1. Introduction
The flavor precursors present in grapes and wine comprise a heterogeneous blend of mono- and disaccharide glycosides of volatile aglycones, which include monoterpenes, C
13-norisoprenoids, benzene derivatives, and long-chain aliphatic alcohols [
1]. The compositional analysis of glycoconjugates has been undertaken by several procedures [
2].
At the analytical level, a method using Fourier-transform infrared (FTIR) spectrometry and chemometric techniques allowed the rapid determination of C
13-norisoprenoidic and monoterpene glycoconjugates, with predictive errors of 14% and 15%, respectively [
3]. No improvement seems to have been published to date. Early experiments carried out the identification of glycoconjugates by exhaustive trifluoroacetylation of sugar hydroxyls followed by GC-MS analysis and subsequent comparisons of retention times (
tR) and mass spectra with synthetic standards [
4]. Recently, the high sensitivity and selectivity of the high-performance liquid chromatography (HPLC) system interfaced to the high-resolution mass spectrometry (HRMS) unit was exploited to characterize the GAPs in grapes [
5]. The experiments disclosed 20 monoterpene-diglycoside derivatives but could not confirm the structure of isobaric aglycones and their sugar residues.
At the preparative level, liquid chromatography is the recommended analytical technique. Countercurrent chromatography has been widely used and its applications reviewed [
6]. However, even in this case, purification still requires peracetylation of the glycosidic compounds. HPLC purification using reverse phase (RP) columns offers an effective alternative and this approach allowed isolation of 12 GAPs from “Moscato Giallo” grapes [
7]. Hydrophilic interaction liquid chromatography (HILIC) is a technique that uses a hydrophilic stationary phase and an aqueous–organic solvent as the mobile phase. HILIC provides a particularly powerful method for the separation of polar compounds and it has been successfully applied to the isolation of aroma precursors [
8].
The treatment of grape juice with lead (II) acetate precipitates phenol compounds such as cinnamic acid derivatives, flavonoids, and anthocyanins, without modifying the chemical structure of flavor glycoconjugates. This isolation protocol could be optimized by combining it with the HILIC method in sequence with reversed phase (RP) elution. This study therefore aimed to: (1) isolate and elucidate GAPs from “Moscato Rosa” grapes; (2) investigate the differences and synergies between HILIC and RP-HPLC stationary phases; and (3) confirm the effectiveness of lead acetate in precipitating phenols.
2. Materials and Methods
2.1. Chemicals
Analytical grade solvents were used for extraction and flash chromatography (FC); ethanol was HPLC-grade (VWR International, Fontenay-sous-Bois, France). Amberlite® XAD-2 resin was purchased from Fluka Chemie (Büchs, Switzerland) and lead acetate Pb(OAc)2 from Merck (Steinheim, Germany). Two stationary phases, cyanopropyl silica (CN, LiChroprep® CN 40–63 µm, Merck, Darmstadt, Germany) and octadecyl silica (ODS, LiChroprep® RP-18 40–63 µm, Merck, Darmstadt, Germany), were employed for FC.
2.2. Apparatus and Method
The preparative HPLC system consisted of a Merck Hitachi model L-7100 pump, an L-7400 UV detector, a D-7500 integrator, and a Rheodyne manual injector equipped with a 200 µL loop. Analytical separations were carried out using an Agilent 1100 series LC system consisting of a binary pump, a vacuum degasser, an autosampler with standard analytical head (100 µL), a column thermostat, and a 1200 series diode array detector (DAD) and evaporative light scattering detector (ELSD). The column oven temperature was fixed at 25 °C and the UV or DAD detectors were set at λ = 210 nm. The ELSD was connected in series with the DAD; the drift tube temperature was set at 50 °C and the pressure of nitrogen gas at 3.5 bar.
The analyses of components in RP mode were performed on a Synergi Hydro column (150 × 4.6 mm or 150 × 10.0 mm, 4 µm, 80 Å; Phenomenex, Torrance, CA, USA). The HILIC column was a ZIC SeQuant (150 × 10 mm, 5 µm, 200 Å; Merck). The injection volume was 10 µL.
Nuclear magnetic resonance (NMR) spectra were acquired using a Bruker Avance 400 spectrometer (1H at 400 MHz) and a 5 mm BBI probe. Chemical shifts were reported in ppm on the δ scale with the residual solvent signal used as an internal reference (CD3OD = 49.0, CD2HOD = 3.31, DHO = 4.72), proton coupling constants (J values in Hz) and multiplicities from 1D spectra, peak assignments from 1H,1H-COSY, and 1JCH (edited HSQC) and nJCH (HMBC) heterocorrelation experiments. NOESY data are reported as correlation map(s) between protons 1H ↔ 1H; HMBC data are reported as 13C→correlated to 1H.
LC-MS analyses were performed using an Agilent 1100 series LC system interfaced to a Bruker model Esquire_LC multiple ion trap mass spectrometer equipped with an atmospheric pressure interface electrospray (API-ES) chamber. Conditions for electrospray ionization mass spectrometry (ESI-MS) analysis of HPLC peaks in both positive and negative ion mode included a capillary voltage of 4000 V, a nebulizing pressure of 30.0 psi, a drying gas flow of 7 mL/min, and a temperature of 300 °C.
2.3. Plant Material
Vitis vinifera L. cultivar “Moscato Rosa” grapes (5.8 kg) were harvested in 2012 at the vineyard of Azienda Agricola ZENI, Sorni di Lavis, Trentino. After harvesting, the grapes (~35.0 °Brix) were crushed and sodium metabisulfite Na2S2O5 (300 mg/L) was added to the juice, which was promptly extracted.
2.4. Sample Preparation
Fresh grape juice (2 L) was centrifuged at 3000 rpm for 15 min at room temperature (RT). The supernatant (in batches of ~1 L) was passed three times through a column (25 cm × 4 cm i.d.) packed with Amberlite® XAD-2. The column was washed with water (2 L) and eluted with ethanol (2 L). The alcoholic eluate was desiccated in vacuo to give ~1.4 g of adsorbed organic material.
The dry extract was subjected to flash chromatography (8 cm × 4 cm i.d.) on CN with stepwise elution of 50% ethyl acetate in hexane (100 mL, fr. 1, 88 mg), 100% ethyl acetate (300 mL, fr. 2, 410 mg), and 100% ethanol (200 mL, fr. 3, 210 mg).
Aliquots of the pooled FC fractions 2 and 3 (190 mg) were dissolved in 6 mL of methanol and lead (II) acetate (0.8 g) was added. The mixture was stirred for two hours at RT then centrifuged at 3000 rpm for 15 min. The precipitate was washed with methanol and discarded. The ODS stationary phase (1 spoon) was added to the clear solution and the solvent was evaporated. The desiccated slurry was applied to an ODS column (8 cm × 4 cm i.d.) and chromatographed with a gradient of methanol in water (15:85, 30:70, 45:55, 60:40, 100:0).
2.5. Isolation of the Glycosidic Fractions
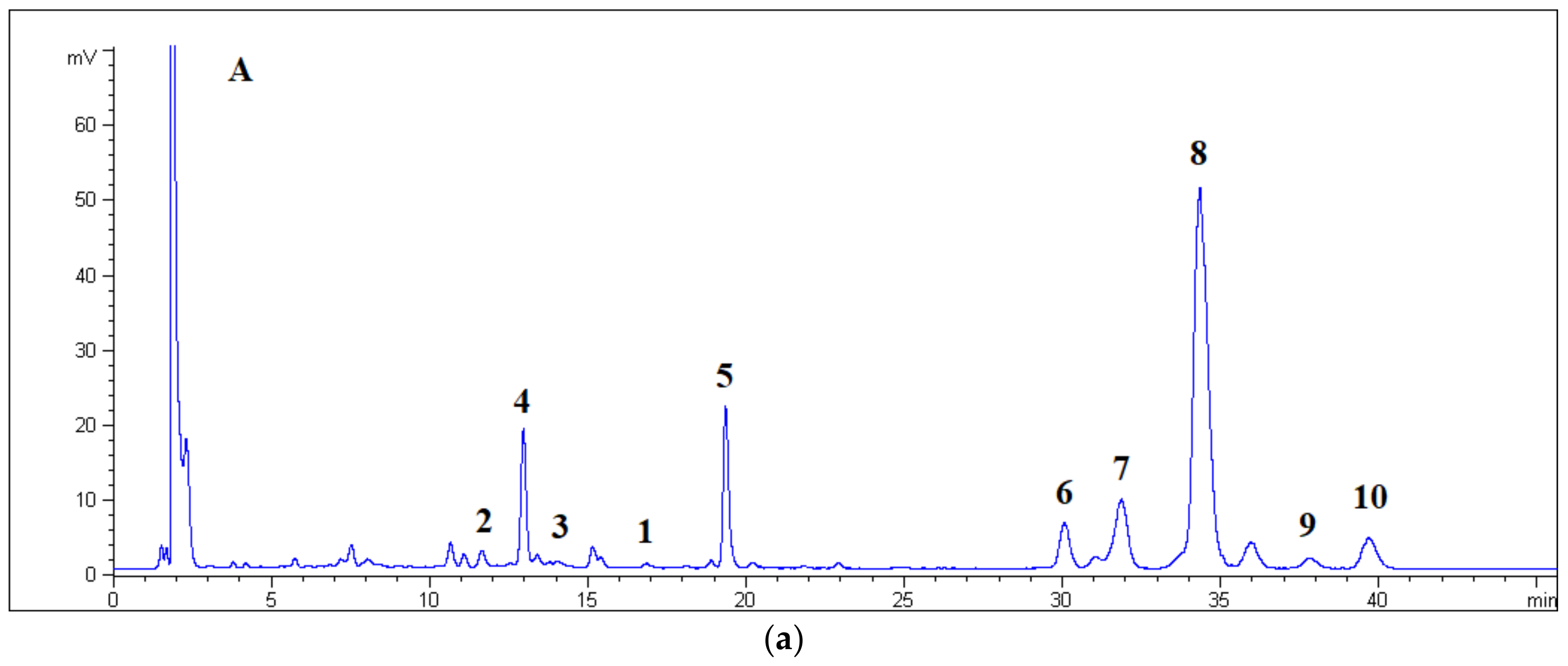
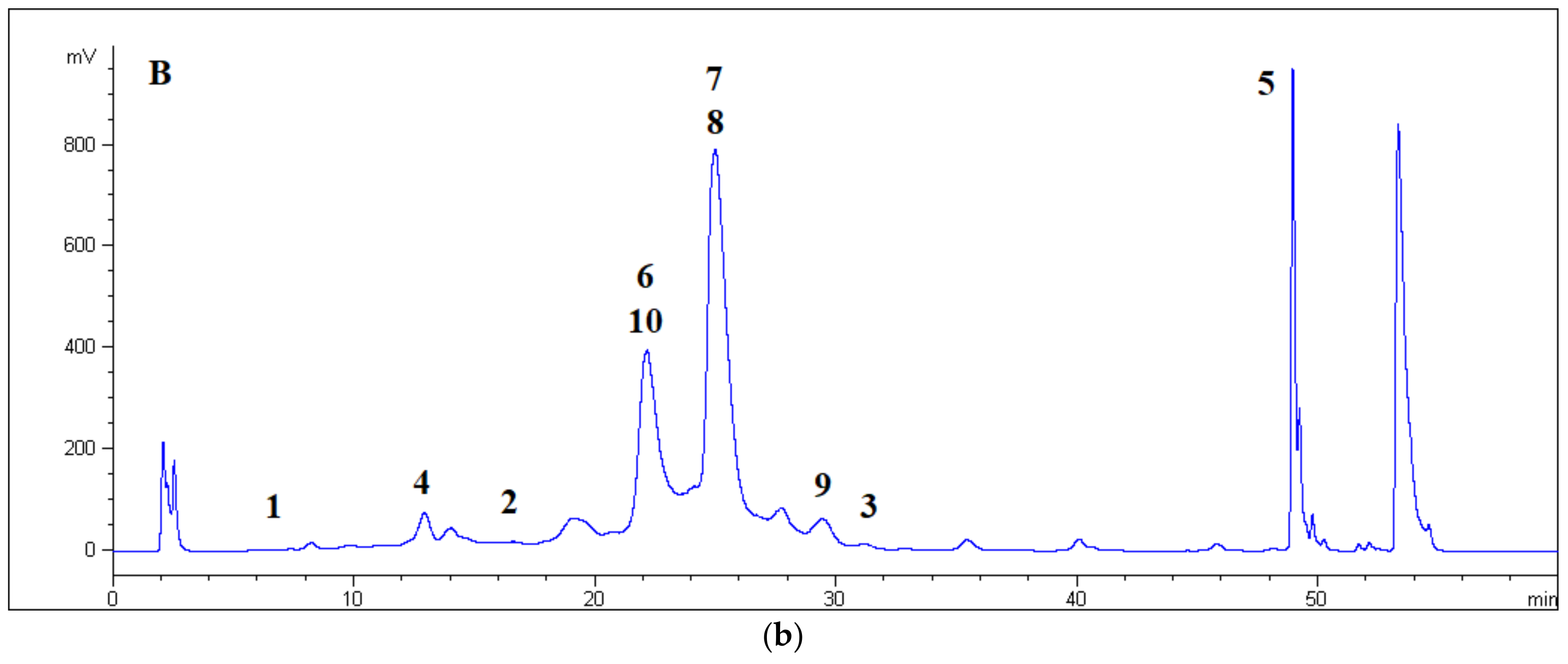
Central fractions (40 mg), eluted with methanol ranging from 30% to 45%, were dissolved in ethanol (1 mL) and purified by RP-HPLC, with the detection wavelength set at λ = 210 nm and the flux at 5 mL/min. The mobile phase consisted of ethanol/water 1:1 (A) and water (B) applied in the following stepped linear gradient: A = 40% at t = 0, A = 70% at t = 10, A = 70% at t = 30, A = 100% at t = 40, A = 40% at t = 42 min, and 13 min equilibration time.
The peaks of compounds 4, 5, 6, 7, 8, 9, and 10 were eluted at tR = 6.0 (2.8 mg), 10.4 (1.1 mg), 18.0 (1.6 mg), 19.2 (2.9 mg), 21.1 (4.9 mg), 23.7 (1.2 mg), and 24.8 (1.5 mg) min, respectively.
2.6. Comparative Chromatography of the Aroma Precursors
The analyses of components in RP mode were performed at a flux of 0.8 mL/min. The mobile phase consisted of ethanol/water 10:90 (A) and ethanol/water 50:50 (B) applied in the following stepped linear gradient: A = 90% at t = 0, A = 40% at t = 18, A = 40% at t = 40, A = 90% at t = 41 min, and 9 min as equilibration time. The analyses of components in normal phase (HILIC) mode were performed at a flux of 0.2 mL/min. The mobile phase consisted of acetonitrile (A) and water (1% v/v HCOOH) (B) applied in the following stepped linear gradient: A = 98% at t = 0, A = 94% at t = 40, A = 50% at t = 48, A = 50% at t = 55, A = 98% at t = 56 min, and 9 min equilibration time.
2.7. Precipitation of Model Phenols by Lead (II) Acetate
Three classes of compounds were selected for treatment with lead acetate: (A) simple phenols; (B) flavonoids; and (C) anthocyanins. Group A included resveratrol (0.1 mmol), cinnamic acid (0.1 mmol), coumaric acid (0.1 mmol), caffeic acid (0.1 mmol), and ferulic acid (0.1 mmol). Group B included naringin (0.025 mmol), diosmin (0.025 mmol), kaempferol (0.025 mmol), luteolin 7-O-glucoside (0.01 mmol), and quercetin (0.015 mmol). Group C included kaempferol 3-O-glucoside (0.005 mmol), cyanidin (0.01 mmol), myrtillin (0.005 mmol), and malvin (0.005 mmol).
For each group, a stock solution (10 mL) was divided into 10 identical aliquots (1.0 mL), one of which was used as a reference. Six aliquots were then treated with lead acetate at three different concentrations (0.5, 1.0, and 2.0 molar equivalents) and two pH levels. The acidic level was represented by the initial pH value of each solution, while the basic level was obtained by adjusting three aliquots (for each group) to a pH of ~8.0 with 0.1 M aqueous NaOH.
The appropriate amount of lead acetate was dissolved in methanol (0.5 mL), then mixed with an aliquot, stirred for 15 min at RT, and finally centrifuged at 2500 rpm for 15 min. The supernatant was passed through a 0.45 mm filter, evaporated to dryness, dissolved in methanol (or methanol/water 1:1) to a volume of 1.0 mL, and chromatographed by RP-HPLC. The detection wavelength was set at λ = 280 nm. For Group A, elution started at 0.25% v/v acetonitrile in water (1% v/v FA), then acetonitrile was increased to 50% over 30 min and maintained at 50% for 10 min with the flux at 1 mL/min. For Group B, elution started at 0.25% v/v acetonitrile in water (1% v/v FA), then acetonitrile was increased to 50% over 20 min and maintained at 50% for 20 min with the flux at 1 mL/min. For Group C, elution started at 10% v/v acetonitrile in water (0.2% v/v TFA), then acetonitrile was increased as follows: to 10% at t = 5, 15% at t = 20, 15% at t = 25, 18% at t = 30, and 40% at t = 50 min, and maintained for 10 min with the flux at 0.5 mL/min.
4. Discussion
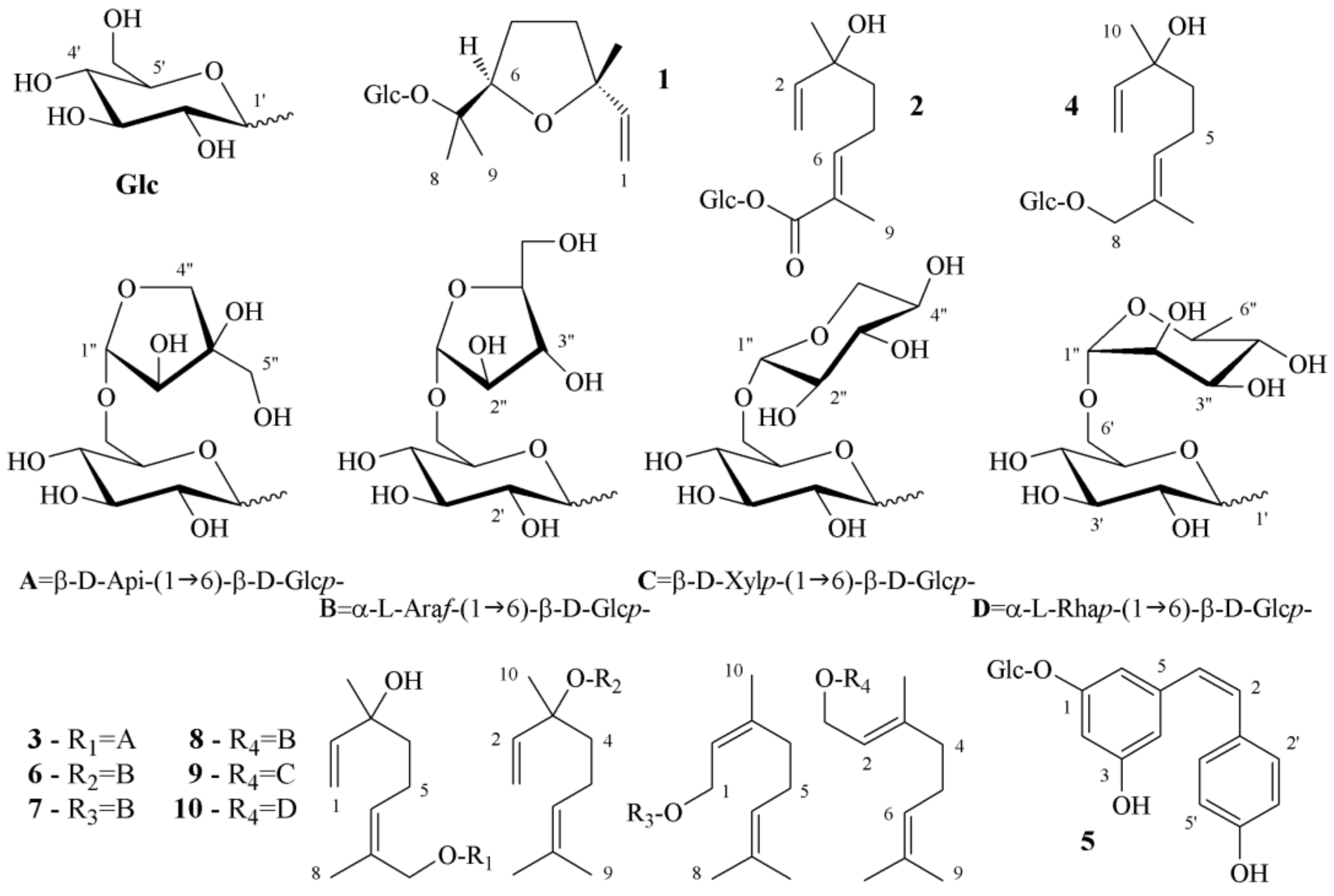
Aim 1. In this paper, the structural formula of seven GAPs were characterized: three (5, 6, and 8) were previously identified in grapes, two (7, 10) were obtained by synthesis, whereas 4 was isolated from Viburnum orientale and 9 from Zingiber officinale. Herein, we report four compounds (4, 7, 9, and 10) for the first time in a grape juice. Geraniol was found to be linked to several disaccharides (8, 9, and 10) whereas arabinose occurs as furanoside (6, 7, and 8) rather than the common vicianoside. Notably, the primeverose and rutinose sugars were also present. Finally, (E)-hydroxylinalool (4) was identified in “Moscato Rosa”, whilst the (Z) isomer (3) occurred in “Moscato Giallo”.
Aim 2. The separation of GAP is a consequence of the type and the number of hydrophilic functional groups (HFGs) that can form hydrogen bonds. Few HFGs in the aglycone moiety will increase the tR during RP elution, whereas many HFGs in the carbohydrate moiety will increase the tR during normal phase HILIC elution. In fact, the sugar portion is poorly soluble in the hydrophobic layer of the bonded ODS particles and, vice versa, the aglycone portion is poorly soluble in the aqueous layer covering the bonded ZIC particles. The chemical structure of (Z)-piceid (5) was different so its tR was not comparable to those of GAPs (1–4, 6–10).
The order of elution in the RP mode can be rationalized by examining the chemical structure of the aglycone moiety. With respect to the geraniol molecule (8, 9, and 10), compounds 2, 3, and 4 had an additional hydroxyl group. These three compounds were first eluted, irrespective of their number of monosaccharide units. In fact, 3 (which comprised two sugar units) showed a higher tR than 4 (which only comprised one sugar unit). The discriminating factor between 3 and 4 was therefore the configuration at the double bond of the hydroxylinalool, which was found to be Z and E, respectively. With respect to the geraniol molecule, compound 1 possessed an ether functionality, which acts only as acceptor of hydrogen bonds such that 1 eluted at an intermediate tR. Below were the compounds 6, 7, and 8, which had identical disaccharide moieties, whereas their terpenoid portion comprised linalool, nerol, and geraniol, respectively. Again, it was possible to observe discrimination between geometric isomers. The last eluted compounds were two glycosylated geraniol molecules, which were characterized by the presence of xylopyranose (9) and rhamnose (10) units. The xylopyranose sugar possessed one less hydroxyl group than the arabinofuranose; the rhamnose unit had an apolar methyl group.
The normal-phase HILIC allowed partitioning of the glycosidic moieties, differentiating monosaccharides and disaccharides. The compounds that were glycosylated by one sugar unit were eluted first because they were less soluble in the aqueous layer covering the stationary phase. Moreover, compounds 1, 4, and 2 showed the reverse order of elution with respect to the ODS stationary phase. The hydroxylinalool disaccharide of 3 became the last eluted aroma precursor by HILIC. Focusing on the set of five disaccharides linked to hydrophobic monoterpenes (i.e., 6–10), the least retained compound appeared to again be compound 6. The next eluted compound was 10, with a rhamnose unit. Compounds 7 and 8 had identical disaccharide linkages and eluted together because they differed only in their configuration at the double bond of the terpene.
Aim 3. The use of lead (II) acetate for several purposes has been reported in recent literature [
15] and is considered an official method of analysis [
16]. However, a detailed mechanism of action has not yet been described. Two articles concerning the purification of anthocyanins from cranberry juice [
17] or wine [
18] using lead (II) acetate reported the quantity of precipitated anthocyanins, determined either by a pH differential [
17] or a spectrophotometric method [
18]. In summary, the literature stated that: (i) compounds possessing an
o-dihydroxy benzene moiety are easily precipitated, while simple phenols form lead salts to a low extent; and (ii) basic lead acetate [Pb(OAc)
2·2Pb(OH)
2] is more effective than neutral lead acetate [Pb(OAc)
2].
It is evident from
Table 2 the importance of an
o-dihydroxy benzene moiety. In fact, caffeic acid forms the insoluble lead salt exhaustively even at acidic pH and at half-molar equivalence. In comparison, the complete precipitation of coumaric and ferulic acids can only be achieved at basic pH and with two molar equivalents. The carboxylic functionality of cinnamic acid acts to a certain extent because cinnamic acid does not possess a phenol group. The presence of an
m-dihydroxy benzene moiety is ineffective, so most of the stilbenoid resveratrol remained dissolved. The flavanol naringin and the flavone diosmin formed insoluble salts partially because they lacked the
o-dihydroxy benzene group. Interestingly, the flavonol kaempferol was easily precipitated, suggesting that the hydroxyl at the C-3 position played somewhat of a role. This was confirmed by the decreased precipitation of kaempferol 3-
O-glucoside. The flavone luteolin 7-
O-glucoside and the flavonol quercetin formed insoluble complexes at acidic pH and at half-molar equivalence. Finally, the anthocyans cyanidin and mirtillin could be precipitated at an acidic pH, whereas malvin could only be precipitated at a basic pH. In conclusion, data confirmed our previous statements in the literature and underlined that the effectiveness of precipitation is derived from the structural features of individual phenolic compounds. In addition, a larger extent of precipitation might be achieved by using excess lead acetate rather than basic pH conditions; this avails also because the content of phenolic compounds in crude natural extracts is difficult to establish. The lead acetate procedure enables uninteresting phenolic compounds to be discarded, but they cannot be easily recovered after precipitation.
Preliminary experiments suggest polyvinylpolypyrrolidone (PVPP) has different binding properties compared to lead (II) acetate.





{kind=link}
{kind=link}
{kind=link}