Int. 2011, 94, 201–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanova, N.V.; Kuzmina, M.L.; Braukmann, T.W.A.; Borisenko, A.V.; Zakharov, E.V. Authentication of Herbal Supplements Using Next-Generation Sequencing. PLoS ONE 2016, 11, e0156426. [Google Scholar] [CrossRef]
- Handy, S.M.; Ott, B.M.; Hunter, E.S.; Zhang, S.; Erickson, D.L.; Wolle, M.M.; Conklin, S.D.; Lane, C.E. Suitability of DNA Sequencing Tools for Identifying Edible Seaweeds Sold in the United States. J. Agric. Food Chem. 2020, 68, 15516–15525. [Google Scholar] [CrossRef] [PubMed]
- Handy, S.M.; Pawar, R.S.; Ottesen, A.R.; Ramachandran, P.; Sagi, S.; Zhang, N.; Hsu, E.; Erickson, D.L. HPLC-UV, Metabarcoding and Genome Skims of Botanical Dietary Supplements: A Case Study in Echinacea. Planta Med. 2021, a-1336-1685. [Google Scholar] [CrossRef]
- Zhang, N.; Erickson, D.L.; Ramachandran, P.; Ottesen, A.R.; Timme, R.E.; Funk, V.A.; Luo, Y.; Handy, S.M. An Analysis of Echinacea Chloroplast Genomes: Implications for Future Botanical Identification. Sci. Rep. 2017, 7, 216. [Google Scholar] [CrossRef] [PubMed]
- Wirta, H.; Abrego, N.; Miller, K.; Roslin, T.; Vesterinen, E. DNA Traces the Origin of Honey by Identifying Plants, Bacteria and Fungi. Sci. Rep. 2021, 11, 4798. [Google Scholar] [CrossRef] [PubMed]
- Mutebi, R.R.; Ario, A.R.; Nabatanzi, M.; Kyamwine, I.B.; Wibabara, Y.; Muwereza, P.; Eurien, D.; Kwesiga, B.; Bulage, L.; Kabwama, S.N.; et al. Large Outbreak of Jimsonweed (Datura Stramonium) Poisoning Due to Consumption of Contaminated Humanitarian Relief Food: Uganda, March–April 2019. 2021; In Review. [Google Scholar]
- Literman, R.; Ott, B.M.; Wen, J.; Grauke, L.; Schwartz, R.; Handy, S.M. Reference-Free Discovery of Millions of SNPs Permits Species and Hybrid Identification in Carya (Hickory). In prep.
- Soffritti, G.; Busconi, M.; Sánchez, R.A.; Thiercelin, J.-M.; Polissiou, M.; Roldán, M.; Fernández, J.A. Genetic and Epigenetic Approaches for the Possible Detection of Adulteration and Auto-Adulteration in Saffron (Crocus Sativus L.) Spice. Molecules 2016, 21, 343. [Google Scholar] [CrossRef]
- Böhme, K.; Calo-Mata, P.; Barros-Velázquez, J.; Ortea, I. Recent Applications of Omics-Based Technologies to Main Topics in Food Authentication. TrAC Trends Anal. Chem. 2019, 110, 221–232. [Google Scholar] [CrossRef]
- Sakamoto, W.; Takami, T. Chloroplast DNA Dynamics: Copy Number, Quality Control and Degradation. Plant Cell Physiol. 2018, 59, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Golczyk, H.; Greiner, S.; Wanner, G.; Weihe, A.; Bock, R.; Börner, T.; Herrmann, R.G. Chloroplast DNA in Mature and Senescing Leaves: A Reappraisal. Plant Cell 2014, 26, 847–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodsworth, S. Genome Skimming for Next-Generation Biodiversity Analysis. Trends. Plant Sci. 2015, 20, 525–527. [Google Scholar] [CrossRef]
- Straub, S.C.K.; Parks, M.; Weitemier, K.; Fishbein, M.; Cronn, R.C.; Liston, A. Navigating the Tip of the Genomic Iceberg: Next-Generation Sequencing for Plant Systematics. Am. J. Bot. 2012, 99, 349–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Merwe, M.; McPherson, H.; Siow, J.; Rossetto, M. Next-Gen Phylogeography of Rainforest Trees: Exploring Landscape-Level CpDNA Variation from Whole-Genome Sequencing. Mol. Ecol. Resour. 2014, 14, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Dong, W.; Zhang, J.; Guo, X.; Chen, J.; Wang, Z.; Lin, Z.; Tang, H.; Zhang, L. The Sequenced Angiosperm Genomes and Genome Databases. Front. Plant Sci. 2018, 9, 418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitwieser, F.P.; Lu, J.; Salzberg, S.L. A Review of Methods and Databases for Metagenomic Classification and Assembly. Brief. Bioinform. 2019, 20, 1125–1136. [Google Scholar] [CrossRef]
- Cook, D.; Lee, S.T.; Gardner, D.R.; Molyneux, R.J.; Johnson, R.L.; Taylor, C.M. Use of Herbarium Voucher Specimens To Investigate Phytochemical Composition in Poisonous Plant Research. J. Agric. Food Chem. 2021, 69, 4037–4047. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Ramachandran, P.; Wen, J.; Duke, J.; Metzman, H.; McLaughlin, W.; Ottesen, A.; Timme, R.; Handy, S. Development of a Reference Standard Library of Chloroplast Genome Sequences, GenomeTrakrCP. Planta Med. 2017, 83, 1420–1430. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.S.; Harkins, K.M.; Stone, A.C.; Cartwright, R.A. A Composite Genome Approach to Identify Phylogenetically Informative Data from Next-Generation Sequencing. BMC Bioinform. 2015, 16, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Windsor, A.M.; Ott, B.M.; Zhang, N.; Wen, J.; Hsu, E.; Handy, S.M. Full Chloroplast Genome Sequence of the Economically Important Dietary Supplement and Spice Curcuma Longa. Microbiol. Resour. Announc. 2019, 8, e00576-19. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.-J.; Yu, W.-B.; Yang, J.-B.; Song, Y.; dePamphilis, C.W.; Yi, T.-S.; Li, D.-Z. GetOrganelle: A Fast and Versatile Toolkit for Accurate de Novo Assembly of Organelle Genomes. Genome Biol. 2020, 21, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 30 November 2019).
- Castro, M.; Castro, S.; Loureiro, J. Genome Size Variation and Incidence of Polyploidy in Scrophulariaceae Sensu Lato from the Iberian Peninsula. AoB Plants 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Boisvert, S.; Raymond, F.; Godzaridis, É.; Laviolette, F.; Corbeil, J. Ray Meta: Scalable de Novo Metagenome Assembly and Profiling. Genome Biol. 2012, 13, R122. [Google Scholar] [CrossRef] [Green Version]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Leys, C.; Ley, C.; Klein, O.; Bernard, P.; Licata, L. Detecting Outliers: Do Not Use Standard Deviation around the Mean, Use Absolute Deviation around the Median. J. Exp. Soc. Psychol. 2013, 49, 764–766. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- Ounit, R.; Wanamaker, S.; Close, T.J.; Lonardi, S. CLARK: Fast and Accurate Classification of Metagenomic and Genomic Sequences Using Discriminative k-Mers. BMC Genom. 2015, 16, 236. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and Sensitive Classification of Metagenomic Sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madden, T. The BLAST Sequence Analysis Tool; National Center for Biotechnology Information: Bethesda, MD, USA, 2003.
- von Meijenfeldt, F.A.B.; Arkhipova, K.; Cambuy, D.D.; Coutinho, F.H.; Dutilh, B.E. Robust Taxonomic Classification of Uncharted Microbial Sequences and Bins with CAT and BAT. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Mirdita, M.; Steinegger, M.; Breitwieser, F.; Karin, E.L. Fast and Sensitive Taxonomic Assignment to Metagenomic Contigs. Bioinformatics 2021, btab184. [Google Scholar] [CrossRef]
- Ramachandran, P.; Mammel, M.; Ottesen, A.; Pava-Ripoll, M. MitochonTrakr: A Reference Collection of High-Quality Mitochondrial Genomes for Detecting Insect Species in Food Products. Mitochondrial DNA Part B 2019, 4, 292–293. [Google Scholar] [CrossRef]
- Kalyuzhnaya, M.G.; Lapidus, A.; Ivanova, N.; Copeland, A.C.; McHardy, A.C.; Szeto, E.; Salamov, A.; Grigoriev, I.V.; Suciu, D.; Levine, S.R.; et al. High-Resolution Metagenomics Targets Specific Functional Types in Complex Microbial Communities. Nat. Biotechnol. 2008, 26, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Firetti, F.; Zuntini, A.R.; Gaiarsa, J.W.; Oliveira, R.S.; Lohmann, L.G.; Sluys, M.-A.V. Complete Chloroplast Genome Sequences Contribute to Plant Species Delimitation: A Case Study of the Anemopaegma Species Complex. Am. J. Bot. 2017, 104, 1493–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurya, S.; Darshetkar, A.M.; Yi, D.-K.; Kim, J.; Lee, C.; Ali, M.A.; Choi, S.; Choudhary, R.K.; Kim, S.-Y. Plastome Comparison and Evolution within the Tribes of Plantaginaceae: Insights from an Asian Gypsyweed. Saudi J. Biol. Sci. 2020, 27, 3489–3498. [Google Scholar] [CrossRef] [PubMed]
- Kreis, W. The Foxgloves (Digitalis) Revisited. Planta Med. 2017, 83, 962–976. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hunter, E.S.; Literman, R.; Handy, S.M. Utilizing Big Data to Identify Tiny Toxic Components: Digitalis. Foods 2021, 10, 1794. https://doi.org/10.3390/foods10081794
Hunter ES, Literman R, Handy SM. Utilizing Big Data to Identify Tiny Toxic Components: Digitalis. Foods. 2021; 10(8):1794. https://doi.org/10.3390/foods10081794
Chicago/Turabian StyleHunter, Elizabeth Sage, Robert Literman, and Sara M. Handy. 2021. "Utilizing Big Data to Identify Tiny Toxic Components: Digitalis" Foods 10, no. 8: 1794. https://doi.org/10.3390/foods10081794
APA StyleHunter, E. S., Literman, R., & Handy, S. M. (2021). Utilizing Big Data to Identify Tiny Toxic Components: Digitalis. Foods, 10(8), 1794. https://doi.org/10.3390/foods10081794