
Comparative Metagenomic and Metatranscriptomic Analyses Reveal the Response of Black Soldier Fly (Hermetia illucens) Larvae Intestinal Microbes and Reduction Mechanisms to High Concentrations of Tetracycline

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Effect of Tetracycline on the BSFL Growth
2.2. Overview of Metagenomic and Metatranscriptomic Raw Data
2.3. Composition and Diversity of Microbial Communities in BSFL Gut
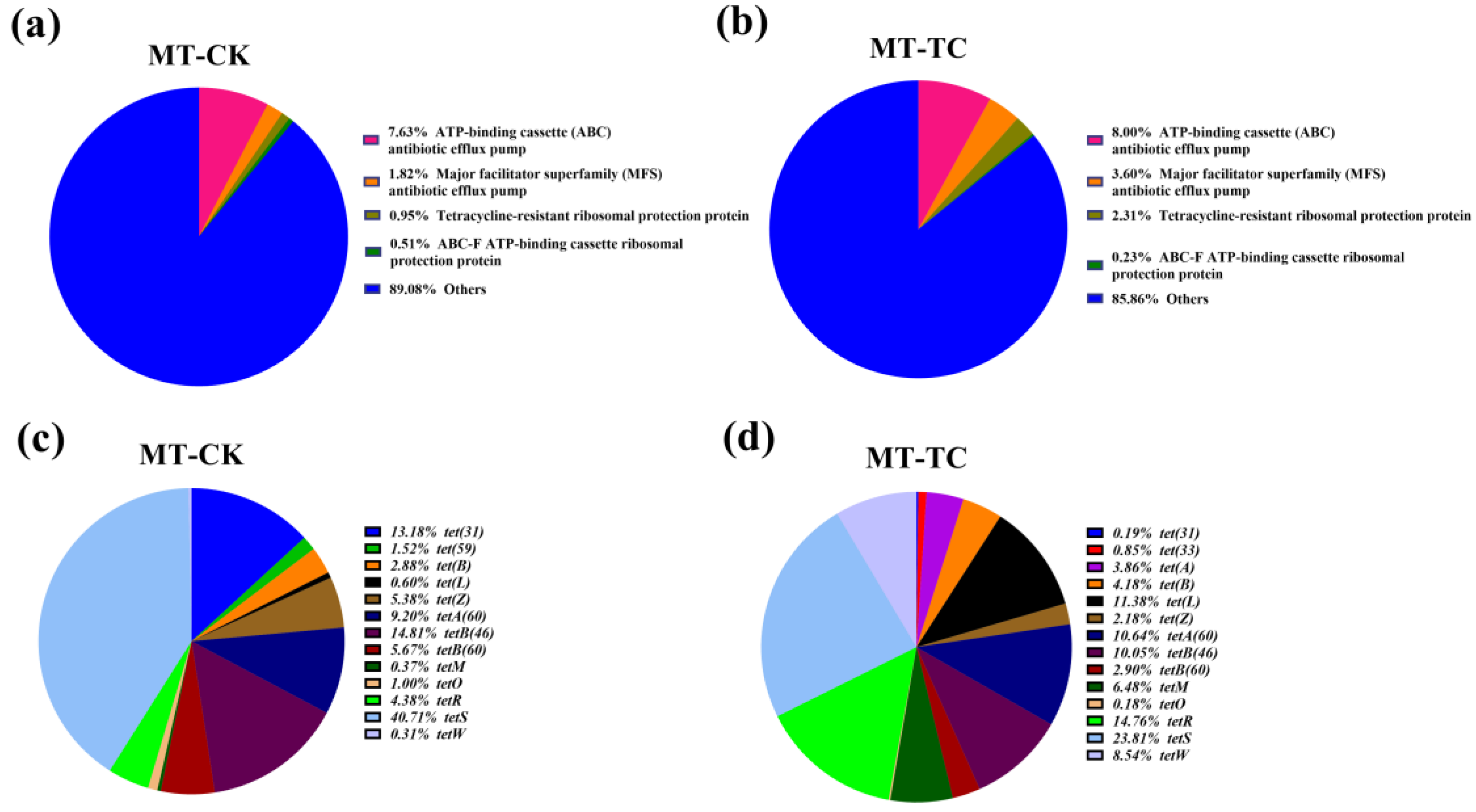
2.4. Antimicrobial Resistance (AMR) Gene Analysis
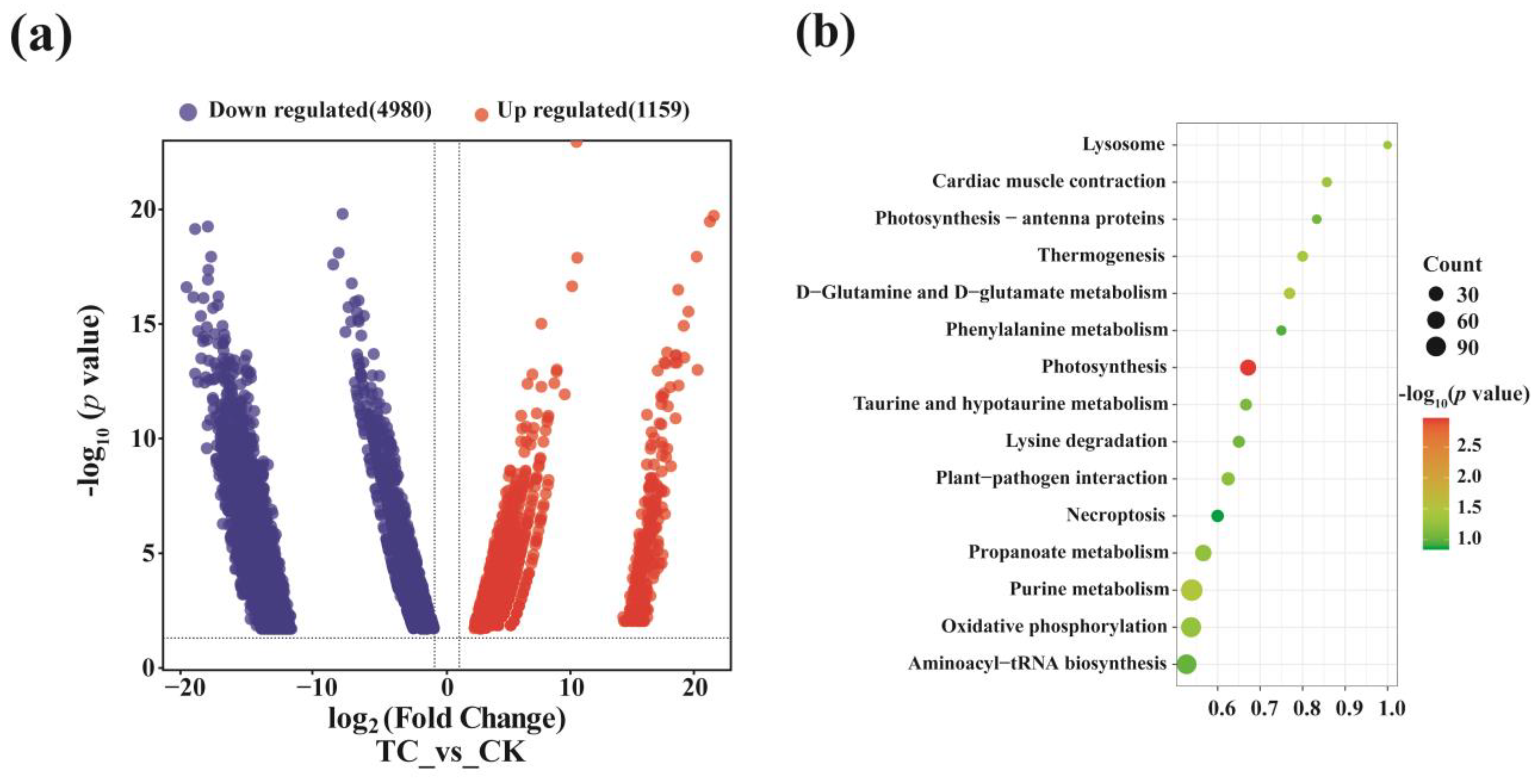
2.5. Differentially Expressed Genes in Metatranscriptomic Library
2.6. Potential Tetracycline Degradation Genes
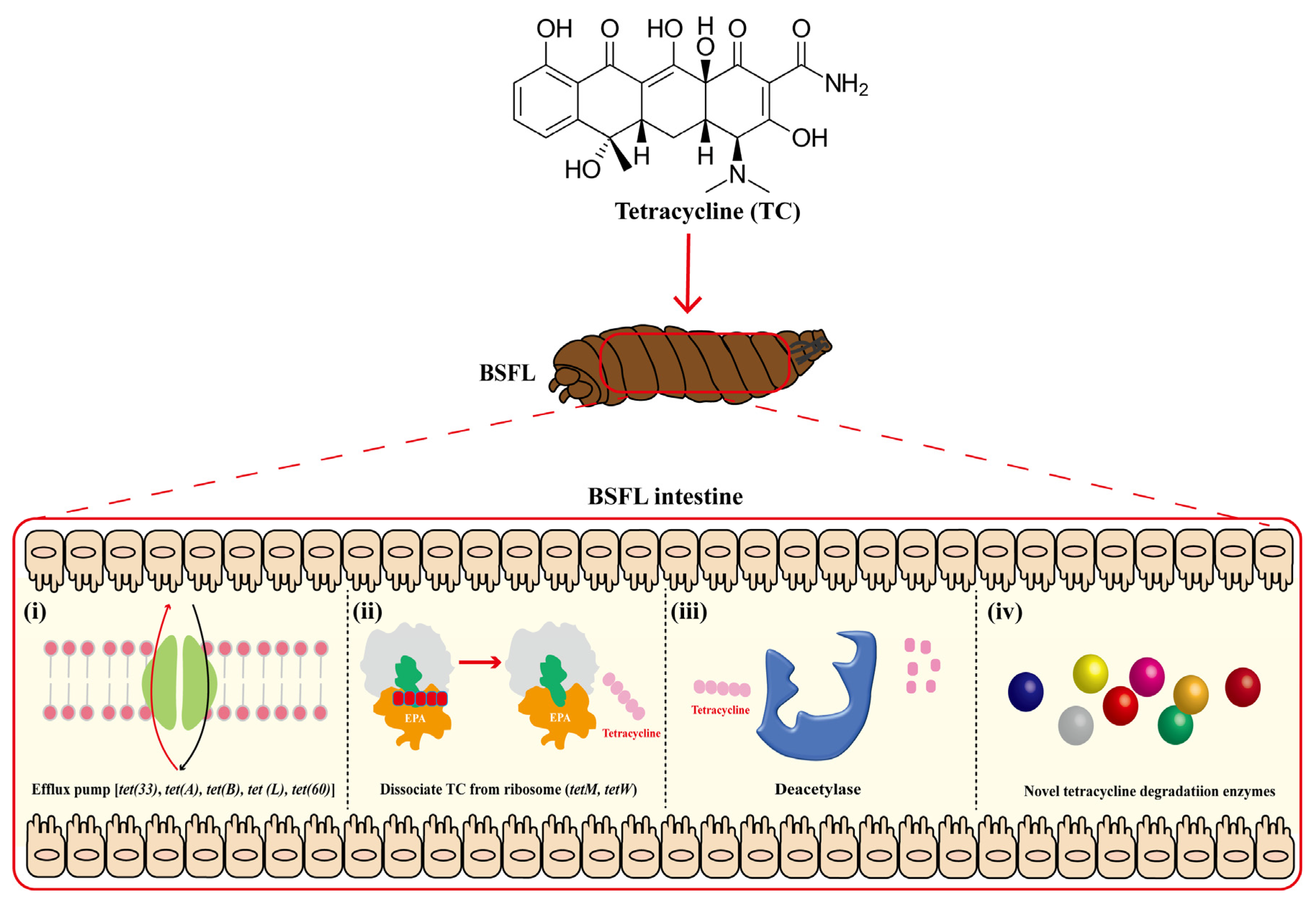
2.7. Possible Mechanisms of BSFL Intestinal Microbiome Response to High Tetracycline Concentration Stress
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. BSFL Husbandry and Substrate
- BSFL’s survival rate (%) = (N2/N1) × 100%, where N1 and N2 are the initial and final number of BSFL in each group, respectively. Ten larvae were randomly selected and weighed on the 8th day of rearing to determine their biomass.
- BSFL’s substrate consumption rate (%) = [(W1 − W2)/W1] × 100%, where W1 and W2 denote the initial and final dry weights of the wheat bran substrate during 8 days of rearing, respectively.
5.2. Germ-Free and Sterile BSFL Model Construction
5.3. Tetracycline Determination
5.4. DNA and RNA Extraction, Library Construction, Sequencing
5.5. Data Sequencing Analysis
5.6. Data Availability
5.7. Statistical Analysis
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kang, Y.; Li, Q.; Xia, D.; Shen, M.; Mei, L.; Hu, J. Short-term thermophilic treatment cannot remove tetracycline resistance genes in pig manures but exhibits controlling effects on their accumulation and spread in soil. J. Hazard. Mater. 2017, 340, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.; Yang, L.; Zhang, L.; Ye, B.; Wang, L. Antibiotics in soil and water in China–a systematic review and source analysis. Environ. Pollut. 2020, 266, 115147. [Google Scholar] [CrossRef] [PubMed]
- Ben, Y.; Fu, C.; Hu, M.; Liu, L.; Wong, M.H.; Zheng, C. Human health risk assessment of antibiotic resistance associated with antibiotic residues in the environment: A review. Environ. Res. 2019, 169, 483–493. [Google Scholar] [CrossRef]
- Li, W.; Zhang, G. Detection and various environmental factors of antibiotic resistance gene horizontal transfer. Environ. Res. 2022, 212, 113267. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yao, H.; Wang, C. Black soldier fly larvae can effectively degrade oxytetracycline bacterial residue by means of the gut bacterial community. Front. Microbiol. 2021, 12, 663972. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lin, H.; Ma, J.; Zhu, R.; Sun, W.; Lin, X.; Zhang, J.; Zheng, H.; Zhang, X. Degradation of tetracycline antibiotics by Arthrobacter nicotianae OTC-16. J. Hazard. Mater. 2021, 403, 123996. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y. Tetracycline degradation by Klebsiella sp. strain TR5: Proposed degradation pathway and possible genes involved. Chemosphere 2020, 253, 126729. [Google Scholar]
- Huang, X.; Zhang, X.; Feng, F.; Xu, X. Biodegradation of tetracycline by the yeast strain Trichosporon mycotoxinivorans XPY-10. Prep. Biochem. Biotechnol. 2016, 46, 15–22. [Google Scholar] [CrossRef]
- Leng, Y.; Bao, J.; Chang, G.; Zheng, H.; Li, X.; Du, J.; Snow, D.; Li, X. Biotransformation of tetracycline by a novel bacterial strain Stenotrophomonas maltophilia DT1. J. Hazard. Mater. 2016, 318, 125–133. [Google Scholar] [CrossRef]
- Bhatt, P.; Jeon, C.-H.; Kim, W. Tetracycline bioremediation using the novel Serratia marcescens strain WW1 isolated from a wastewater treatment plant. Chemosphere 2022, 298, 134344. [Google Scholar] [CrossRef]
- Chen, X.; Shen, W.; Chen, J.; Zhu, Y.; Chen, C.; Xie, S. Tetracycline biotransformation by a novel bacterial strain Alcaligenes sp. T17. Sci. Total Environ. 2022, 832, 155130. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Jia, Y.; Li, J. Degradation of tetracycline and oxytetracycline by crude lignin peroxidase prepared from Phanerochaete chrysosporium—A white rot fungus. Chemosphere 2009, 75, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Awasthi, M.K.; Chen, H.; Duan, Y.; Awasthi, S.K.; Zhang, Z. Performance of black soldier fly larvae (Diptera: Stratiomyidae) for manure composting and production of cleaner compost. J. Environ. Manag. 2019, 251, 109593. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Wang, X.; Mao, Z.; Liang, J.; Liu, X.; Xu, X. Bioconversion of chicken meat and bone meal by black soldier fly larvae: Effects of straw addition on the quality and microbial profile of larval frass. J. Environ. Manag. 2022, 307, 114579. [Google Scholar] [CrossRef]
- Raksasat, R.; Lim, J.W.; Kiatkittipong, W.; Kiatkittipong, K.; Ho, Y.C.; Lam, M.K.; Font-Palma, C.; Zaid, H.F.Z.; Cheng, C.K. A review of organic waste enrichment for inducing palatability of black soldier fly larvae: Wastes to valuable resources. Environ. Pollut. 2020, 267, 115488. [Google Scholar] [CrossRef]
- Yang, C.; Ma, S.; Li, F.; Zheng, L.; Tomberlin, J.K.; Yu, Z.; Zhang, J.; Yu, C.; Fan, M.; Cai, M. Characteristics and mechanisms of ciprofloxacin degradation by black soldier fly larvae combined with associated intestinal microorganisms. Sci. Total Environ. 2021, 811, 151371. [Google Scholar] [CrossRef]
- Luo, X.; Yang, Q.; Lin, Y.; Tang, Z.; Tomberlin, J.K.; Liu, W.; Huang, Y. Black soldier fly larvae effectively degrade lincomycin from pharmaceutical industry wastes. J. Environ. Manag. 2022, 307, 114539. [Google Scholar] [CrossRef]
- Liu, C.; Yao, H.; Chapman, S.J.; Su, J.; Wang, C. Changes in gut bacterial communities and the incidence of antibiotic resistance genes during degradation of antibiotics by black soldier fly larvae. Environ. Int. 2020, 142, 105834. [Google Scholar] [CrossRef]
- Xiao, K.Q.; Li, L.G.; Ma, L.P.; Zhang, S.Y.; Bao, P.; Zhang, T.; Zhu, Y.G. Metagenomic analysis revealed highly diverse microbial arsenic metabolism genes in paddy soils with low-arsenic contents. Environ. Pollut. 2016, 211, 1–8. [Google Scholar] [CrossRef]
- Menezes, A.D.; Clipson, N.; Doyle, E. Comparative metatranscriptomics reveals widespread community responses during phenanthrene degradation in soil. Environ. Microbiol. 2012, 14, 2577–2588. [Google Scholar] [CrossRef]
- Zhao, X.-L.; Chen, Z.-G.; Yang, T.-C.; Jiang, M.; Wang, J.; Cheng, Z.-X.; Yang, M.-J.; Zhu, J.-X.; Zhang, T.-T.; Li, H. Glutamine promotes antibiotic uptake to kill multidrug-resistant uropathogenic bacteria. Sci. Transl. Med. 2021, 13, eabj0716. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Ma, S.; Hu, R.; Tomberlin, J.K.; Yu, C.; Huang, Y.; Zhan, S.; Li, W.; Zheng, L.; Yu, Z.; et al. Systematic characterization and proposed pathway of tetracycline degradation in solid waste treatment by Hermetia illucens with intestinal microbiota. Environ. Pollut. 2018, 242, 634–642. [Google Scholar] [CrossRef]
- Mei, H.; Li, C.; Li, X.; Hu, B.; Lu, L.; Tomberlin, J.K.; Hu, W. Characteristics of tylosin and enrofloxacin degradation in swine manure digested by black soldier fly (Hermetia illucens L.) larvae. Environ. Pollut. 2022, 293, 118495. [Google Scholar] [CrossRef]
- Yun, J.H.; Roh, S.W.; Whon, T.W.; Jung, M.J.; Bae, J.W. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl. Environ. Microbiol. 2014, 80, 5254–5264. [Google Scholar] [CrossRef]
- Peng, D.; Feng, Y.Z.; Wang, Y.M.; Lin, X.G. Enrichment of the antibiotic resistance gene tet(L) in an alkaline soil fertilized with plant derived organic manure. Front. Microbiol. 2018, 9, 1140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.J.; Shen, J.G.; Wang, H.; Liu, M.; Wu, L.H.; Ping, F.; He, Q.; Li, H.Y.; Zheng, C.F.; Xu, X.H. Attenuation of veterinary antibiotics in full-scale vermicomposting of swine manure via the housefly larvae (Musca domestica). Sci. Rep. 2013, 4, 6844. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.Y.; Ahn, Y.; Khare, S.; Gokulan, K.; Piñeiro, S.A.; Cerniglia, C.E. An in vitro study to assess the impact of tetracycline on the human intestinal microbiome. Anaerobe 2018, 49, 85–94. [Google Scholar] [CrossRef]
- Cauwerts, K.; Decostere, A.; Graef, E.M.D.; Haesebrouck, F.; Pasmans, F. High prevalence of tetracycline resistance in Enterococcus isolates from broilers carrying the erm(B) gene. Avian Pathol. 2007, 36, 395–399. [Google Scholar] [CrossRef]
- Niu, S.-H.; Liu, S.; Deng, W.-K.; Wu, R.-T.; Cai, Y.-F.; Liao, X.-D.; Xing, S.-C. A sustainable and economic strategy to reduce risk antibiotic resistance genes during poultry manure bioconversion by black soldier fly Hermetia illucens larvae: Larval density adjustment. Ecotoxicol. Environ. Saf. 2022, 232, 113294. [Google Scholar] [CrossRef]
- Li, K.; Cao, R.; Mo, S.; Yao, R.; Wu, J. Swine manure composting with compound microbial inoculants: Removal of antibiotic resistance genes and their associations with microbial community. Front. Microbiol. 2020, 11, 592592. [Google Scholar] [CrossRef]
- Khaira, M.B.; Yusuf, M.B.; Khan, F. Insights to antimicrobial resistance: Heavy metals can inhibit antibiotic resistance in bacteria isolated from wastewater. Environ. Monit. Assess. 2022, 194, 252. [Google Scholar] [CrossRef] [PubMed]
- Webber, M.A.; Piddock, L.J.V. The importance of efflux pumps in bacterial antibiotic resistance. J. Antimicrob. Chemother. 2003, 51, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Molale, L.G.; Bezuidenhout, C.C. Antibiotic resistance, efflux pump genes and virulence determinants in Enterococcus spp. from surface water systems. Environ. Sci. Pollut. Res. 2016, 23, 21501–21510. [Google Scholar] [CrossRef] [PubMed]
- Jordi Vila, J.L.M. Clinical impact of the over-expression of efflux pump in nonfermentative Gram-negative bacilli, development of efflux pump inhibitors. Curr. Drug Targets 2008, 9, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Ss, A.; Assg, A.; Jvp, B. Evaluation of the inhibitory effect of caffeic acid and gallic acid on tetR and tetM efflux pumps mediating tetracycline resistance in Streptococcus sp., using computational approach. J. King Saud Univ. Sci. 2020, 32, 904–909. [Google Scholar]
- Scott, L.J. Eravacycline: A review in complicated intra-abdominal infections. Drugs 2019, 79, 315–324. [Google Scholar] [CrossRef]
- Tian, B.; Fadhil, N.H.; Powell, J.E.; Kwong, W.K.; Moran, N.A. Long-term exposure to antibiotics has caused accumulation of resistance determinants in the gut microbiota of honeybees. MBio 2012, 3, e00312–e00377. [Google Scholar] [CrossRef]
- Liu, B.; Chen, C. Translation elongation factor 4 (LepA) contributes to tetracycline susceptibility by stalling elongating ribosomes. Antimicrob. Agents Chemother. 2018, 62, 10-1128. [Google Scholar] [CrossRef]
- Yang, J.; Lin, Y.; Yang, X.; Ng, T.B.; Ye, X.; Lin, J. Degradation of tetracycline by immobilized laccase and the proposed transformation pathway. J. Hazard. Mater. 2017, 322, 525–531. [Google Scholar] [CrossRef]
- Wen, X.; Zeng, Z.; Du, C.; Huang, D.; Zeng, G.; Xiao, R.; Lai, C.; Xu, P.; Zhang, C.; Wan, J. Immobilized laccase on bentonite-derived mesoporous materials for removal of tetracycline. Chemosphere 2019, 222, 865–871. [Google Scholar] [CrossRef]
- Wen, X.; Jia, Y.; Li, J. Enzymatic degradation of tetracycline and oxytetracycline by crude manganese peroxidase prepared from Phanerochaete chrysosporium. J. Hazard. Mater. 2010, 177, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Leng, Y.; Wan, D.; Chang, F.; Huang, Y.; Li, Z.; Xiong, W.; Wang, J. Transformation of tetracycline by manganese peroxidase from Phanerochaete chrysosporium. Molecules 2021, 26, 6803. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.H.; Kim, S.J.; Lee, H.S.; Cha, S.S.; Lee, J.H.; Yoon, S.H.; Koo, B.S.; Lee, C.M.; Choi, S.H.; Lee, S.H. Novel metagenome-derived carboxylesterase that hydrolyzes β-lactam antibiotics. Appl. Environ. Microbiol. 2011, 77, 7830–7836. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; He, Y.; Zhang, H. Screening and 16S rRNA Analysis of the Bacteria of Producing Chitin Deacetylase. In Proceedings of the 4th International Conference on Bioinformatics and Biomedical Engineering, Chengdu, China, 18–20 June 2010; pp. 1–4. [Google Scholar] [CrossRef]
- Pei, Y.; Zhao, S.; Chen, X.; Zhang, J.; Ni, H.; Sun, M.; Lin, H.; Liu, X.; Chen, H.; Yang, S. Bacillus velezensis EEAM 10B strengthens nutrient metabolic process in black soldier fly larvae (Hermetia illucens) via changing gut microbiome and metabolic pathways. Front. Nutr. 2022, 9, 880488. [Google Scholar] [CrossRef]
- Pei, Y.; Mamtimin, T.; Ji, J.; Khan, A.; Kakade, A.; Zhou, T.; Yu, Z.; Zain, H.; Yang, W.; Ling, Z.; et al. The guanidine thiocyanate-high EDTA method for total microbial RNA extraction from severely heavy metal-contaminated soils. Microb. Biotechnol. 2021, 14, 465–478. [Google Scholar] [CrossRef]
- Hyatt, D.; LoCascio, P.F.; Hauser, L.J.; Uberbacher, E.C. Gene and translation initiation site prediction in metagenomic sequences. Bioinformatics 2012, 28, 2223–2230. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | CK-MG | TC-MG | CK-MT | TC-MT |
|---|---|---|---|---|
| Raw Reads | 113499814 | 121370872 | 115167960 | 117241914 |
| Raw Bases | 17024972100 | 18205630800 | 17275194000 | 17586287100 |
| Clean Reads | 89154368 | 104098312 | 106840552 | 94504924 |
| Clean Bases | 13264807930 | 15490976419 | 15927374626 | 14103693095 |
| Contigs | 41,586 | 43,057 | 9526 | 2500 |
| Clean Reads in Raw Reads (%) | 78.55% | 85.77% | 92.77% | 80.61% |
| Clean Bases in Raw Bases (%) | 77.91% | 85.09% | 92.20% | 80.20% |
| Q20 | 97.63% | 97.48% | 98.25% | 98.73% |
| Q30 | 95.23% | 94.89% | 94.06% | 95.60% |
| Family | CK-MG | TC-MG | CK-MT | TC-MT | Description |
|---|---|---|---|---|---|
| AA1 | 0.50222 | 1.76443 | - | - | Laccase/p-diphenol:oxygen oxidoreductase/ferroxidase (EC 1.10.3.2); ferroxidase (EC 1.10.3.-); Laccase-like multicopper oxidase (EC 1.10.3.-) |
| AA2 | 2.22735 | 9.84698 | - | - | Manganese peroxidase (EC 1.11.1.13); versatile peroxidase (EC 1.11.1.16); lignin peroxidase (EC 1.11.1.14); peroxidase (EC 1.11.1.-) |
| AA3 | 334.669 | 310.651 | 177.314 | 153.316 | Cellobiose dehydrogenase (EC 1.1.99.18); glucose 1-oxidase (EC 1.1.3.4); aryl alcohol oxidase (EC 1.1.3.7); alcohol oxidase (EC 1.1.3.13); pyranose oxidase (EC 1.1.3.10) |
| AA4 | 189.943 | 294.33 | - | - | Vanillyl-alcohol oxidase (EC 1.1.3.38) |
| AA6 | 199.316 | 380.512 | 4541.03 | 4518.02 | 1,4-benzoquinone reductase (EC. 1.6.5.6) |
| AA7 | 65.8805 | 125.302 | - | - | Glucooligosaccharide oxidase (EC 1.1.3.-); chitooligosaccharide oxidase (EC 1.1.3.-) |
| AA10 | 75.5844 | 146.551 | - | - | AA10 (formerly CBM33) proteins are copper-dependent lytic polysaccharide mono-oxygenases (LPMOs); some proteins have been shown to act on chitin, while others act on cellulose. |
| AA12 | 2.68267 | 5.62463 | - | - | The pyrroloquinoline quinone-dependent oxidoreductase activity was demonstrated as present for the CC1G_09525 protein of Coprinopsis cinerea. |
| CE1 | 1464.2 | 1816.33 | 864.206 | 910.166 | Acetyl xylan esterase (EC 3.1.1.72); cinnamoyl esterase (EC 3.1.1.-); feruloyl esterase (EC 3.1.1.73); carboxylesterase (EC 3.1.1.1); S-formylglutathione hydrolase (EC 3.1.2.12) |
| CE2 | 37.8945 | 30.416 | - | - | Acetyl xylan esterase (EC 3.1.1.72). |
| CE3 | 180.225 | 156.923 | 25.0276 | 29.0947 | Acetyl xylan esterase (EC 3.1.1.72). |
| CE4 | 501.018 | 768.334 | 1443.96 | 1964.17 | Acetyl xylan esterase (EC 3.1.1.72); chitin deacetylase (EC 3.5.1.41); chito-oligosaccharide deacetylase (EC 3.5.1.-); peptidoglycan GlcNAc deacetylase (EC 3.5.1.-); peptidoglycan N-acetylmuramic acid deacetylase (EC 3.5.1.-). |
| CE5 | 0.96599 | 3.93697 | - | - | Acetyl xylan esterase (EC 3.1.1.72); cutinase (EC 3.1.1.74) |
| CE6 | 39.7137 | 77.6012 | 133.101 | 948.039 | Acetyl xylan esterase (EC 3.1.1.72). |
| CE7 | 61.6056 | 177.511 | 69.4047 | 81.6384 | Acetyl xylan esterase (EC 3.1.1.72); cephalosporin-C deacetylase (EC 3.1.1.41). |
| CE8 | 43.027 | 80.0709 | 18.1287 | 25.6374 | Pectin methylesterase (EC 3.1.1.11). |
| CE9 | 294.834 | 212.118 | 474.72 | 395.33 | N-acetylglucosamine 6-phosphate deacetylase (EC 3.5.1.25); N-acetylgalactosamine-6-phosphate deacetylase (EC 3.5.1.80). |
| CE10 | 596.732 | 1009.64 | 225.639 | 135.508 | Arylesterase (EC 3.1.1.-); carboxyl esterase (EC 3.1.1.3); acetylcholinesterase (EC 3.1.1.7); cholinesterase (EC 3.1.1.8); sterol esterase (EC 3.1.1.13); brefeldin A esterase (EC 3.1.1.-). |
| CE11 | 376.726 | 322.36 | 228.196 | 301.047 | UDP-3-0-acyl N-acetylglucosamine deacetylase (EC 3.5.1.-). |
| CE12 | 98.2928 | 251.826 | 203.141 | 122.558 | Pectin acetylesterase (EC 3.1.1.-); rhamnogalacturonan acetylesterase (EC 3.1.1.-); acetyl xylan esterase (EC 3.1.1.72) |
| CE14 | 46.9864 | 308.344 | - | - | N-acetyl-1-D-myo-inosityl-2-amino-2-deoxy-alpha-D-glucopyranoside deacetylase (EC 3.5.1.89); diacetylchitobiose deacetylase (EC 3.5.1.-); mycothiol S-conjugate amidase (EC 3.5.1.-) |
| CE15 | 5.14113 | 25.4286 | - | - | 4-O-methyl-glucuronoyl methylesterase (EC 3.1.1.-) |
| CE16 | 22.5685 | 51.4155 | - | - | Acetylesterase (EC 3.1.1.6) is active on various carbohydrate acetyl esters |
| Others | 20,856.9 | 26,276.4 | 21,728.5 | 25,421 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pei, Y.; Sun, M.; Zhang, J.; Lei, A.; Chen, H.; Kang, X.; Ni, H.; Yang, S. Comparative Metagenomic and Metatranscriptomic Analyses Reveal the Response of Black Soldier Fly (Hermetia illucens) Larvae Intestinal Microbes and Reduction Mechanisms to High Concentrations of Tetracycline. Toxics 2023, 11, 611. https://doi.org/10.3390/toxics11070611
Pei Y, Sun M, Zhang J, Lei A, Chen H, Kang X, Ni H, Yang S. Comparative Metagenomic and Metatranscriptomic Analyses Reveal the Response of Black Soldier Fly (Hermetia illucens) Larvae Intestinal Microbes and Reduction Mechanisms to High Concentrations of Tetracycline. Toxics. 2023; 11(7):611. https://doi.org/10.3390/toxics11070611
Chicago/Turabian StylePei, Yaxin, Mengxiao Sun, Jiran Zhang, Aojie Lei, Hongge Chen, Xiangtao Kang, Hongyuhang Ni, and Sen Yang. 2023. "Comparative Metagenomic and Metatranscriptomic Analyses Reveal the Response of Black Soldier Fly (Hermetia illucens) Larvae Intestinal Microbes and Reduction Mechanisms to High Concentrations of Tetracycline" Toxics 11, no. 7: 611. https://doi.org/10.3390/toxics11070611
APA StylePei, Y., Sun, M., Zhang, J., Lei, A., Chen, H., Kang, X., Ni, H., & Yang, S. (2023). Comparative Metagenomic and Metatranscriptomic Analyses Reveal the Response of Black Soldier Fly (Hermetia illucens) Larvae Intestinal Microbes and Reduction Mechanisms to High Concentrations of Tetracycline. Toxics, 11(7), 611. https://doi.org/10.3390/toxics11070611