
Characteristics and Correlation of the Microbial Communities and Flavor Compounds during the First Three Rounds of Fermentation in Chinese Sauce-Flavor Baijiu

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Flavor Compound Analysis
2.3. Microbial Composition Analysis
2.4. Correlation Analysis
2.5. Data Analysis
3. Results and Discussion
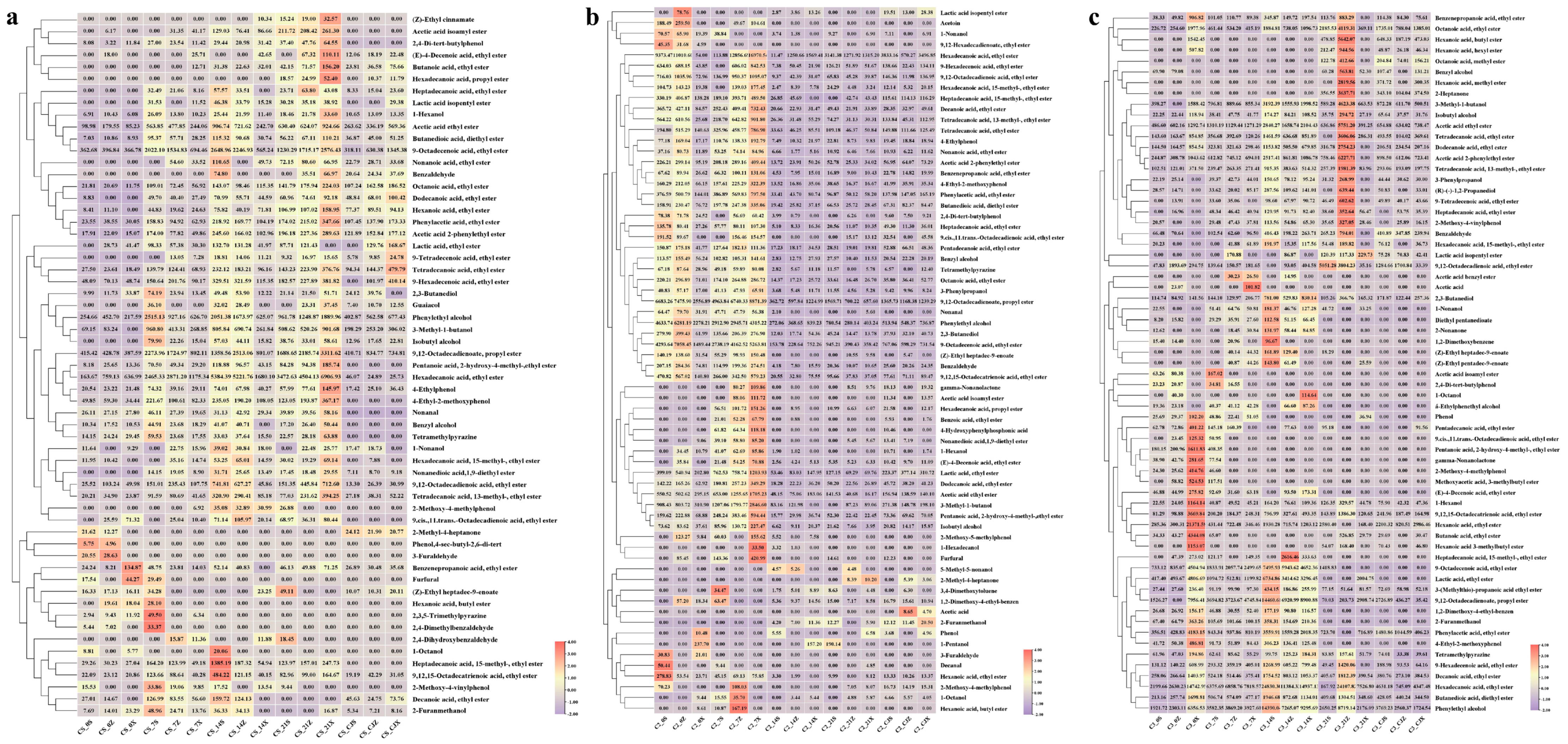
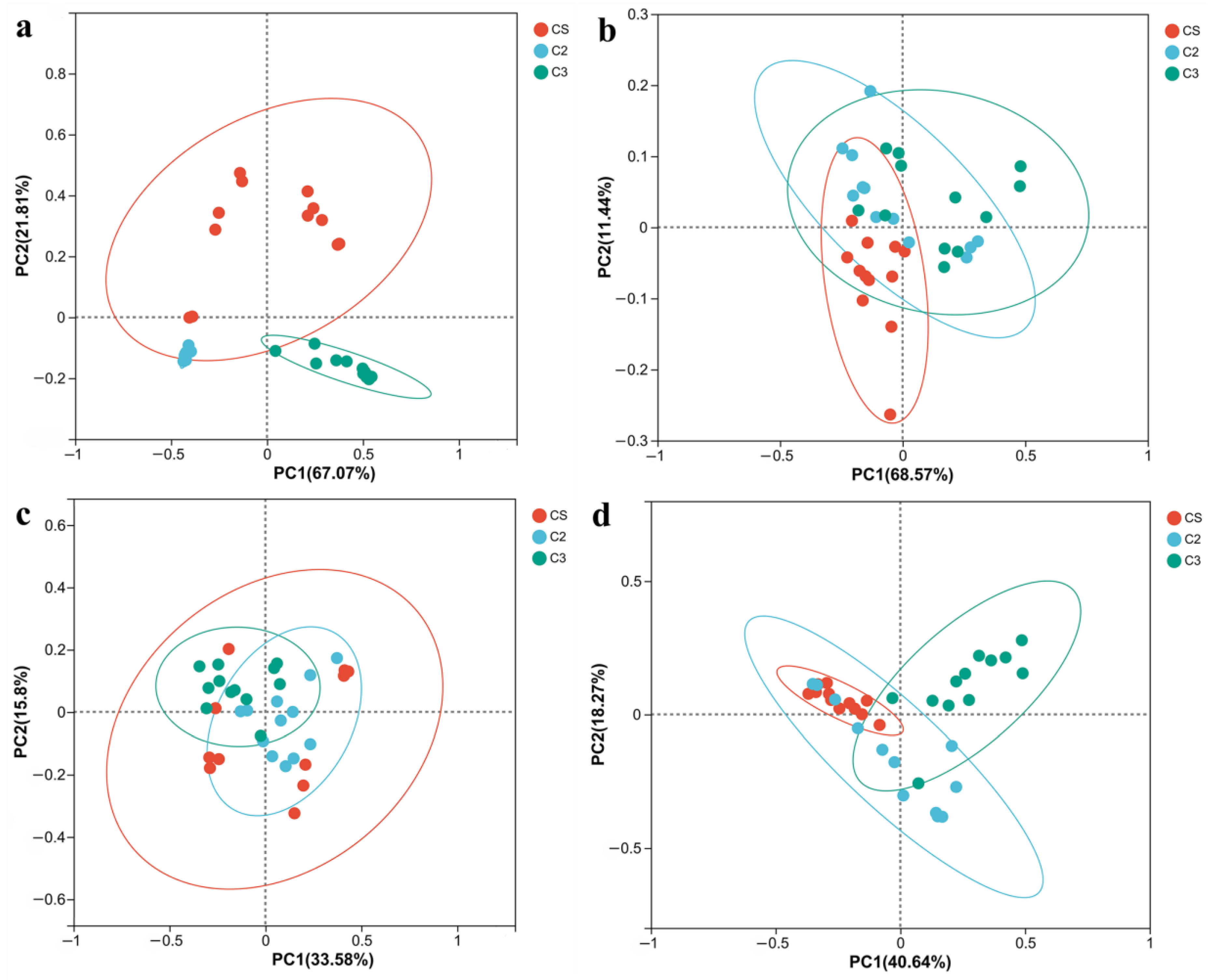
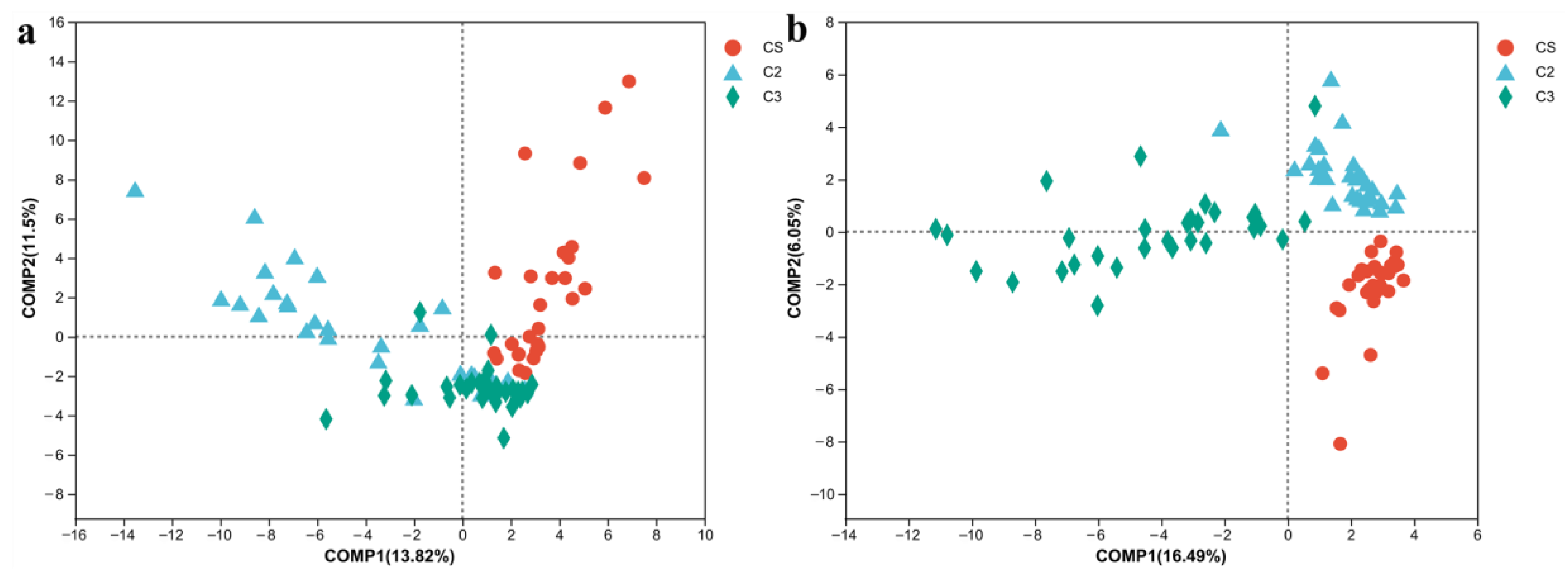
3.1. Flavor Analysis of the Samples in the First Three Rounds of Sauce-Flavor Baijiu
3.2. Microbial Community Analysis
3.2.1. The Dynamic Composition of Microbial Communities during Fermentation
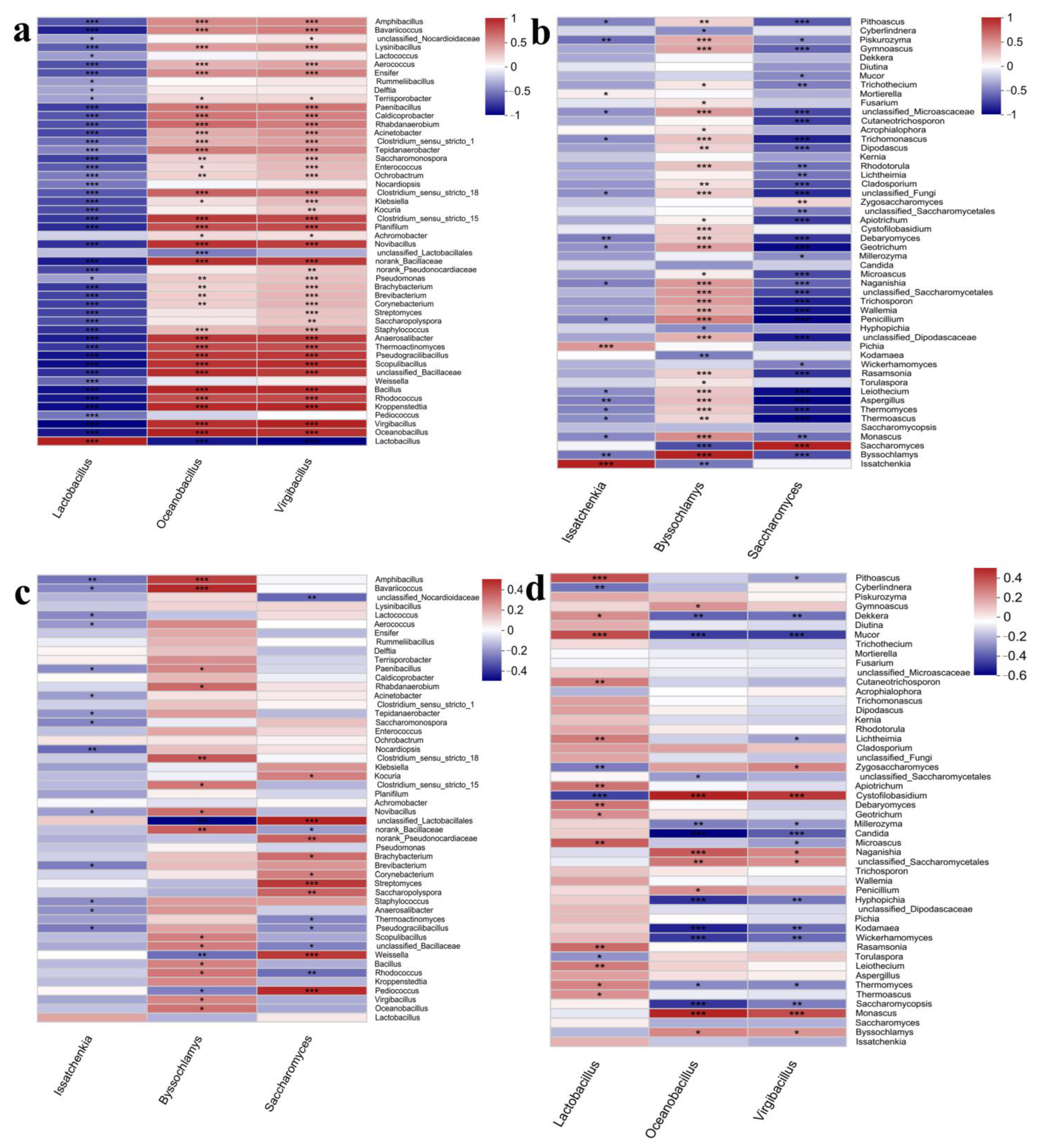
3.2.2. Microbial Interactions during Fermentation
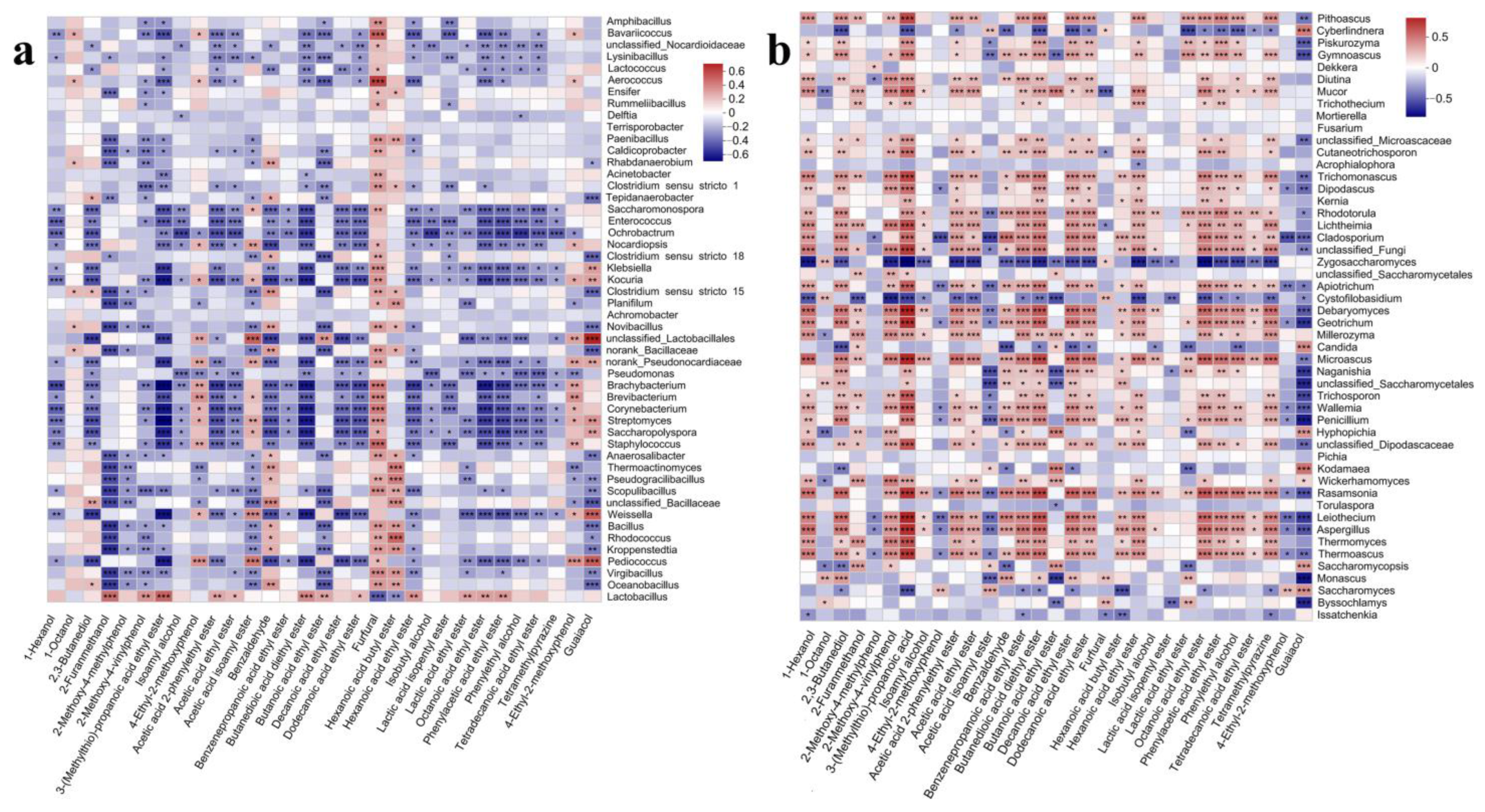
3.3. Correlation Analysis of Flavor Chemicals and Microbial Communities
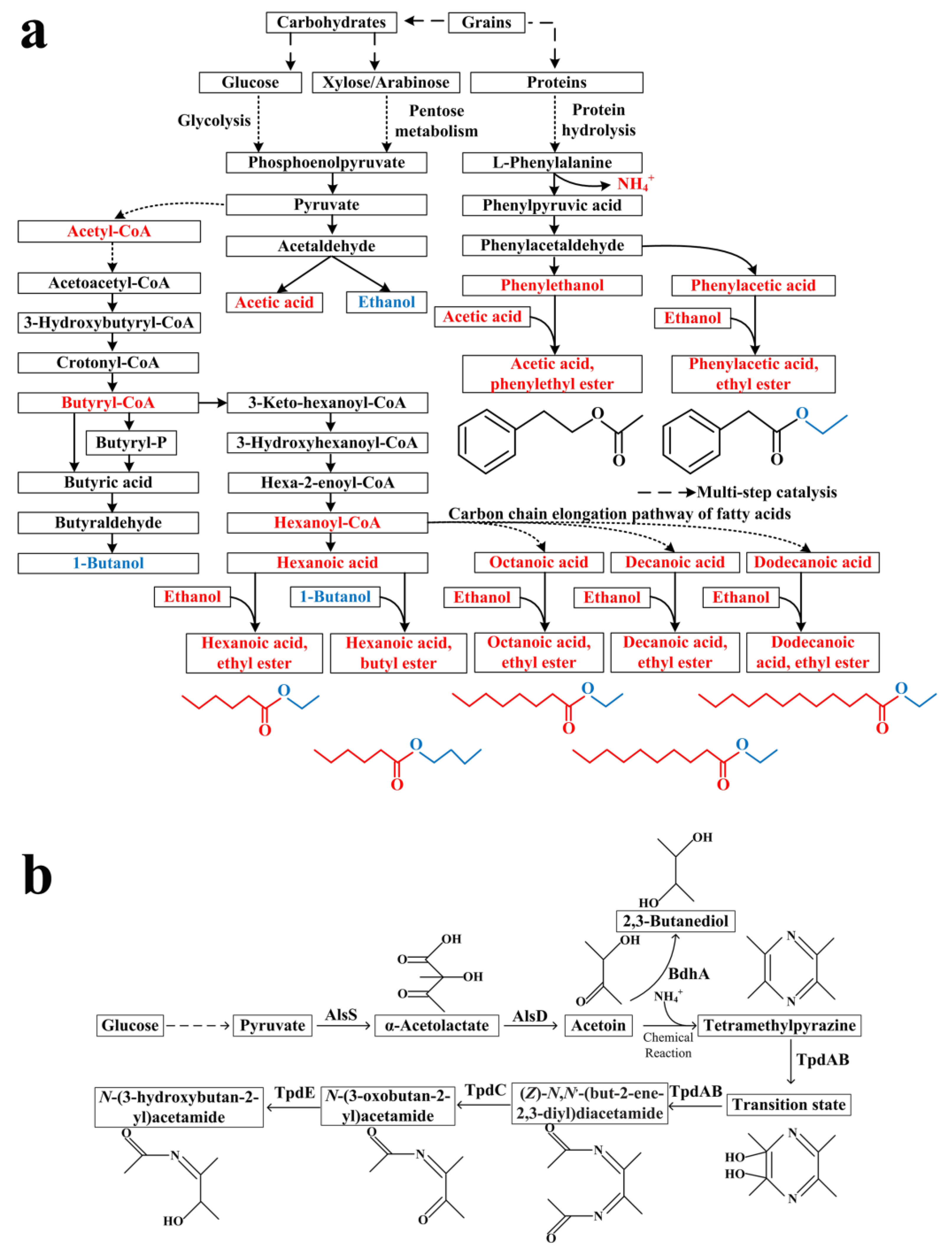
3.4. The Synthesizing and Degrading Metabolic Pathways of Key Flavor Chemicals
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, L. Research trends in Jiang-flavor baijiu fermentation: From fermentation microecology to environmental ecology. J. Food Sci. 2022, 87, 1362–1374. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhao, J.; Liu, X.; Zhang, C.; Zhao, Z.; Li, X.; Sun, B. Flavor mystery of Chinese traditional fermented baijiu: The great contribution of ester compounds. Food Chem. 2022, 369, 130920. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, Y.; Huang, H.; Pang, Z.; Fu, Z.; Niu, J.; Zhang, C.; Li, W.; Li, X.; Sun, B. Correlation between microbial communities and flavor compounds during the fifth and sixth rounds of sauce-flavor baijiu fermentation. Food Res. Int. 2021, 150, 110741. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Huang, H.; Lu, H.; Wu, M.; Lin, M.; Zhang, C.; Zhao, Z.; Li, W.; Zhang, C.; Li, X.; et al. Characterization of an Aspergillus niger for efficient fatty acid ethyl ester synthesis in aqueous phase and the molecular mechanism. Front. Microbiol. 2021, 12, 820380. [Google Scholar] [CrossRef] [PubMed]
- Gan, S.H.; Yang, F.; Sahu, S.K.; Luo, R.Y.; Liao, S.L.; Wang, H.Y.; Jin, T.; Wang, L.; Zhang, P.F.; Liu, X.; et al. Deciphering the composition and functional profile of the microbial communities in Chinese moutai liquor Starters. Front. Microbiol. 2019, 10, 1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Xu, Y.; Chen, L. Diversity of yeast species during fermentative process contributing to Chinese Maotai-flavour liquor making. Lett. Appl. Microbiol. 2012, 55, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Tian, Z.; Meng, W.; Li, Z. Microbial diversity and physicochemical characteristics of the maotai-flavored liquor fermentation process. J. Nanosci. Nanotechnol. 2020, 20, 4097–4109. [Google Scholar] [CrossRef]
- Wang, M.Y.; Yang, J.G.; Zhao, Q.S.; Zhang, K.Z.; Su, C. Research progress on flavor compounds and microorganisms of maotai flavor Baijiu. J. Food Sci. 2019, 84, 6–18. [Google Scholar] [CrossRef] [Green Version]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Sun, F.; Lin, W.; Zhang, S. AC-PCoA: Adjustment for confounding factors using principal coordinate analysis. PLoS Comput. Biol. 2022, 18, e1010184. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.C.; Liong, C.Y.; Jemain, A.A. Partial least squares-discriminant analysis (PLS-DA) for classification of high-dimensional (HD) data: A review of contemporary practice strategies and knowledge gaps. Analyst 2018, 143, 3526–3539. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Hu, G.; Lei, L.; Lin, L.; Wang, D.; Wu, J. Identification and aroma impact of volatile terpenes in moutai liquor. Int. J. Food Prop. 2016, 19, 1335–1352. [Google Scholar] [CrossRef]
- Xu, Y.; Jiang, Y.; Li, X.; Sun, B.; Teng, C.; Yang, R.; Xiong, K.; Fan, G.; Wang, W. Systematic characterization of the metabolism of acetoin and its derivative ligustrazine in Bacillus subtilis under micro-oxygen conditions. J. Agric. Food Chem. 2018, 66, 3179–3187. [Google Scholar] [CrossRef]
- Wei, Y.; Zou, W.; Shen, C.H.; Yang, J.G. Basic flavor types and component characteristics of Chinese traditional liquors: A review. J. Food Sci. 2020, 85, 4096–4107. [Google Scholar] [CrossRef]
- Milheiro, J.; Filipe-Ribeiro, L.; Cosmea, F. A simple, cheap and reliable method for control of 4-ethylphenol and 4-ethylguaiacol in red wines. Screening of fining agents for reducing volatile phenols levels in red wines. J. Chromatogr. B 2017, 1041–1042, 183–190. [Google Scholar] [CrossRef]
- Zhao, D.; Shi, D.; Sun, J.; Li, H.; Zhao, M.; Sun, B. Quantification and cytoprotection by vanillin, 4-methylguaiacol and 4-ethylguaiacol against AAPH-induced abnormal oxidative stress in HepG2 cells. RSC Adv. 2018, 8, 35474–35484. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Ao, Z.; Chui, W.; Shen, C.; Tao, W.; Zhang, S. Characterization of the aroma-active compounds in Daqu: A tradition Chinese liquor starter. Eur. Food Res. Technol. 2012, 234, 69–76. [Google Scholar] [CrossRef]
- Jiang, Q.; Wu, X.; Xu, Y.; Zhang, Y.; Wang, Z.; Shen, L.; Yang, W.; Sun, J.; Liu, Y. Microbial composition and dynamic succession during the Daqu production process of Northern Jiang-flavored liquor in China. 3 Biotech 2021, 11, 224. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.L.; Yaylayan, V.A. Model studies on the oxygen-induced formation of benzaldehyde from phenylacetaldehyde using pyrolysis GC-MS and FTIR. J. Agric. Food Chem. 2008, 56, 10697–10704. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Chen, L.; Peng, Z.; Zhang, Q.; Huang, W.; Yang, F.; Du, G.; Zhang, J.; Wang, L. Analysis of environmental driving factors on core functional community during Daqu fermentation. Food Res. Int. 2022, 157, 111286. [Google Scholar] [CrossRef] [PubMed]
- Bardou, P.; Mariette, J.; Escudie, F.; Djemiel, C.; Klopp, C. jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Du, H.; Zhang, Y.; Xu, Y. Environmental microbiota drives microbial succession and metabolic profiles during Chinese liquor fermentation. Appl. Environ. Microbiol. 2018, 84, aem.02369-17. [Google Scholar] [CrossRef] [Green Version]
- Hölker, U.; Lenz, J. Solid-state fermentation—Are there any biotechnological advantages? Curr. Opin. Microbiol. 2005, 8, 301–306. [Google Scholar] [CrossRef]
- Tang, J.; Liu, Y.; Lin, B.; Zhu, H.; Jiang, W.; Yang, Q.; Chen, S. Effects of ultra-long fermentation time on the microbial community and flavor components of light-flavor Xiaoqu Baijiu based on fermentation tanks. World J. Microbiol. Biotechnol. 2021, 38, 3. [Google Scholar] [CrossRef]
- Lin, B.; Tang, J.; Yang, Q.; Su, Z.; Zhu, L.; Li, Q.; Jiang, W.; Zhang, L.; Liu, Y.; Chen, S. Microbial succession and its effect on key aroma components during light-aroma-type Xiaoqu Baijiu brewing process. World J. Microbiol. Biotechnol. 2022, 38, 166. [Google Scholar] [CrossRef]
- Ma, S.; Luo, H.; Zhao, D.; Qiao, Z.; Zheng, J.; An, M.; Huang, D. Environmental factors and interactions among microorganisms drive microbial community succession during fermentation of Nongxiangxing daqu. Bioresour. Technol. 2022, 345, 126549. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, L.; Tan, Y.; Wang, H.; Yang, F.; Chen, L.; Hao, F.; Lv, X.; Du, H.; Xu, Y. Effect of Pichia on shaping the fermentation microbial community of sauce-flavor Baijiu. Int. J. Food Microbiol. 2021, 336, 108898. [Google Scholar] [CrossRef]
- Wu, Q.; Zhu, Y.; Fang, C.; Wijffels, R.H.; Xu, Y. Can we control microbiota in spontaneous food fermentation?—Chinese liquor as a case example. Trends Food Sci. Technol. 2021, 110, 321–331. [Google Scholar] [CrossRef]
- Wolfe, B.E.; Button, J.E.; Santarelli, M.; Dutton, R.J. Cheese rind communities provide tractable systems for in situ and in vitro studies of microbial diversity. Cell 2014, 158, 422–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Ji, M.; Xing, M.; Wang, X.; Xu, Y. The effects of dynamic bacterial succession on the flavor metabolites during Baijiu fermentation. Food Res. Int. 2021, 140, 109860. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Huang, Z.; Wu, L.; Wu, Q.; Guo, W.; Zhao, W.; Liu, B.; Zhang, W.; Rao, P.; Lv, X.; et al. Microbial diversity and flavor of Chinese rice wine (Huangjiu): An overview of current research and future prospects. Curr. Opin. Food Sci. 2021, 42, 37–50. [Google Scholar] [CrossRef]
- Zhao, J.; Xu, Y.; Lu, H.; Zhao, D.; Zheng, J.; Lin, M.; Liang, X.; Ding, Z.; Dong, W.; Yang, M.; et al. Molecular mechanism of LIP05 derived from Monascus purpureus YJX-8 for synthesizing fatty acid ethyl esters under aqueous phase. Front. Microbial. 2023, 14, 1107104. [Google Scholar]
- Dellomonaco, C.; Clomburg, J.M.; Miller, E.N.; Gonzalez, R. Engineered reversal of the β-oxidation cycle for the synthesis of fuels and chemicals. Nature 2011, 476, 355–361. [Google Scholar] [CrossRef]
- Kruis, A.J.; Bohnenkamp, A.C.; Patinios, C.; van Nuland, Y.M.; Levisson, M.; Mars, A.E.; van den Berg, C.; Kengen, S.W.M.; Weusthuis, R.A. Microbial production of short and medium chain esters: Enzymes, pathways, and applications. Biotechnol. Adv. 2019, 37, 107407. [Google Scholar] [CrossRef]
- Lai, J.; Huang, H.; Lin, M.; Xu, Y.; Li, X.; Sun, B. Enzyme catalyzes ester bond synthesis and hydrolysis: The key step for sustainable usage of plastics. Front. Microbiol. 2023, 14, 1113705. [Google Scholar]
- Shi, X.; Zhao, S.; Chen, S.; Han, X.; Yang, Q.; Zhang, L.; Xia, X.; Tu, J.; Hu, Y. Tetramethylpyrazine in Chinese baijiu: Presence, analysis, formation, and regulation. Front. Nutr. 2022, 9, 1004435. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, C.; Li, X.; Sun, B.; Eldin, A.A.; Jia, Y. A combinational optimization method for efficient synthesis of tetramethylpyrazine by the recombinant Escherichia coli. Biochem. Eng. J. 2018, 129, 33–43. [Google Scholar] [CrossRef]
- Kutanovas, S.; Stankeviciute, J.; Urbelis, G.; Tauraite, D.; Rutkiene, R.; Meskys, R. Identification and characterization of a tetramethylpyrazine catabolic pathway in Rhodococcus jostii TMP1. Appl. Environ. Microbiol. 2013, 79, 3649–3657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Wang, Z.; Sun, B. Low quantity but critical contribution to flavor: Review of the current understanding of volatile sulfur-containing compounds in Baijiu. J. Food Comp. Anal. 2021, 103, 104079. [Google Scholar] [CrossRef]
- Devanthi, P.V.P.; Gkatzionis, K. Soy sauce fermentation: Microorganisms, aroma formation, and process modification. Food Res. Int. 2019, 120, 364–374. [Google Scholar] [CrossRef] [PubMed]
- van Hijum, S.A.; Vaughan, E.E.; Vogel, R.F. Application of state-of-art sequencing technologies to indigenous food fermentations. Curr. Opin. Biotechnol. 2013, 24, 178–186. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Number | The Data and Position of Samples | Sample Number for Sequencing in Duplicate |
|---|---|---|
| CS_0S | Round 1b, the 0 day, fermented grains in upper layer of Jiaochi | CS_0S_1, CS_0S_2 |
| CS_0Z | Round 1b, the 0 day, fermented grains in middle layer of Jiaochi | CS_0Z_1, CS_0Z_2 |
| CS_0X | Round 1b, the 0 day, fermented grains in bottom layer of Jiaochi | CS_0X_1, CS_0X_2 |
| CS_7S | Round 1b, the 7th day, fermented grains in upper layer of Jiaochi | CS_7S_1, CS_7S_2 |
| CS_7Z | Round 1b, the 7th day, fermented grains in middle layer of Jiaochi | CS_7Z_1, CS_7Z_2 |
| CS_7X | Round 1b, the 7th day, fermented grains in bottom layer of Jiaochi | CS_7X_1, CS_7X_2 |
| CS_14S | Round 1b, the 14th day, fermented grains in upper layer of Jiaochi | CS_14S_1, CS_14S_2 |
| CS_14Z | Round 1b, the 14th day, fermented grains in middle layer of Jiaochi | CS_14Z_1, CS_14Z_2 |
| CS_14X | Round 1b, the 14th day, fermented grains in bottom layer of Jiaochi | CS_14X_1, CS_14X_2 |
| CS_21S | Round 1b, the 21st day, fermented grains in upper layer of Jiaochi | CS_21S_1, CS_21S_2 |
| CS_21Z | Round 1b, the 21st day, fermented grains in middle layer of Jiaochi | CS_21Z_1, CS_21Z_2 |
| CS_21X | Round 1b, the 21st day, fermented grains in bottom layer of Jiaochi | CS_21X_1, CS_21X_2 |
| CS_CJS | Round 1b, fermented grains in up layer of Jiaochi at the end of fermentation | CS_CJS_1, CS_CJS_2 |
| CS_CJZ | Round 1b, fermented grains in middle layer of Jiaochi at the end of fermentation | CS_CJZ_1, CS_CJZ_2 |
| CS_CJX | Round 1b, fermented grains in bottom layer of Jiaochi at the end of fermentation | CS_CJX_1, CS_CJX_2 |
| C2_0S | Round 2, the 0 day, fermented grains in up layer of Jiaochi | C2_0S_1, C2_0S_2 |
| C2_0Z | Round 2, the 0 day, fermented grains in middle layer of Jiaochi | C2_0Z_1, C2_0Z_2 |
| C2_0X | Round 2, the 0 day, fermented grains in bottom layer of Jiaochi | C2_0X_1, C2_0X_2 |
| C2_7S | Round 2, the 7th day, fermented grains in up layer of Jiaochi | C2_7S_1, C2_7S_2 |
| C2_7Z | Round 2, the 7th day, fermented grains in middle layer of Jiaochi | C2_7Z_1, C2_7Z_2 |
| C2_7X | Round 2, the 7th day, fermented grains in bottom layer of Jiaochi | C2_7X_1, C2_7X_2 |
| C2_14S | Round 2, the 14th day, fermented grains in up layer of Jiaochi | C2_14S_1, C2_14S_2 |
| C2_14Z | Round 2, the 14th day, fermented grains in middle layer of Jiaochi | C2_14Z_1, C2_14Z_2 |
| C2_14X | Round 2, the 14th day, fermented grains in bottom layer of Jiaochi | C2_14X_1, C2_14X_2 |
| C2_21S | Round 2, the 21st day, fermented grains in up layer of Jiaochi | C2_21S_1, C2_21S_2 |
| C2_21Z | Round 2, the 21st day, fermented grains in middle layer of Jiaochi | C2_21Z_1, C2_21Z_2 |
| C2_21X | Round 2, the 21st day, fermented grains in bottom layer of Jiaochi | C2_21X_1, C2_21X_2 |
| C2_CJS | Round 2, fermented grains in up layer of Jiaochi at the end of fermentation | C2_CJS_1, C2_CJS_2 |
| C2_CJZ | Round 2, fermented grains in middle layer of Jiaochi at the end of fermentation | C2_CJZ_1, C2_CJZ_2 |
| C2_CJX | Round 2, fermented grains in bottom layer of Jiaochi at the end of fermentation | C2_CJX_1, C2_CJX_2 |
| C3_0S | Round 3, the 0 day, fermented grains in up layer of Jiaochi | C3_0S_1, C3_0S_2 |
| C3_0Z | Round 3, the 0 day, fermented grains in middle layer of Jiaochi | C3_0Z_1, C3_0Z_2 |
| C3_0X | Round 3, the 0 day, fermented grains in bottom layer of Jiaochi | C3_0X_1, C3_0X_2 |
| C3_7S | Round 3, the 7th day, fermented grains in up layer of Jiaochi | C3_7S_1, C3_7S_2 |
| C3_7Z | Round 3, the 7th day, fermented grains in middle layer of Jiaochi | C3_7Z_1, C3_7Z_2 |
| C3_7X | Round 3, the 7th day, fermented grains in bottom layer of Jiaochi | C3_7X_1, C3_7X_2 |
| C3_14S | Round 3, the 14th day, fermented grains in up layer of Jiaochi | C3_14S_1, C3_14S_2 |
| C3_14Z | Round 3, the 14th day, fermented grains in middle layer of Jiaochi | C3_14Z_1, C3_14Z_2 |
| C3_14X | Round 3, the 14th day, fermented grains in bottom layer of Jiaochi | C3_14X_1, C3_14X_2 |
| C3_21S | Round 3, the 21st day, fermented grains in up layer of Jiaochi | C3_21S_1, C3_21S_2 |
| C3_21Z | Round 3, the 21st day, fermented grains in middle layer of Jiaochi | C3_21Z_1, C3_21Z_2 |
| C3_21X | Round 3, the 21st day, fermented grains in bottom layer of Jiaochi | C3_21X_1, C3_21X_2 |
| C3_CJS | Round 3, fermented grains in up layer of Jiaochi at the end of fermentation | C3_CJS_1, C3_CJS_2 |
| C3_CJZ | Round 3, fermented grains in middle layer of Jiaochi at the end of fermentation | C3_CJZ_1, C3_CJZ_2 |
| C3_CJX | Round 3, fermented grains in bottom layer of Jiaochi at the end of fermentation | C3_CJX_1, C3_CJX_2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Wu, M.; Niu, J.; Lin, M.; Zhu, H.; Wang, K.; Li, X.; Sun, B. Characteristics and Correlation of the Microbial Communities and Flavor Compounds during the First Three Rounds of Fermentation in Chinese Sauce-Flavor Baijiu. Foods 2023, 12, 207. https://doi.org/10.3390/foods12010207
Xu Y, Wu M, Niu J, Lin M, Zhu H, Wang K, Li X, Sun B. Characteristics and Correlation of the Microbial Communities and Flavor Compounds during the First Three Rounds of Fermentation in Chinese Sauce-Flavor Baijiu. Foods. 2023; 12(1):207. https://doi.org/10.3390/foods12010207
Chicago/Turabian StyleXu, Youqiang, Mengqin Wu, Jialiang Niu, Mengwei Lin, Hua Zhu, Kun Wang, Xiuting Li, and Baoguo Sun. 2023. "Characteristics and Correlation of the Microbial Communities and Flavor Compounds during the First Three Rounds of Fermentation in Chinese Sauce-Flavor Baijiu" Foods 12, no. 1: 207. https://doi.org/10.3390/foods12010207
APA StyleXu, Y., Wu, M., Niu, J., Lin, M., Zhu, H., Wang, K., Li, X., & Sun, B. (2023). Characteristics and Correlation of the Microbial Communities and Flavor Compounds during the First Three Rounds of Fermentation in Chinese Sauce-Flavor Baijiu. Foods, 12(1), 207. https://doi.org/10.3390/foods12010207