

Effect of Coffee on the Bioavailability of Sterols
 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Preparation of the Coffee Extracts
2.3. Caco-2 Cell Culture
2.4. Cell Viability Assays
2.5. Absorptive Permeability Assays
2.6. Quantitative Analysis of LY
2.7. Quantitative Analysis of DHE
2.8. Evaluation of the Apparent Permeability Coefficient
2.9. Confocal Fluorescence Microscopy and Image Analysis
2.10. Statistical Analysis
3. Results and Discussion
3.1. Study of the Effect of Bile Salt and Coffee Composition and Concentration on the Intestinal Epithelium Model
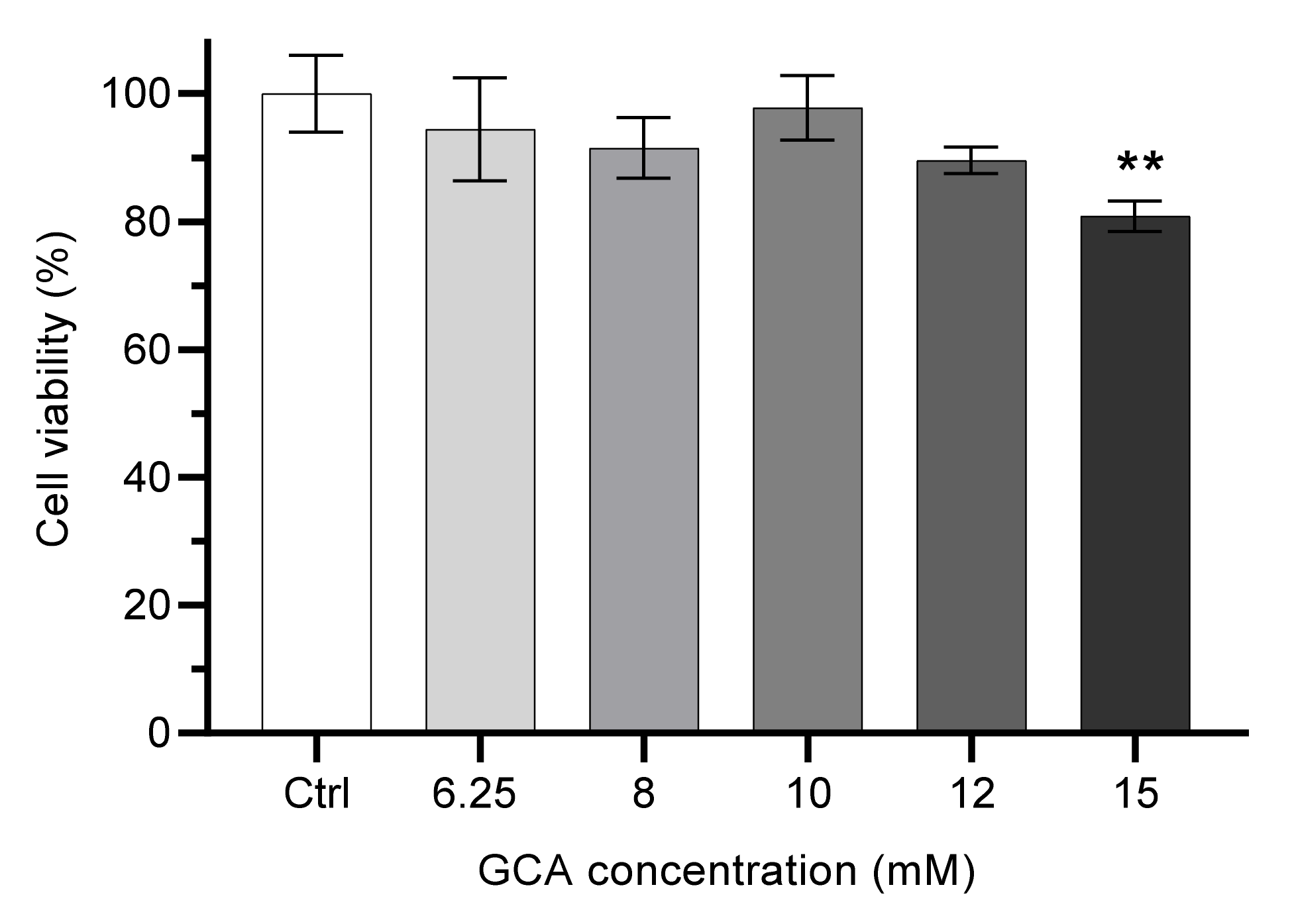
3.1.1. Effect of Bile Salt Composition and Concentration on Caco-2 Monolayers Viability
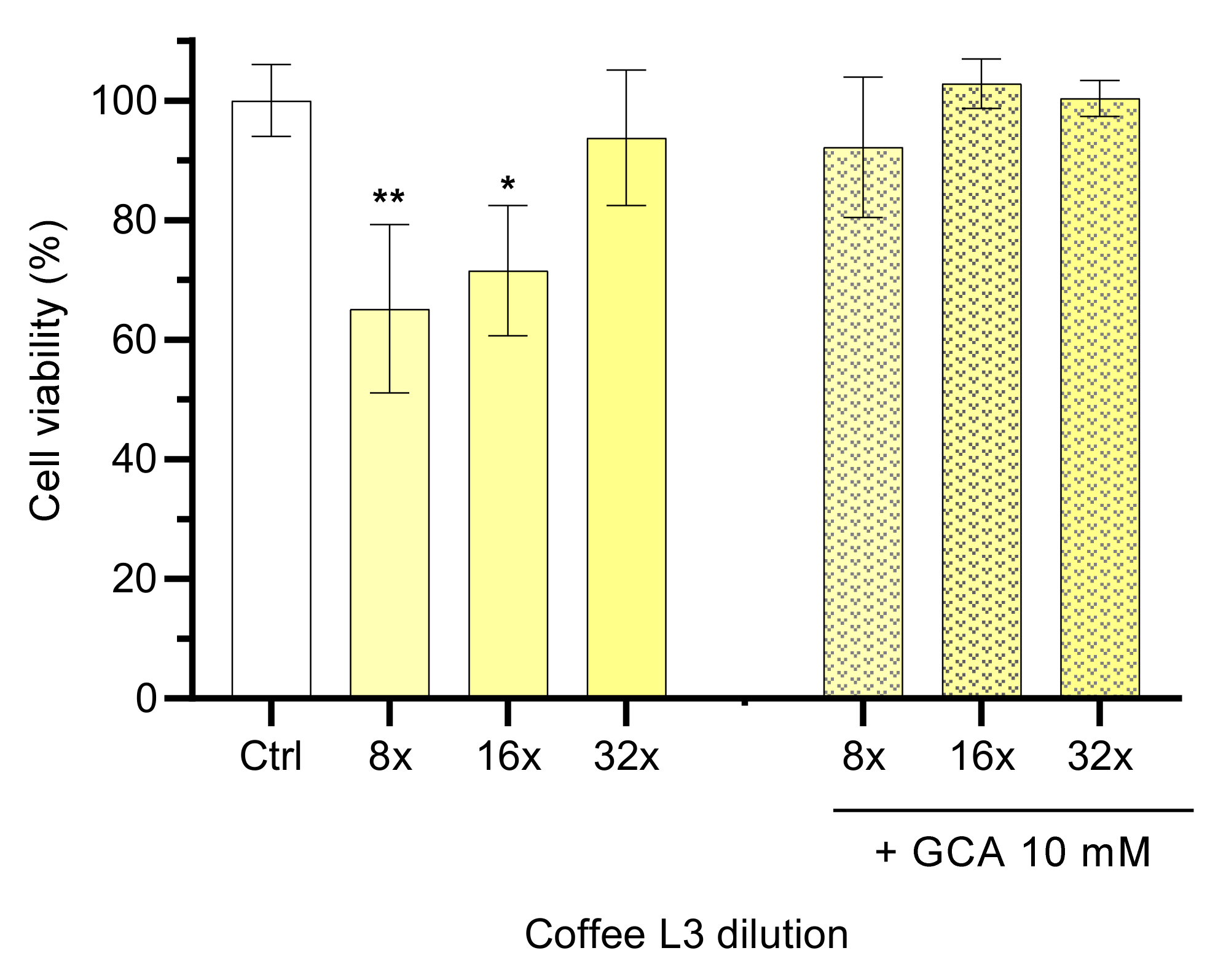
3.1.2. Effect of Coffee Composition and Concentration on Caco-2 Monolayers Viability
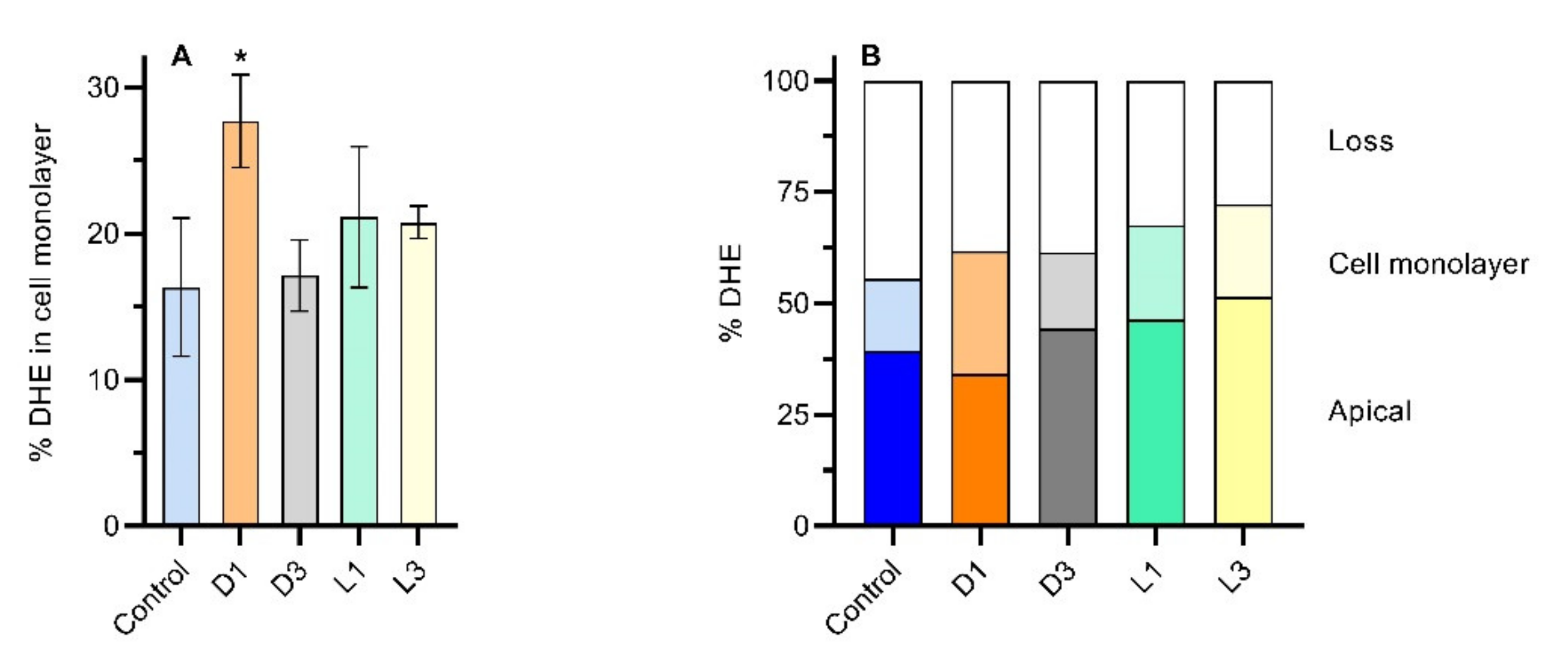
3.2. Effect of Emulsified DHE and Coffee on Cell Monolayer Integrity
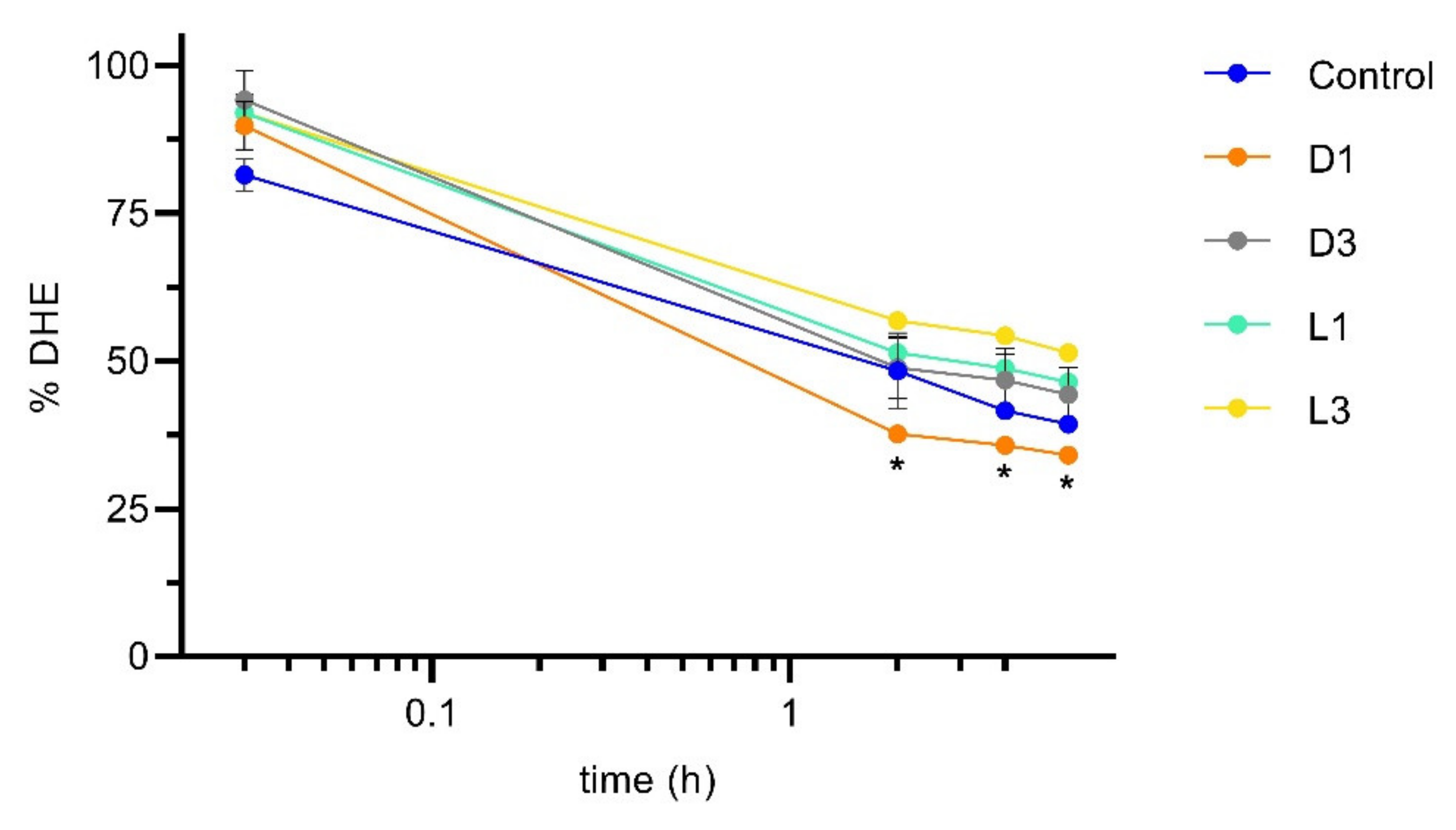
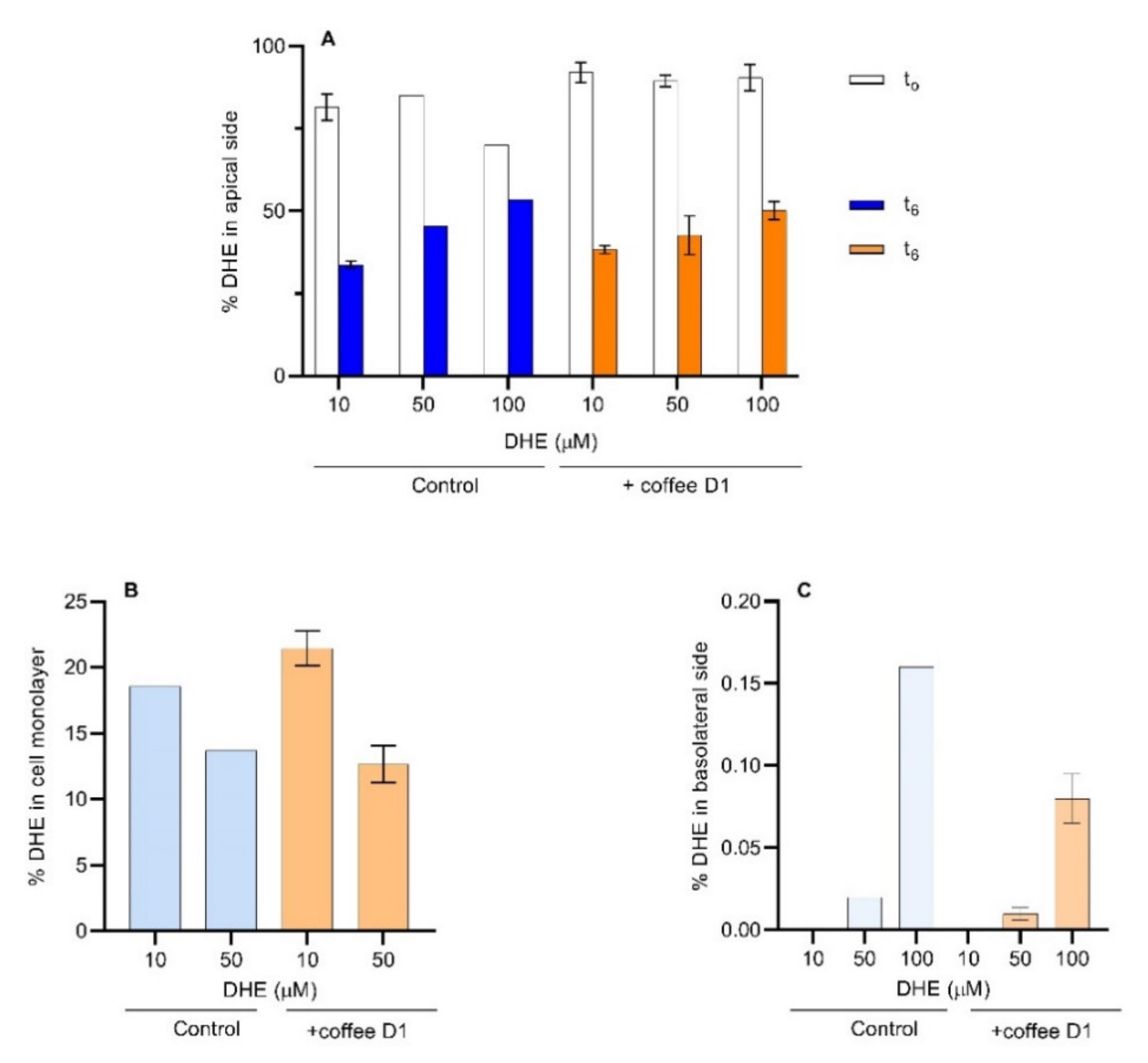
3.3. Influence of Coffee in DHE Bioaccessibility and Bioavailability
3.4. DHE Bioaccessibility and Bioavailability at High Concentrations
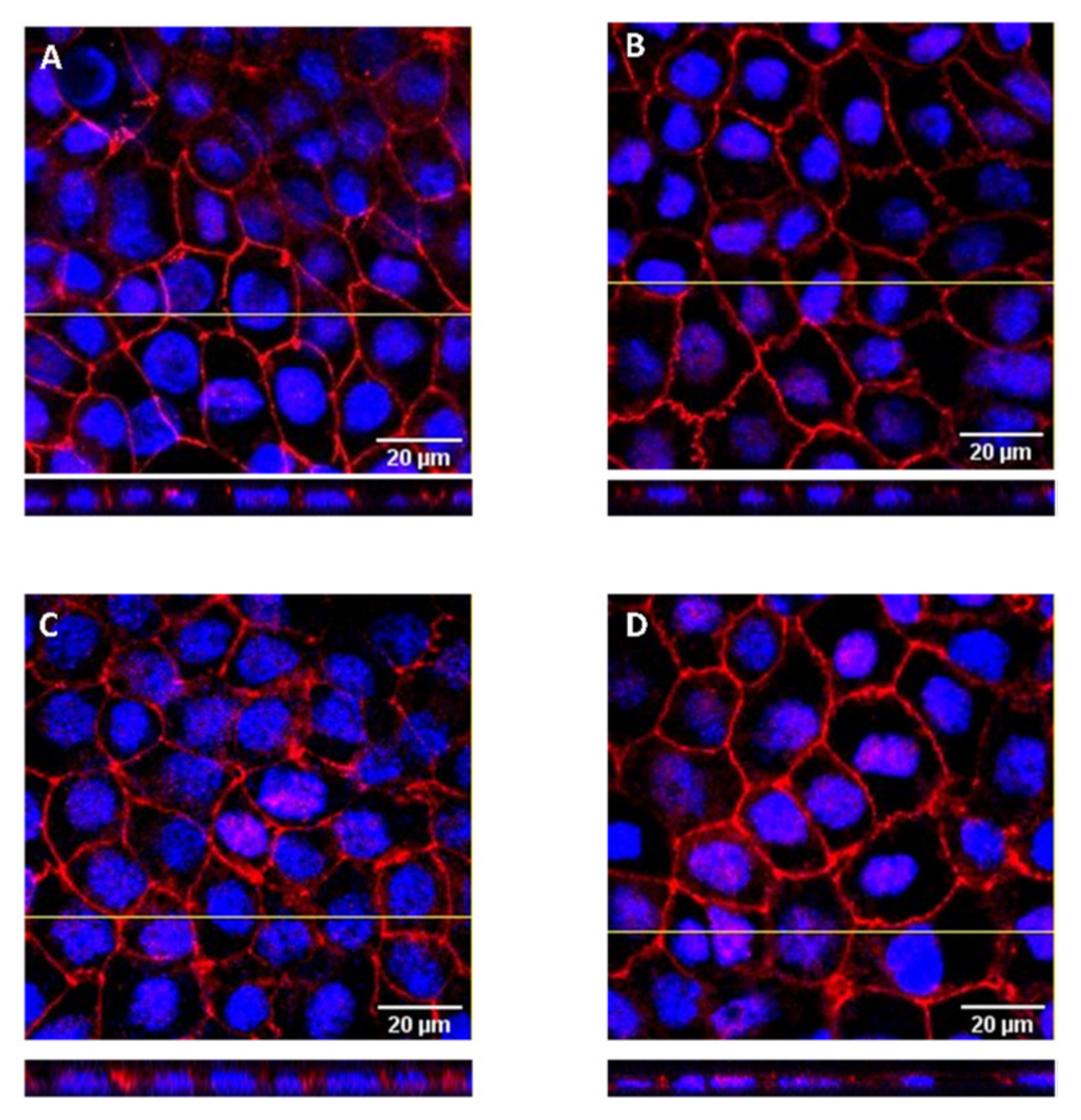
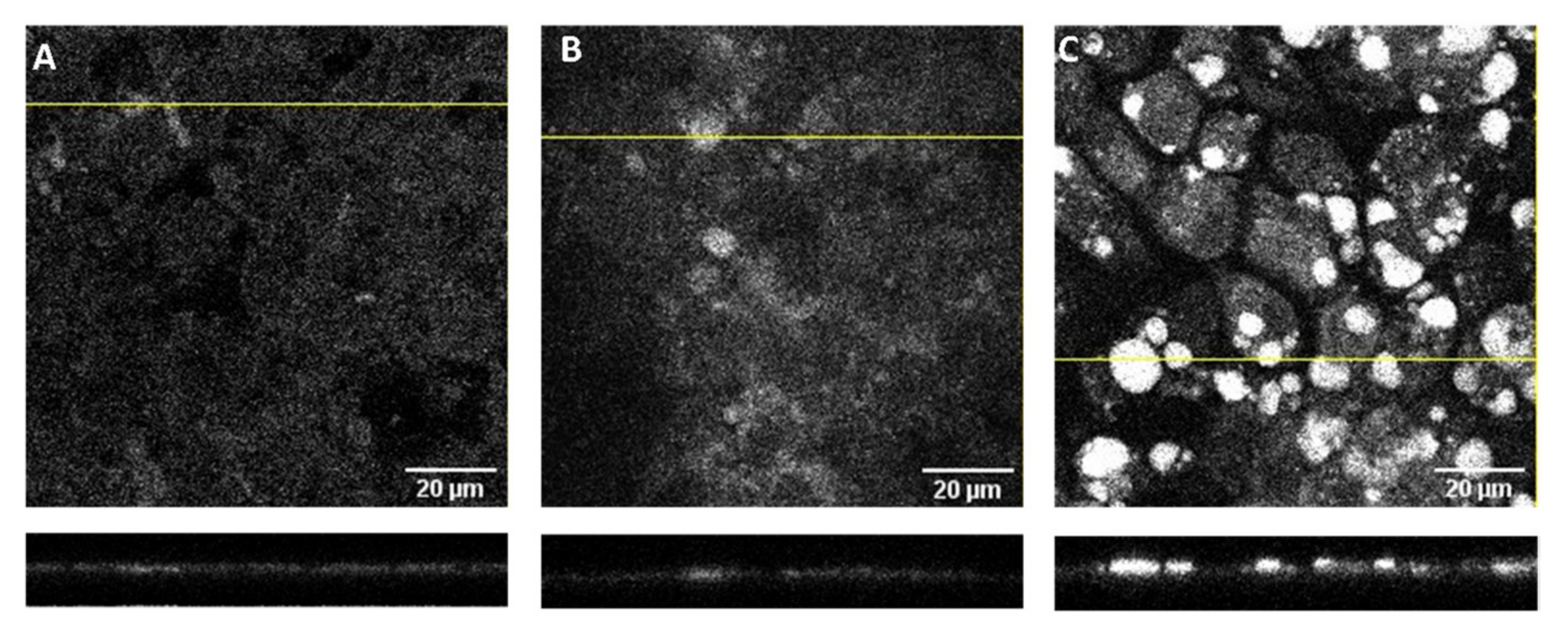
3.5. Localization of DHE in the Caco-2 Monolayers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. World Health Statistics 2020; WHO: Geneva, Switzerland, 2020; Volume 5. [Google Scholar]
- West, R.S.C. Pathogenesis of Atherosclerosis. J. Am. Coll. Cardiol. 2006, 47, C7–C12. [Google Scholar]
- Hui, D.Y.; Howles, P.N. Molecular mechanisms of cholesterol absorption and transport in the intestine. Semin. Cell Dev. Biol. 2005, 16, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. A Century of Cholesterol and Coronaries: From Plaques to Genes to Statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Betters, J.L.; Yu, L. Niemann-Pick C1-Like 1 (NPC1L1) Protein in Intestinal and Hepatic Cholesterol Transport. Annu. Rev. Physiol. 2011, 73, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, P.V.; Moreno, M.J.; Serra, A.C.; Simões, S.; Coelho, J.F.J. Synthesis of tailor-made bile acid sequestrants by supplemental activator and reducing agent atom transfer radical polymerization. RSC Adv. 2016, 6, 52143–52153. [Google Scholar] [CrossRef]
- Insull, W. Clinical Utility of Bile Acid Sequestrants in the Treatment of Dyslipidemia: A Scientific Review. South. Med. J. 2006, 99, 257–273. [Google Scholar] [CrossRef]
- Silva, I.M.V.; Machado, F.; Moreno, M.J.; Nunes, C.; Coimbra, M.A.; Coreta-Gomes, F. Polysaccharide structures and their hypocholesterolemic potential. Molecules 2021, 26, 4559. [Google Scholar] [CrossRef]
- Coreta-Gomes, F.M.; Lopes, G.R.; Passos, C.P.; Vaz, I.M.; Machado, F.; Geraldes, C.F.G.C.; Moreno, M.J.; Nyström, L.; Coimbra, M.A. In vitro hypocholesterolemic effect of coffee compounds. Nutrients 2020, 12, 437. [Google Scholar] [CrossRef]
- Coreta-Gomes, F.M.; Vaz, W.L.C.; Wasielewski, E.; Geraldes, C.F.G.; Moreno, M.J. Quantification of cholesterol solubilized in dietary micelles: Dependence on human bile salt variability and the presence of dietary food ingredients. Langmuir 2016, 32, 4564–4574. [Google Scholar] [CrossRef] [PubMed]
- Gylling, H.; Plat, J.; Turley, S.; Ginsberg, H.N.; Ellegård, L.; Jessup, W.; Jones, P.J.; Lütjohann, D.; Maerz, W.; Masana, L.; et al. Plant sterols and plant stanols in the management of dyslipidaemia and prevention of cardiovascular disease. Atherosclerosis 2014, 232, 346–360. [Google Scholar] [CrossRef]
- Lopes, M.; Coimbra, M.A.; Costa, M.C.; Ramos, F. Food supplement vitamins, minerals, amino-acids, fatty acids, phenolic and alkaloid-based substances: An overview of their interaction with drugs. Crit. Rev. Food Sci. Nutr. 2021, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Carey, M.C.; Montet, J.C.; Phillips, M.C.; Armstrong, M.J.; Mazer, N.A. Thermodynamic and molecular basis for dissimilar cholesterol-solubilizing capacities by micellar solutions of bile salts: Cases of sodium chenodeoxycholate and sodium ursodeoxycholate and their glycine and taurine conjugates. Biochemistry 1981, 20, 3637–3648. [Google Scholar] [CrossRef] [PubMed]
- Coreta-Gomes, F.M.; Vaz, W.L.C.; Wasielewski, E.; Geraldes, C.F.G.C.; Moreno, M.J. Quantification of cholesterol solubilized in bile salt micellar aqueous solutions using 13C nuclear magnetic resonance. Anal. Biochem. 2012, 427, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Nunes, F.M.; Coimbra, M.A.; Duarte, A.C.; Delgadillo, I. Foamability, Foam Stability, and Chemical Composition of Espresso Coffee As Affected by the Degree of Roast. J. Agric. Food Chem. 1997, 45, 3238–3243. [Google Scholar] [CrossRef]
- Bekedam, E.K.; Loots, M.J.; Schols, H.A.; Van Boekel, M.A.J.S.; Smit, G. Roasting effects on formation mechanisms of coffee brew melanoidins. J. Agric. Food Chem. 2008, 56, 7138–7145. [Google Scholar] [CrossRef]
- Bekedam, E.K.; Schols, H.A.; Van Boekel, M.A.J.S.; Smit, G. High molecular weight melanoidins from coffee brew. J. Agric. Food Chem. 2006, 54, 7658–7666. [Google Scholar] [CrossRef]
- Moreira, A.S.P.; Nunes, F.M.; Simões, C.; Maciel, E.; Domingues, P.; Domingues, M.R.M.; Coimbra, M.A. Transglycosylation reactions, a main mechanism of phenolics incorporation in coffee melanoidins: Inhibition by Maillard reaction. Food Chem. 2017, 227, 422–431. [Google Scholar] [CrossRef]
- Moreira, A.S.P.; Coimbra, M.A.; Nunes, F.M.; Passos, C.P.; Santos, S.A.O.; Silvestre, A.J.D.; Silva, A.M.N.; Rangel, M.; Domingues, M.R.M. Chlorogenic acid-arabinose hybrid domains in coffee melanoidins: Evidences from a model system. Food Chem. 2015, 185, 135–144. [Google Scholar] [CrossRef]
- Lopes, G.R.; Passos, C.P.; Rodrigues, C.; Teixeira, J.A.; Coimbra, M.A. Modulation of infusion processes to obtain coffee-derived food ingredients with distinct composition. Eur. Food Res. Technol. 2019, 245, 2133–2146. [Google Scholar] [CrossRef]
- Cordoba, N.; Pataquiva, L.; Osorio, C.; Moreno, F.L.M.; Ruiz, R.Y. Effect of grinding, extraction time and type of coffee on the physicochemical and flavour characteristics of cold brew coffee. Sci. Rep. 2019, 9, 8440. [Google Scholar] [CrossRef]
- Artursson, P.; Karlsson, J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem. Biophys. Res. Commun. 1991, 175, 880–885. [Google Scholar] [CrossRef]
- Yu, H.; Huang, Q. Improving the oral bioavailability of curcumin using novel organogel-based nanoemulsions. J. Agric. Food Chem. 2012, 60, 5373–5379. [Google Scholar] [CrossRef] [PubMed]
- Punt, A.; Peijnenburg, A.A.C.M.; Hoogenboom, R.L.A.P.; Bouwmeester, H. Non-animal approaches for toxicokinetics in risk evaluations of food chemicals. ALTEX 2017, 34, 501–514. [Google Scholar] [CrossRef]
- Tran, V.N.; Viktorová, J.; Ruml, T. Mycotoxins: Biotransformation and bioavailability assessment using caco-2 cell monolayer. Toxins 2020, 12, 628. [Google Scholar] [CrossRef] [PubMed]
- McClements, D.J.; Decker, E.A. Designing Functional Foods: Measuring and Controlling Food Structure Breakdown and Nutrient Absorption; Woodhead Publishing: Sawston, UK, 2009; ISBN 9781845694326. [Google Scholar]
- Glahn, R. The use of Caco-2 cells in defining nutrient bioavailability: Application to iron bioavailability of foods. In Designing Functional Foods: Measuring and Controlling Food Structure Breakdown and Nutrient Absorption; Woodhead Publishing: Sawston, UK, 2009; pp. 340–361. ISBN 9781845694326. [Google Scholar]
- Turley, S.D.; Dietschy, J.M. Sterol absorption by the small intestine. Curr. Opin. Lipidol. 2003, 14, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, F.; Jefferson, J.R.; Kier, A.B.; Knittel, J.; Scallen, T.J.; Wood, W.G.; Hapala, I. Membrane Cholesterol Dynamics: Cholesterol Domains and Kinetic Pools. Proc. Soc. Exp. Biol. Med. 1991, 196, 235–252. [Google Scholar] [CrossRef]
- Estronca, L.M.B.B.; Filipe, H.A.L.; Salvador, A.; Moreno, M.J.; Vaz, W.L.C. Homeostasis of free cholesterol in the blood: A preliminary evaluation and modeling of its passive transport. J. Lipid Res. 2014, 55, 1033–1043. [Google Scholar] [CrossRef]
- Pires, C.L.; Praça, C.; Martins, P.A.T.; Batista de Carvalho, A.L.M.; Ferreira, L.; Marques, M.P.M.; Moreno, M.J. Re-use of caco-2 monolayers in permeability assays—Validation regarding cell monolayer integrity. Pharmaceutics 2021, 13, 1563. [Google Scholar] [CrossRef]
- Fotakis, G.; Timbrell, J.A. In vitro cytotoxicity assays: Comparison of LDH, neutral red, MTT and protein assay in hepatoma cell lines following exposure to cadmium chloride. Toxicol. Lett. 2006, 160, 171–177. [Google Scholar] [CrossRef]
- Hubatsch, I.; Ragnarsson, E.G.E.; Artursson, P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat. Protoc. 2007, 2, 2111–2119. [Google Scholar] [CrossRef]
- Haberland, M.E.; Reynolds, J.A. Self association of cholesterol in aqueous solution. Proc. Natl. Acad. Sci. USA 1973, 70, 2313–2316. [Google Scholar] [CrossRef] [Green Version]
- Fischer, R.T.; Dempsey, M.E.; Schroeder, F.; Cowlen, M.S. Fluorescence of Δ5′7,9(11)’22-Ergostatetraen-3β-ol in Micelles, Sterol Carrier Protein Complexes, and Plasma Membranesδ. Biochemistry 1985, 24, 3322–3331. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.B.; Staples, E.M. Plasma protein concentrations in interstitial fluid from human aortas. Proc. R. Soc. Lond. Biol. Sci. 1982, 217, 59–75. [Google Scholar]
- Estronca, L.M.B.B.; Moreno, M.J.; Vaz, W.L.C. Kinetics and Thermodynamics of the Association of Dehydroergosterol with Lipid Bilayer Membranes. Biophys. J. 2007, 93, 4244–4253. [Google Scholar] [CrossRef]
- Broeders, J.J.W.; Van Eijkeren, J.C.H.; Blaauboer, B.J.; Hermens, J.L.M. Transport of chlorpromazine in the Caco-2 cell permeability assay: A kinetic study. Chem. Res. Toxicol. 2012, 25, 1442–1451. [Google Scholar] [CrossRef] [PubMed]
- Van Breemen, R.B.; Li, Y. Caco-2 cell permeability assays to measure drug absorption. Expert Opin. Drug Metab. Toxicol. 2005, 1, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Coreta-Gomes, F.M.; Martins, P.A.T.; Velazquez-Campoy, A.; Vaz, W.L.C.; Geraldes, C.F.G.; Moreno, M.J. Interaction of Bile Salts with Model Membranes Mimicking the Gastrointestinal Epithelium: A Study by Isothermal Titration Calorimetry. Langmuir 2015, 31, 9097–9104. [Google Scholar] [CrossRef] [PubMed]
- Heuman, D.M.; Pandak, W.M.; Hylemon, P.B.; Vlahcevic, Z.R. Conjugates of ursodeoxycholate protect against cytotoxicity of more hydrophobic bile salts: In vitro studies in rat hepatocytes and human erythrocytes. Hepatology 1991, 14, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Bedoya-Ramírez, D.; Cilla, A.; Contreras-Calderón, J.; Alegría-Torán, A. Evaluation of the antioxidant capacity, furan compounds and cytoprotective/cytotoxic effects upon Caco-2 cells of commercial Colombian coffee. Food Chem. 2017, 219, 364–372. [Google Scholar] [CrossRef]
- Hidalgo, I.J. Assessing the Absorption of New Pharmaceuticals. Curr. Top. Med. Chem. 2005, 1, 385–401. [Google Scholar] [CrossRef]
- Konsoula, R.; Barile, F.A. Correlation of in vitro cytotoxicity with paracellular permeability in Caco-2 cells. Toxicol. Vitr. 2005, 19, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Corazza, F.G.; Ernesto, J.V.; Nambu, F.A.N.; de Carvalho, L.R.; Leite-Silva, V.R.; Varca, G.H.C.; Calixto, L.A.; Vieira, D.P.; Andréo-Filho, N.; Lopes, P.S. Papain-cyclodextrin complexes as an intestinal permeation enhancer: Permeability and in vitro safety evaluation. J. Drug Deliv. Sci. Technol. 2020, 55, 101413. [Google Scholar] [CrossRef]
- Hernell, O.; Staggers, J.E.; Carey, M.C. Physical-chemical behavior of dietary and biliary lipids during intestinal digestion and absorption. 2. Phase analysis and aggregation states of luminal lipids during duodenal fat digestion in healthy adult human beings. Biochemistry 1990, 29, 2041–2056. [Google Scholar] [CrossRef]
- Moreno, M.J.; Martins, P.A.T.; Bernardino, E.F.; Abel, B.; Ambudkar, S.V. Characterization of the lipidome and biophysical properties of membranes from high five insect cells expressing mouse P-glycoprotein. Biomolecules 2021, 11, 426. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, G. The Composition of Biological Membranes. Arch. Intern. Med. 1972, 129, 194–201. [Google Scholar] [CrossRef]
- Sampaio, J.L.; Gerl, M.J.; Klose, C.; Ejsing, C.S.; Beug, H.; Simons, K.; Shevchenko, A. Membrane lipidome of an epithelial cell line. Proc. Natl. Acad. Sci. USA 2011, 108, 1903–1907. [Google Scholar] [CrossRef]
- Kaiser, H.J.; Lingwood, D.; Levental, I.; Sampaio, J.L.; Kalvodova, L.; Rajendran, L.; Simons, K. Order of lipid phases in model and plasma membranes. Proc. Natl. Acad. Sci. USA 2009, 106, 16645–16650. [Google Scholar] [CrossRef]
- Meder, D.; Moreno, M.J.; Verkade, P.; Vaz, W.L.C.; Simons, K. Phase coexistence and connectivity in the apical membrane of polarized epithelial cells. Proc. Natl. Acad. Sci. USA 2006, 103, 329–334. [Google Scholar] [CrossRef]
- Lange, Y.; Tabei, S.M.A.; Ye, J.; Steck, T.L. Stability and stoichiometry of bilayer phospholipid-cholesterol complexes: Relationship to cellular sterol distribution and homeostasis. Biochemistry 2013, 52, 6950–6959. [Google Scholar] [CrossRef]
- Yi, J.; Knudsen, T.A.; Nielsen, A.L.; Duelund, L.; Christensen, M.; Hervella, P.; Needham, D.; Mouritsen, O.G. Inhibition of cholesterol transport in an intestine cell model by pine-derived phytosterols. Chem. Phys. Lipids 2016, 200, 62–73. [Google Scholar] [CrossRef]
- Ressaissi, A.; Attia, N.; Falé, P.L.; Pacheco, R.; Victor, B.L.; Machuqueiro, M.; Serralheiro, M.L.M. Isorhamnetin derivatives and piscidic acid for hypercholesterolemia: Cholesterol permeability, HMG-CoA reductase inhibition, and docking studies. Arch. Pharm. Res. 2017, 40, 1278–1286. [Google Scholar] [CrossRef]
- Field, F.J.; Born, E.; Mathur, S.N. Effect of micellar β-sitosterol on cholesterol metabolism in CaCo-2 cells. J. Lipid Res. 1997, 38, 348–360. [Google Scholar] [CrossRef]
- Lin, D.S.; Steiner, R.D.; Merkens, L.S.; Pappu, A.S.; Connor, W.E. The effects of sterol structure upon sterol esterification. Atherosclerosis 2010, 208, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Temel, R.E.; Gebre, A.K.; Parks, J.S.; Rudel, L.L. Compared with Acyl-CoA:Cholesterol O-Acyltransferase (ACAT) 1 and Lecithin: Cholesterol Acyltransferase, ACAT2 Displays the Greatest Capacity to Differentiate Cholesterol from Sitosterol. J. Biol. Chem. 2003, 278, 47594–47601. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| % Transport of LY 0–2 h | % Transport of LY 2–4 h | % Transport of LY 4–6 h | Papp of LY (×10−6) a | |
|---|---|---|---|---|
| LY | 1.6 | 0.9 | 1.3 | 0.64 |
| DHE/GCA | 0.84 ± 0.45 | 1.4 ± 0.20 | 0.81 ± 0.21 | 0.40 ± 0.09 |
| Coffee extracts | ||||
| D1 | 0.41 ± 0.005 | 0.54 ± 0.26 | 0.60 ± 0.38 | 0.30 ± 0.19 |
| D3 | 0.45 ± 0.12 | 0.46 ± 0.04 | 0.60 ± 0.37 | 0.30 ± 0.18 |
| L1 | 0.51 ± 0.34 | 0.64 ± 0.14 | 0.85 ± 0.42 | 0.42 ± 0.21 |
| L3 | 0.46 ± 0.07 | 0.19 ± 0.12 | 0.38 ± 0.02 | 0.19 ± 0.01 |
| % Transport of LY 6 h | Papp of LY (×10−6) | ||
|---|---|---|---|
| LY | 1.6 | 0.27 | |
| [µM] | |||
| DHE/GCA | 10 | 0.7 | 0.11 |
| 50 | 1.1 | 0.18 | |
| 100 | 0.6 | 0.11 | |
| Coffee extract | |||
| D1 | 10 | 0.66 ± 0.13 | 0.11 ± 0.02 |
| 50 | 0.64 ± 0.05 | 0.11 ± 0.01 | |
| 100 | 0.40 ± 0.12 | 0.07 ± 0.02 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pires, C.L.; Silva, I.M.V.; Coimbra, M.A.; Moreno, M.J.; Coreta-Gomes, F. Effect of Coffee on the Bioavailability of Sterols. Foods 2022, 11, 2935. https://doi.org/10.3390/foods11192935
Pires CL, Silva IMV, Coimbra MA, Moreno MJ, Coreta-Gomes F. Effect of Coffee on the Bioavailability of Sterols. Foods. 2022; 11(19):2935. https://doi.org/10.3390/foods11192935
Chicago/Turabian StylePires, Cristiana L., Inês M. V. Silva, Manuel A. Coimbra, Maria João Moreno, and Filipe Coreta-Gomes. 2022. "Effect of Coffee on the Bioavailability of Sterols" Foods 11, no. 19: 2935. https://doi.org/10.3390/foods11192935
APA StylePires, C. L., Silva, I. M. V., Coimbra, M. A., Moreno, M. J., & Coreta-Gomes, F. (2022). Effect of Coffee on the Bioavailability of Sterols. Foods, 11(19), 2935. https://doi.org/10.3390/foods11192935