Comparison of Targeted (HPLC) and Nontargeted (GC-MS and NMR) Approaches for the Detection of Undeclared Addition of Protein Hydrolysates in Turkey Breast Muscle

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Sampling and Adulteration of Turkey Breast
2.3. Amino Acid Analysis Using HPLC-UV/VIS
2.3.1. Sample Preparation for Amino Acid Analysis
2.3.2. Amino Acid Content Determination
2.4. GC-MS-Based Metabolomics Analyses
2.4.1. Sample Preparation for Metabolomics Study of White Breast Meat
2.4.2. GC-MS Measurements and Data Processing
2.4.3. Amino Acid Quantification
2.5. 1H NMR-Based Metabolomics Analyses
2.5.1. Sample Preparation for Metabolomics Study of White Breast Meat
2.5.2. 1H NMR Spectroscopy, Data Processing and Identification of the Signals
2.6. Statistical Analysis
3. Results and Discussion
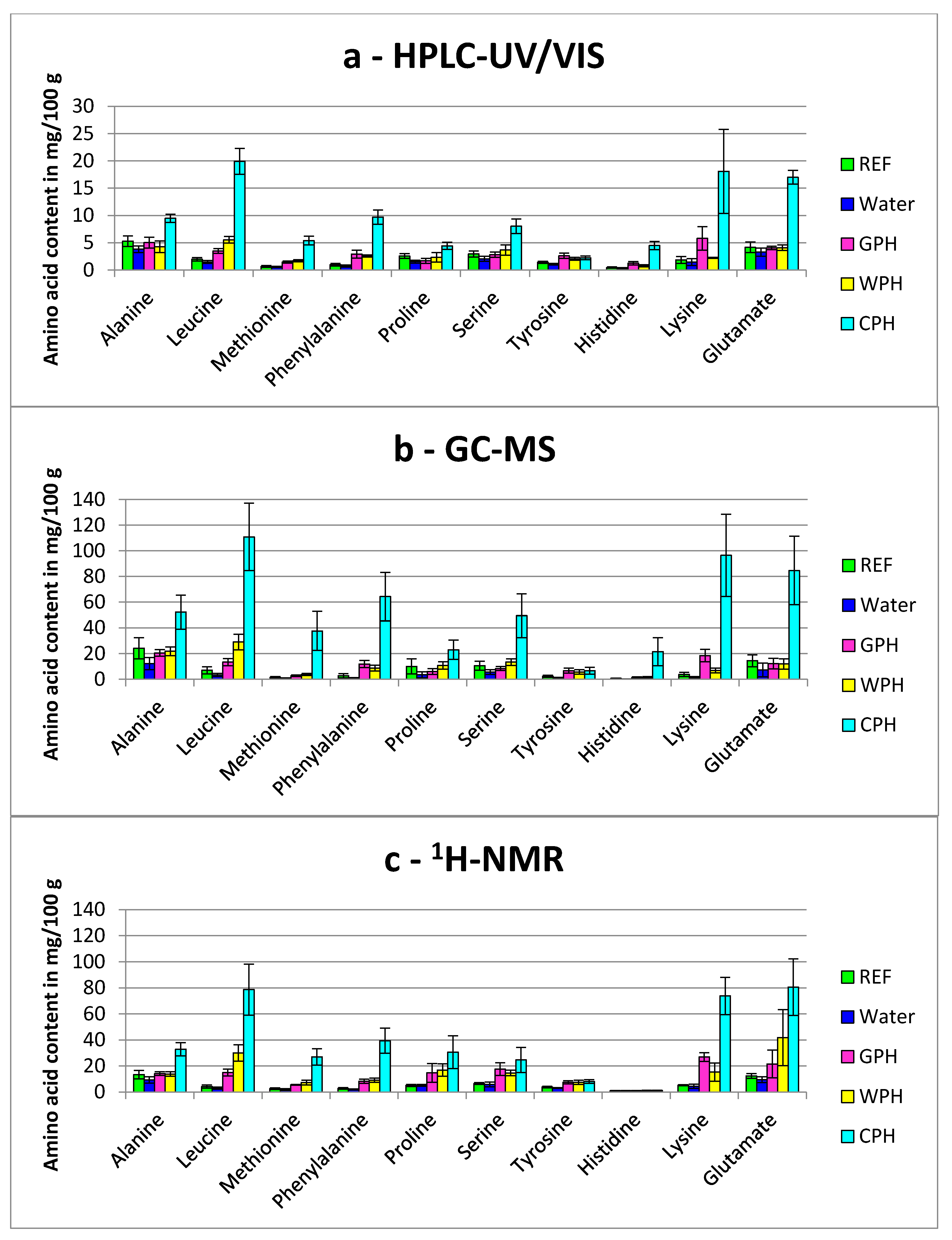
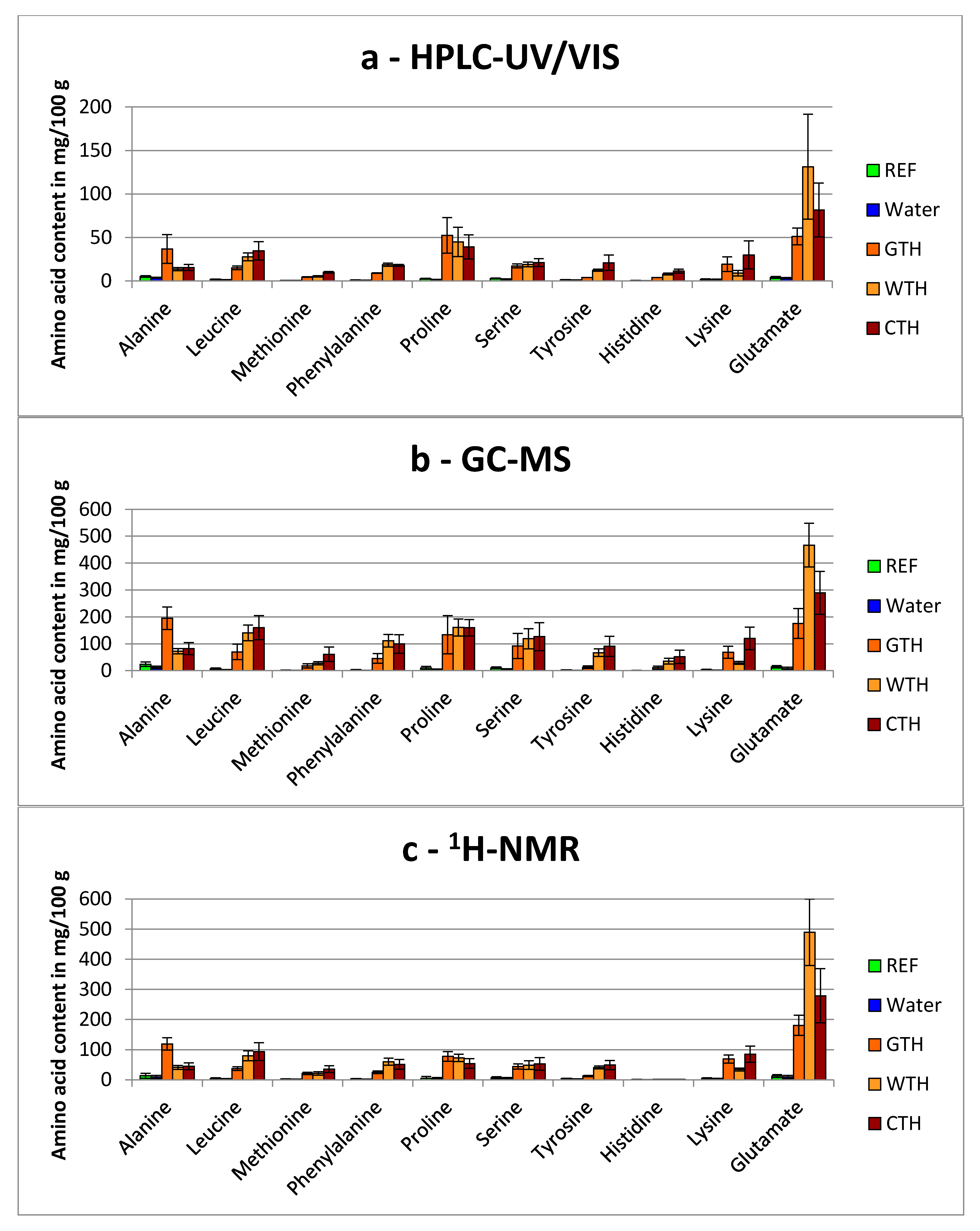
3.1. Capability of Amino Acid Profiling for the Detection of Added Protein Hydrolysates
3.1.1. Comparison of Partial Hydrolyzed Wheat-, Gelatin- and Casein-Hydrolysates
3.1.2. Comparison of Total Hydrolyzed Wheat-, Gelatin- and Casein-Hydrolysates
3.1.3. General Aspects Regarding the Detection of Free Amino Acids in Treated Breast Muscles
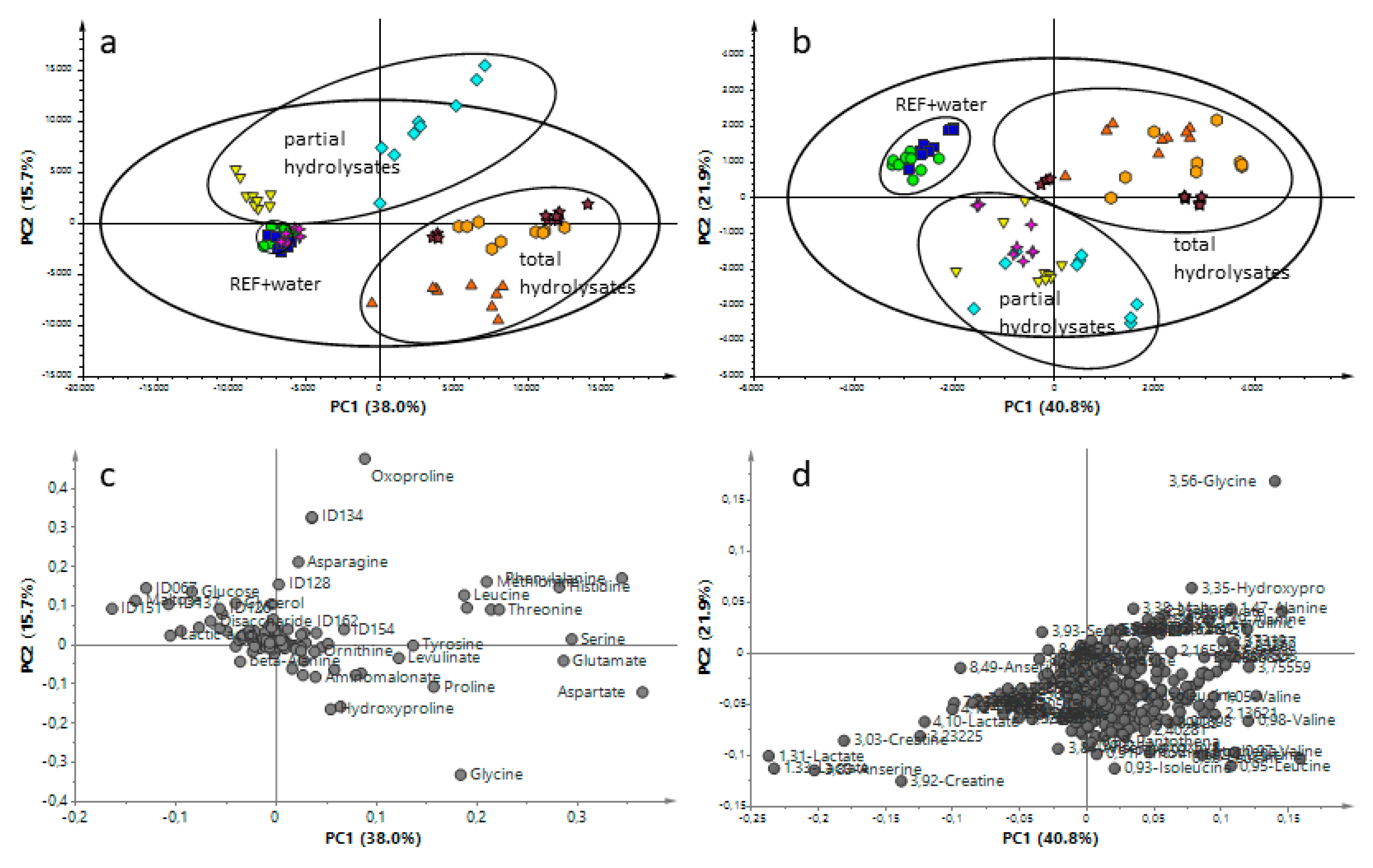
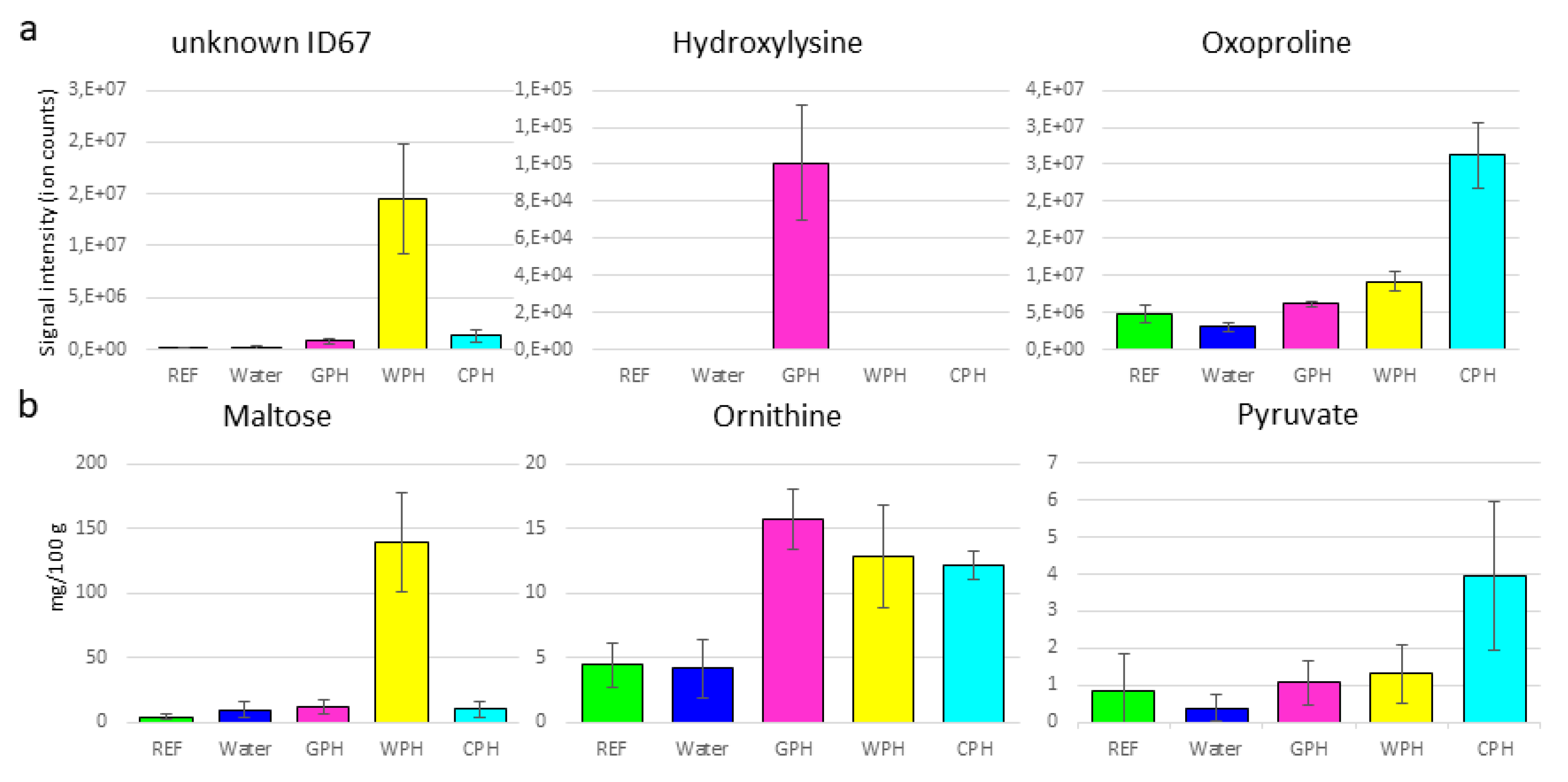
3.2. Metabolomics Approaches to Obtain Additional Information Regarding Hydrolysate-Treated Samples Independently of the Hydrolyzation Degree
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Trivedi, D.K.; Hollywood, K.A.; Rattray, N.J.W.; Ward, H.; Trivedi, D.K.; Greenwood, J.; Ellis, D.I.; Goodacre, R. Meat, the metabolites: An integrated metabolite profiling and lipidomics approach for the detection of the adulteration of beef with pork. Analyst 2016, 141, 2155–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campmajó, G.; Saez-Vigo, R.; Saurina, J.; Nunez, O. High-performance liquid chromatography with fluorescence detection fingerprinting combined with chemometrics for nut classification and the detection and quantitation of almond-based product adulterations. Food Control 2020, 114, 107265. [Google Scholar] [CrossRef]
- Ballin, N.Z. Authentication of meat and meat products. Meat Sci. 2010, 86, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Kamruzzaman, M.; Makino, Y.; Oshita, S. Hyperspectral imaging in tandem with multivariate analysis and image processing for non-invasive detection and visualization of pork adulteration in minced beef. Anal. Methods 2015, 7, 7496–7502. [Google Scholar] [CrossRef]
- Hamm, R. Colloid Chemistry of Meat; the Water-Binding Capacity of Muscle Protein in Theory and Practice. In Kolloidchemie des Fleisches; Parey: Singhofen, Germany, 1972. [Google Scholar]
- Hahn, G.; Judas, M.; Berk, J. Forced locomotor activity improves walking ability of male turkeys and modifies carcass characteristics. Br. Poult. Sci. 2020, 61, 107–115. [Google Scholar] [CrossRef]
- ISO (the International Organization for Standardization) 1442:1997(en). Meat and Meat Products—Determination of Moisture Content (Reference Method); International Organization for Standardization: Geneva, Switzerland, 1997. [Google Scholar]
- European Union. Commission regulation (EC) No 543/2008 of 16 June 2008 laying down detailed rules for the application of Council Regulation (EC) No 1234/2007 as regards the marketing standards for poultrymeat. Off. J. Eur. Union 2008, 157. [Google Scholar]
- BVL L 07.00-64 Technische Regel, 2014-08. Untersuchung von Lebensmitteln—Bestimmung von Frei Vorliegenden Aminosäuren in Fleischerzeugnissen—Gaschromatographisches Verfahren (English: Analysing of Food—Determination of Free Amino Acids in Meat Products–Using GC); Beuth Verlag: Berlin, Germany, 2014. [Google Scholar]
- Rodionova, O.Y.; Pomerantsev, A.L. Chemometric tools for food fraud detection: The role of target class in non-targeted analysis. Food Chem. 2020, 317, 7. [Google Scholar] [CrossRef]
- Busch, D.U.; Krenz, O.; Schellenberg, D.A.; Huber, D.I.; Pavlovic, D.M. “Food fraud”—Analytische Herausforderungen von Lebensmittelverfälschungen. Z. Das Gesamte Lebensm. 2017, 424, 424–438. [Google Scholar]
- Wishart, D.S. Metabolomics: Applications to food science and nutrition research. Trends Food Sci. Amp. Technol. 2008, 19, 482–493. [Google Scholar] [CrossRef]
- Gallo, V.; Ragone, R.; Musio, B.; Todisco, S.; Rizzuti, A.; Mastrorilli, P.; Pontrelli, S.; Intini, N.; Scapicchio, P.; Triggiani, M.; et al. A Contribution to the Harmonization of Non-targeted NMR Methods for Data-Driven Food Authenticity Assessment. Food Anal. Methods 2019, 13, 530–541. [Google Scholar] [CrossRef]
- Sentandreu, M.A.; Sentandreu, E. Authenticity of meat products: Tools against fraud. Food Res. Int. 2014, 60, 19–29. [Google Scholar] [CrossRef]
- Böhme, K.; Calo-Mata, P.; Barros-Velázquez, J.; Ortea, I. Recent applications of omics-based technologies to main topics in food authentication. Trac. Trends Anal. Chem. 2019, 110, 221–232. [Google Scholar] [CrossRef]
- Hahn, G.; Spindler, M. Method of dissection of turkey carcases. Worlds Poult. Sci. J. 2002, 58, 179–197. [Google Scholar] [CrossRef]
- Amino Acid Mix Solution; Certificate (according to ISO Guide 31), Product 79248; GmbH Sigma-Aldrich Inc.: Darmstadt, Germany, 2020.
- Wagner, L.; Trattner, S.; Pickova, J.; Gomez-Requeni, P.; Moazzami, A.A. 1H NMR-based metabolomics studies on the effect of sesamin in Atlantic salmon (Salmo salar). Food Chem. 2014, 147, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Castejon, D.; Garcia-Segura, J.M.; Escudero, R.; Herrera, A.; Cambero, M.I. Metabolomics of meat exudate: Its potential to evaluate beef meat conservation and aging. Anal. Chim. Acta 2015, 901, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bankefors, J.; Kaszowska, M.; Schlechtriem, C.; Pickova, J.; Brannas, E.; Edebo, L.; Kiessling, A.; Sandstrom, C. A comparison of the metabolic profile on intact tissue and extracts of muscle and liver of juvenile Atlantic salmon (Salmo salar L.)—Application to a short feeding study. Food Chem. 2011, 129, 1397–1405. [Google Scholar] [CrossRef]
- Tatara, M.R.; Brodzki, A.; Pyz-Lukasik, R.; Pasternak, K.; Szpetnar, M. Sex-related Differences in Skeletal Muscle Amino Acid Concentrations in 20 Week Old Turkeys. J. Poult. Sci. 2012, 49, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Hamano, Y.; Kurimoto, Y. Effects of acetylated wood powder on growth performance, hepatic and muscular free amino acid profiles, and inosine 5′-monophosphate concentration of breast meat in broiler chickens. Br. Poult. Sci. 2016, 57, 643–654. [Google Scholar] [CrossRef]
- Rikimaru, K.; Takahashi, H. Evaluation of the meat from Hinai-jidori chickens and broilers: Analysis of general biochemical components, free amino acids, inosine 5′-monophosphate, and fatty acids. J. Appl. Poult. Res. 2010, 19, 327–333. [Google Scholar] [CrossRef]
- Girisuta, B.; Janssen, L.P.B.M.; Heeres, H.J. Green Chemicals: A Kinetic Study on the Conversion of Glucose to Levulinic Acid. Chem. Eng. Res. Des. 2006, 84, 339–349. [Google Scholar] [CrossRef]
- Shen, J.; Wyman, C.E. Hydrochloric acid-catalyzed levulinic acid formation from cellulose: Data and kinetic model to maximize yields. AIChE J. 2012, 58, 236–246. [Google Scholar] [CrossRef]
- Chen, Y.; Qiao, Y.; Xiao, Y.; Chen, H.; Zhao, L.; Huang, M.; Zhou, G. Differences in Physicochemical and Nutritional Properties of Breast and Thigh Meat from Crossbred Chickens, Commercial Broilers, and Spent Hens. Asian Australas. J. Anim. Sci. 2016, 29, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Gauza-Włodarczyk, M.; Kubisz, L.; Włodarczyk, D. Amino acid composition in determination of collagen origin and assessment of physical factors effects. Int. J. Biol. Macromol. 2017, 104, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Möller, J.; Sjödin, A. Determination of hydroxyproline in meat products by flow injection analysis. Fresenius’ Z. Anal. Chem. 1988, 329, 732–734. [Google Scholar]
- Fey, R. Über die Erfassung des Bindegewebsgehaltes von Fleischwaren mit der Hydroxyprolinmethode. Z. Lebensm. Unters. Forsch. 1977, 164, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Hofman, K.; Hall, B.; Cleaver, H.; Marshall, S. High-throughput quantification of hydroxyproline for determination of collagen. Anal. Biochem. 2011, 417, 289–291. [Google Scholar] [CrossRef]
- Herbert, K.R.; Williams, G.M.; Cooper, G.J.; Brimble, M.A. Synthesis of glycosylated 5-hydroxylysine, an important amino acid present in collagen-like proteins such as adiponectin. Org. Biomol. Chem. 2012, 10, 1137–1144. [Google Scholar] [CrossRef]
- Copley, S.D.; Frank, E.; Kirsch, W.M.; Koch, T.H. Detection and possible origins of aminomalonic acid in protein hydrolysates. Anal. Biochem. 1992, 201, 152–157. [Google Scholar] [CrossRef]
- Van Buskirk, J.J.; Kirsch, W.M.; Kleyer, D.L.; Barkley, R.M.; Koch, T.H. Aminomalonic acid: Identification in Escherichia coli and atherosclerotic plaque. Proc. Natl. Acad. Sci. USA 1984, 81, 722. [Google Scholar] [CrossRef] [Green Version]
- Baer, I.; de la Calle, B.; Taylor, P. 3-MCPD in food other than soy sauce or hydrolysed vegetable protein (HVP). Anal. Bioanal. Chem. 2010, 396, 443–456. [Google Scholar] [CrossRef]


 ), Water (
), Water (  ), GPH (
), GPH (  ), WPH (
), WPH (  ), CPH (
), CPH (  ), GTH (
), GTH (  ), WTH (
), WTH (  ) and CTH (
) and CTH (  ). GC-MS: The first component is explained by 38.0% and the second component by 15.7% of the variation (model parameters: R2X = 98.4%, Q2 = 72.8%, 17 components). 1H-NMR: The first and second components explained 40.8% and 21.9% of variation, respectively (model parameters: R2X = 98.5%, Q2 = 93.4%, 16 components).
), Water ( ), GPH ( ), WPH ( ), CPH ( ), GTH ( ), WTH ( ) and CTH ( ). GC-MS: The first component is explained by 38.0% and the second component by 15.7% of the variation (model parameters: R2X = 98.4%, Q2 = 72.8%, 17 components). 1H-NMR: The first and second components explained 40.8% and 21.9% of variation, respectively (model parameters: R2X = 98.5%, Q2 = 93.4%, 16 components).
). GC-MS: The first component is explained by 38.0% and the second component by 15.7% of the variation (model parameters: R2X = 98.4%, Q2 = 72.8%, 17 components). 1H-NMR: The first and second components explained 40.8% and 21.9% of variation, respectively (model parameters: R2X = 98.5%, Q2 = 93.4%, 16 components).
), Water ( ), GPH ( ), WPH ( ), CPH ( ), GTH ( ), WTH ( ) and CTH ( ). GC-MS: The first component is explained by 38.0% and the second component by 15.7% of the variation (model parameters: R2X = 98.4%, Q2 = 72.8%, 17 components). 1H-NMR: The first and second components explained 40.8% and 21.9% of variation, respectively (model parameters: R2X = 98.5%, Q2 = 93.4%, 16 components).

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wagner, L.; Peukert, M.; Kranz, B.; Gerhardt, N.; Andrée, S.; Busch, U.; Brüggemann, D.A. Comparison of Targeted (HPLC) and Nontargeted (GC-MS and NMR) Approaches for the Detection of Undeclared Addition of Protein Hydrolysates in Turkey Breast Muscle. Foods 2020, 9, 1084. https://doi.org/10.3390/foods9081084
Wagner L, Peukert M, Kranz B, Gerhardt N, Andrée S, Busch U, Brüggemann DA. Comparison of Targeted (HPLC) and Nontargeted (GC-MS and NMR) Approaches for the Detection of Undeclared Addition of Protein Hydrolysates in Turkey Breast Muscle. Foods. 2020; 9(8):1084. https://doi.org/10.3390/foods9081084
Chicago/Turabian StyleWagner, Liane, Manuela Peukert, Bertolt Kranz, Natalie Gerhardt, Sabine Andrée, Ulrich Busch, and Dagmar Adeline Brüggemann. 2020. "Comparison of Targeted (HPLC) and Nontargeted (GC-MS and NMR) Approaches for the Detection of Undeclared Addition of Protein Hydrolysates in Turkey Breast Muscle" Foods 9, no. 8: 1084. https://doi.org/10.3390/foods9081084
APA StyleWagner, L., Peukert, M., Kranz, B., Gerhardt, N., Andrée, S., Busch, U., & Brüggemann, D. A. (2020). Comparison of Targeted (HPLC) and Nontargeted (GC-MS and NMR) Approaches for the Detection of Undeclared Addition of Protein Hydrolysates in Turkey Breast Muscle. Foods, 9(8), 1084. https://doi.org/10.3390/foods9081084