1. Introduction
Chromium (Cr) widely exists in the ecological environment and can be found in pigments, chrome-plated metals, cement, detergents, and industrial Cr waste dumps [
1]. Cr has a variety of oxidation states (−2 to +6), but only trivalent chromium (Cr(III)) and hexavalent chromium (Cr(VI)) are stable. The increase in industrial use, coupled with improper disposal of Cr(VI)-related waste, has led to the serious increase of Cr(VI) levels in air, water, and soil, resulting in the pollution of the environment, even the food chain [
2].
Mitochondria constantly undergo a dynamic fusion/fission process, which is mainly controlled by regulatory proteins such as mitofusins (Mfns) and dynamin-related protein 1 (Drp1) [
3]. This kind of dynamic balance is essential to maintain constant changes in mitochondrial shape, size, and network. Mitochondrial fission process is mainly mediated by Drp1, which exists in the cytosol and translocates to the outer membrane of mitochondrial during fission [
4]. Increasing evidence suggested that the dynamic morphology of the mitochondrial network is very important. In the physiological state, the long, continuous tracks of fused mitochondria and branching networks are dominant and mainly regulated by Mitofusins (Mfns). However, upon exposure to various stresses, mitochondria undergo fission, networks become unraveled, and the fragmented morphology is more prominent (regulated Drp1). Preliminary studies have shown that Drp1 ablation declined Cyt c release and inhibited apoptosis [
5]. In contrast, other studies revealed that blockage of Drp1 partially decreased [
6] or had little effect [
7] on Cyt c release, without affecting apoptosis.
The electron transport chain (ETC), which exists in the folded inner membranes of mitochondria, is mainly composed of four mitochondrial respiratory chain complexes (MRCC; I–IV) and two free-moving electron transfer carriers cytochrome c (cyt c) and ubiquinone. The four complexes are assembled into a specifically configured super-complex, which together with MRCC V (F1F0ATP synthase), becomes the basis of ATP generation during oxidative phosphorylation [
8]. As the largest multi-subunit enzyme complex located in ETC, MRCC I is also called NADH-ubiquinone oxidoreductase, and its key role is to transfer electrons from NADH to ubiquinone [
9]. The ETC is the key component of mitochondria and also known as the most important source of intracellular reactive oxygen species (ROS). ROS include oxygen-free radicals such as hydroxyl radical (OH) and superoxide anion radical (O
2−), and non-radical oxidants such as hydrogen peroxide (H
2O
2). Due to the existence of electron leakage, not all the electrons could be successfully transferred to the final electron acceptor, O
2. Under normal conditions, 0.2–2% of the electrons do not follow the transmission order but directly leak out from ETC, and then interact with oxygen to produce ROS [
10]. As a double-edged sword, ROS plays an important role in intracellular signaling pathways, but ROS accumulation can lead to cytotoxicity and even cell death.
Although it is believed that mitochondrial fragmentation is a common phenomenon in apoptosis, whether excessive fission is crucial for apoptosis progression remains controversial. Our previous studies [
11,
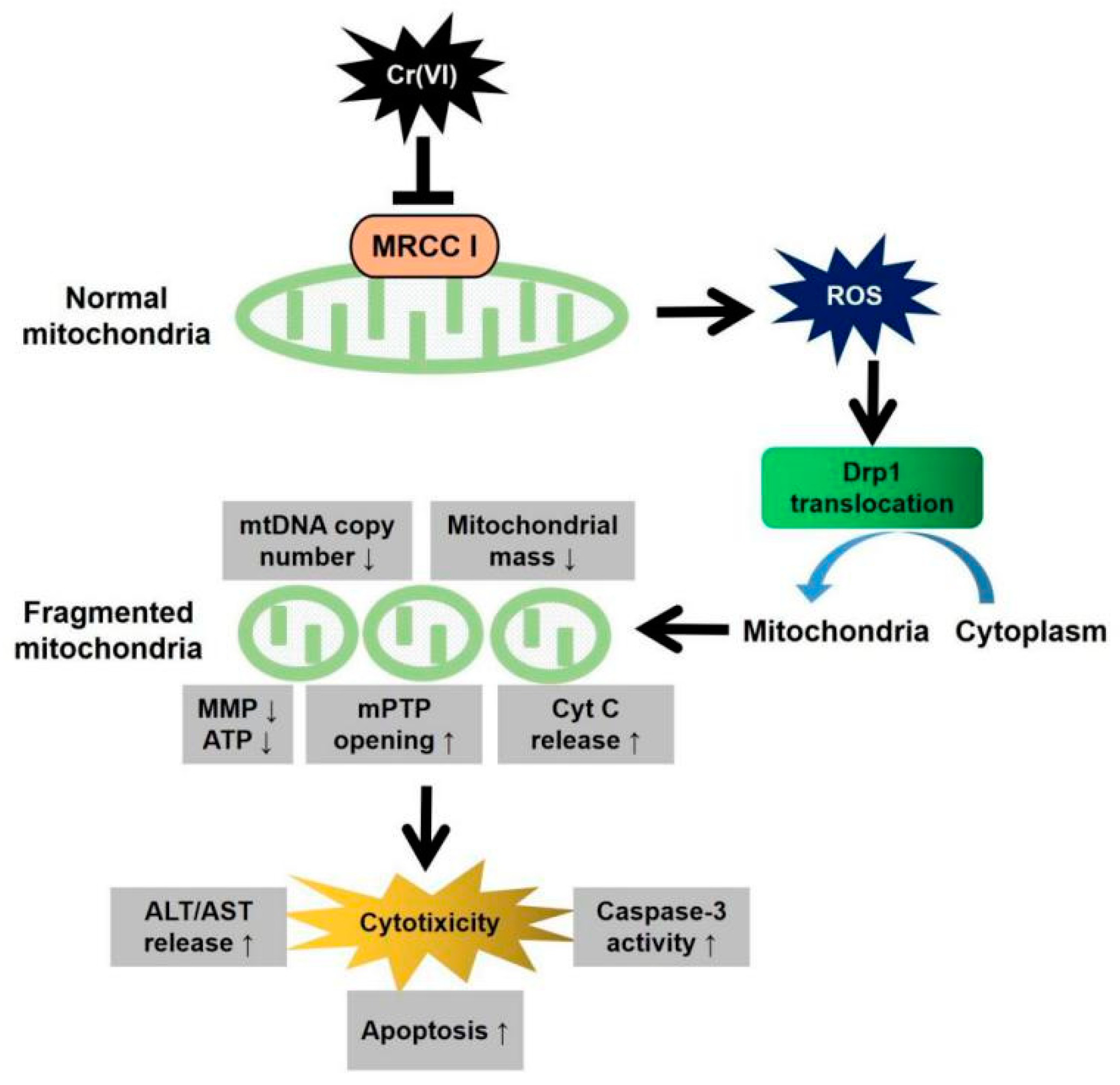
12] have demonstrated that Cr(VI) mainly targeted MRCC I and increased ROS generation to induce cytotoxicity. In addition, Cr(VI) could also cause both mitochondrial damage and apoptotic cell death during ROS-triggered cytotoxicity, but the related mechanism involved in MRCC I-dependent cytotoxicity was unclear. The present study will demonstrate the role of increased mitochondrial fragmentation mediated by Drp1 in Cr(VI)-induced MRCC I-dependent cytotoxicity, which will provide experimental evidence for further elucidating the cytotoxicity of Cr(VI).
2. Materials and Methods
2.1. Cell Culture and Cell Counts
Human L02 hepatocytes, obtained from Experimental Central of Xiangya Hospital of Central South University, Changsha, China, were cultured in 25 cm2 culture flasks in the standard humidified incubator with the set of 5% CO2 and 37 °C. Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum (FBS) (Gibco, Carlsbad, CA, USA) and 1% penicillin-streptomycin (P/S) solution was used.
Under the optimal conditions, the doubling time of L02 hepatocytes was 18–24 h, and the cells were subcultured every 2.5–3 days (d). Cell numbers were measured and recorded everyday using a hemocytometer by trypan blue exclusion method.
2.2. Drp1 SiRNA
The siRNA sequences were designed and synthesized by RibobioCo. Ltd. (Guangzhou, China). The hepatocytes were transfected with siRNA targeting Drp1 (siB121119100350-1-5) and its negative control (siB06525141910-1-5) using lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA). After 4 h of transfection, the hepatocytes were changed with complete medium.
2.3. ROS Level
The intracellular ROS level was determined using 2′, 7′-dichlorofluorescein diacetate (DCFH-DA; Beyotime Institute of Biotechnology, Shanghai, China) by flow cytometry. L02 hepatocytes were treated with indicated chemicals and washed twice by cold PBS and loaded with 10 µM DCFH-DA at 37 °C for 40 min. After the incubation, the hepatocytes were washed again and analyzed by flow cytometry.
2.4. Alanine Aminotransferase (ALT) and Aspartate Aminotransferase (AST) Levels
L-02 hepatocytes were treated with different chemicals, and the supernatants were collected. ALT and AST levels were determined using the commercial kits (Jiancheng, Nanjing, China) according to the instructions. The optical density at 510 nm was measured using the multifunctional microplate reader.
2.5. Caspase-3 Activity
Caspase-3 activity was determined using the commercial colorimetric assay kit. The hepatocytes were treated with indicated chemicals and washed twice with PBS, followed by lysis with the Caspase-3 Assay kit (Beyotime Institute of Biotechnology, China). The centrifugation was performed at 16,000× g at 4 °C for 10 min. The samples were then incubated with Ac-DEVD-pNA (substrate) at 37 °C for 2 h. The absorbance was measured and recorded by a microplate reader at 405 nm.
2.6. Apoptosis
The cell apoptosis was determined using a commercial Annexin V-FITC apoptosis detection kit (Invitrogen, Carlsbad, CA, USA). After the treatment of different chemicals, the hepatocytes were incubated with 100 μL 1× binding buffer containing 5 μL Annexin-V-FITC and 1 μL propidium iodide (PI) for 30 min at room temperature. After the incubation, 400 μL binding buffer was added to the culture to stop the staining. The flow cytometric analysis was then performed. Data was analyzed using Flowjo 7.6 software.
2.7. Mitochondrial Permeability Transition Pore (mPTP) Opening
The mPTP opening was examined using the commercial kit by monitoring the release of calcein from mitochondrial. Briefly, the hepatocytes were treated with 2 µM calcein-AM and 1 mM CoCl2 for 30 min at room temperature, and washed with PBS. The culture was then incubated with 1 mM CoCl2 for an additional 20 min at 37 °C in order to specifically quench the fluorescence of free calcein in the cytosol. The fluorescence intensity of mitochondrial calcein in L02 hepatocytes was determined using a fluorescence microplate reader at 490 nm/515 nm for excitation/emission. The loss of calcein fluorescence suggested the opening of mPTP.
2.8. Mitochondrial Membrane Potential (MMP, Δψm)
The MMP of L02 hepatocytes was detected using JC-1 (Sigma, St. Louis, MO, USA). After the chemicals treatment, the cells were washed with PBS and then incubated with JC-1 for 20 min at 37 °C. The fluorescence intensity was read at the excitation/emission wavelength of 488/530 nm. The MMP is presented as % of control for the fluorescence intensity.
2.9. qRT-PCR
Total RNA was isolated with the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instruction. The RNA quality was verified by spectrophotometry. Later on, the cDNAs were synthesized using the ReverTra Ace qPCR RT kit (Toyobo, Tokyo, Japan). For mitochondrial DNA analyses, total DNA was extracted by a DNA extraction kit (NEP002-1, Dingguo, China) according to the manufacturer’s instructions. qRT-PCR was performed using the Light Cycler®Nano SYBR Green I Master on a Light Cycler® Nano System. The PCR conditions were as follows: 10 min at 95 °C, followed by 40 cycles of 95 °C for 30 s, 56 °C for 30 s and 72 °C for 30 s. The mRNA levels were calculated using the 2−ΔΔCT method normalized to ACTB mRNA.
The Primers used in this study: Drp1, 5′-TAGTGGGCAGGGACCTTCTT-3′ (F) and 5′-TGCTTCAACTCCATTTTCTTCTCC-3′ (R); ACTB, 5′-CACCAGGGCGTGATGGT-3′ (F) and 5′-CTCAAACATGATCTGG GTCAT-3′ (R); NADH dehydrogenase subunit I (ND1), 5′-TACGCAAAGGTTCCCAACG-3′ (F) and 5′-GGTGATGGTGGATGTGGC-3′ (R); cytochrome C Oxidase Subunit IV Isoform 1 (COX4I1), 5-TAGAAACCGTCTGAACTATCC-3′ (F) and 5′- ATGATTATGAGGGCGTGA-3′ (R); β-globin, 5′-GTTACTGCCTG TGGGGCAA-3′ (F) and 5′-CAAAGGTGCCCTTGAGGTT-3′ (R). The qRT-PCR assay was designed and performed in accordance with the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines.
2.10. Mitochondria Mass
Briefly, after the treatment of different chemicals, the hepatocytes were exposed to Mito-Tracker Green (10 μM, 30 min) at 37 °C in the dark. Then the cells were thoroughly washed with pre-warmed PBS, and analyzed with flow cytometer at 490/516 nm for excitation/emission wavelengths.
2.11. ATP Level
The ATP level was examined using a luciferase-based luminescence enhanced ATP assay kit (Beyotime Institute of Biotechnology, China) following the manufacturer’s instruction. The hepatocytes were washed with ice-cold PBS, lysed with 200 μL lysis buffer, and then centrifuged at 12,000× g for 5 min at 4 °C. ATP content in cell lysates was then determined using a luminescence plate reader.
2.12. Western Blotting
Mitochondrial fraction was isolated using the Mitochondria Isolation Kit for Cultured Cells according to the manufacturer’s instructions (Beyotime Institute of Biotechnology, Nanjing, China). For total protein extraction, cells were washed twice with PBS and lysed with RIPA buffer (Beyotime Institute of Biotechnology, Nanjing, China). The homogenates were centrifuged at 12,000× g for 15 min at 4 °C, the supernatant was then collected. Protein concentrations were evaluated using the BCA method. Then the samples were denatured by boiling with sample buffer for 10 min, loaded to SDS-PAGE gel for separation, and then transferred onto the PVDF membrane. The membranes were blocked with 5% skim milk for 1 h at room temperature and incubated with different primary antibodies overnight at 4 °C. The membranes were then washed and incubated with secondary antibodies for 1 h at room temperature. The protein bands were visualized using the enhanced chemiluminescence (ECL) kit (Thermo, Waltham, MA, USA) and quantitated using Image J software (National Institutes of Health, USA). The band density of different proteins was normalized to the control.
ND1 (DF4214) antibody was obtained from Affinity Biosciences (Cincinnati, OH, USA). VDAC1 (55259-1-AP) antibody was purchased from Proteintech Group Inc. (Wuhan, China). Drp1 (A2586), AIF (A19536), cyt c (A4912), and caspase-3 (A11021) antibodies were purchased from ABclonal Technology (Wuhan, China). Antibody against β-actin (70-ab008-040) was obtained from MultiSciences Biotech Co. (Hangzhou, China).
2.13. MRCCs Activity
After the treatment, L02 hepatocytes were collected and suspended in 0.1 M phosphate buffer (pH 7.2). To ensure cellular disruption and after three cycles of freeze/thawing, the activities of MRCC I–V were determined spectrophotometrically as previously described [
11].
2.14. Confocal Microscope
For mitochondrial morphology examination, the hepatocytes were incubated with 10 nM Mitotracker Red (Invitrogen Life Technologies, Carlsbad, CA, USA) for 45 min at 37 °C. After the indicated chemicals treatment, the hepatocytes were incubated with DAPI for another 45 min at 37 °C in the dark. The fluorescence images of each group were captured using Leica TCS SP5 II confocal spectral microscope. More than 20 clearly identifiable mitochondria were randomly selected from each treatment group. The length and density of the mitochondria were analyzed using Image J software.
To analyze Drp1 mitochondrial translocation, Cr(VI)-exposed hepatocytes were fixed with 4% paraformaldehyde for 15 min at room temperature following incubation with MitoTracker Red (10 nM, 45 min, 37 °C). The hepatocytes were then permeabilized with 0.5% Triton X-100, blocked with 5% bovine serum albumin (BSA), and incubated with primary Drp1 antibody at a 1:100 dilution at 4 °C overnight, followed by treatment with the secondary antibody. Nuclei were stained with DAPI prior to mounting. Confocal fluorescence images of each treatment group were captured with the Leica TCS SP5 II confocal spectral microscope.
2.15. Statistical Analysis
All experiments were repeated at least three times. Representative experiments or mean ± SD are shown in the figures. Statistical analyses were performed using one-way of variance (ANOVA), Student’s t-test, using SPSS 17.0 software. A significant difference was taken as p < 0.05.
4. Discussion
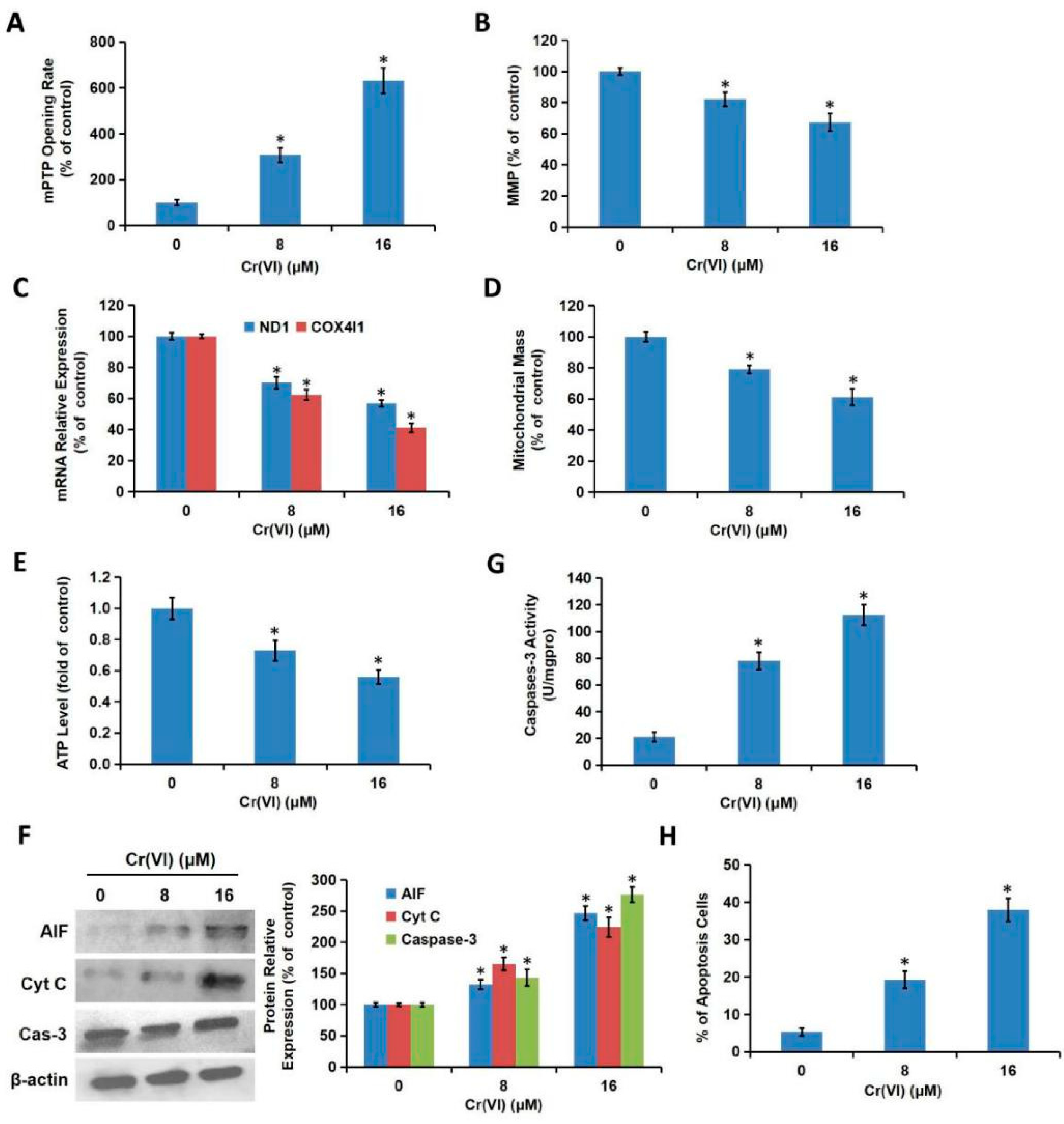
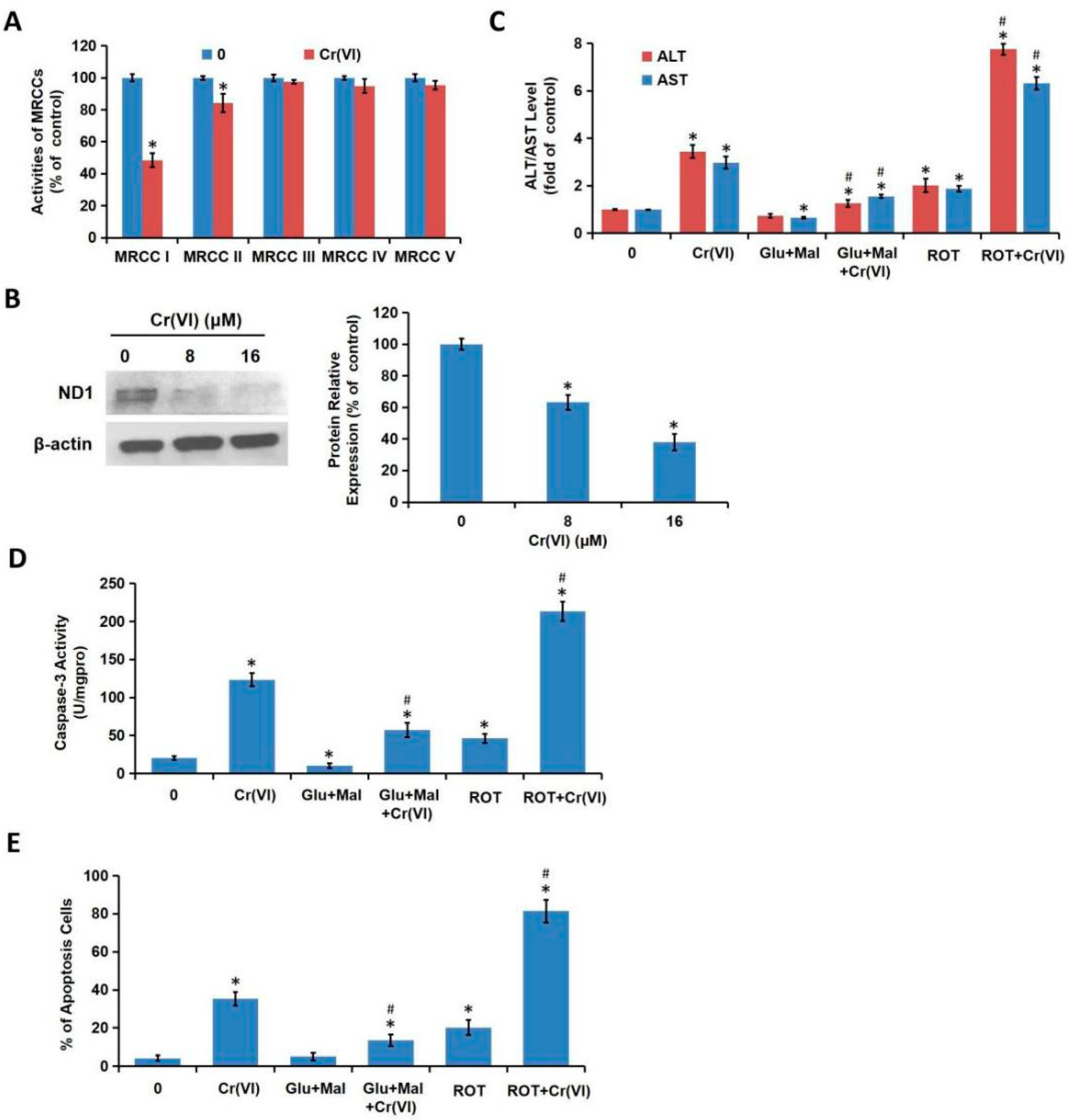
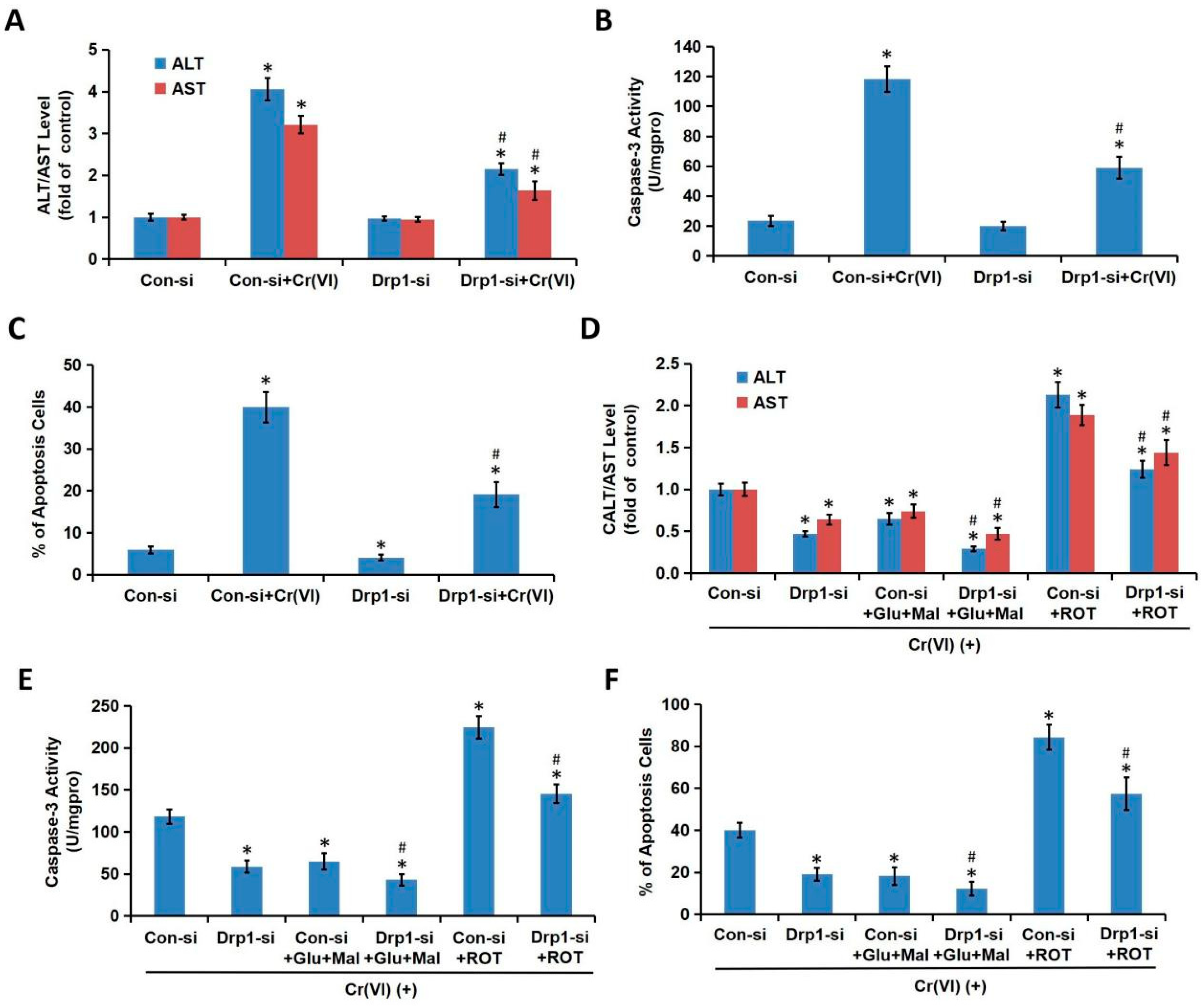
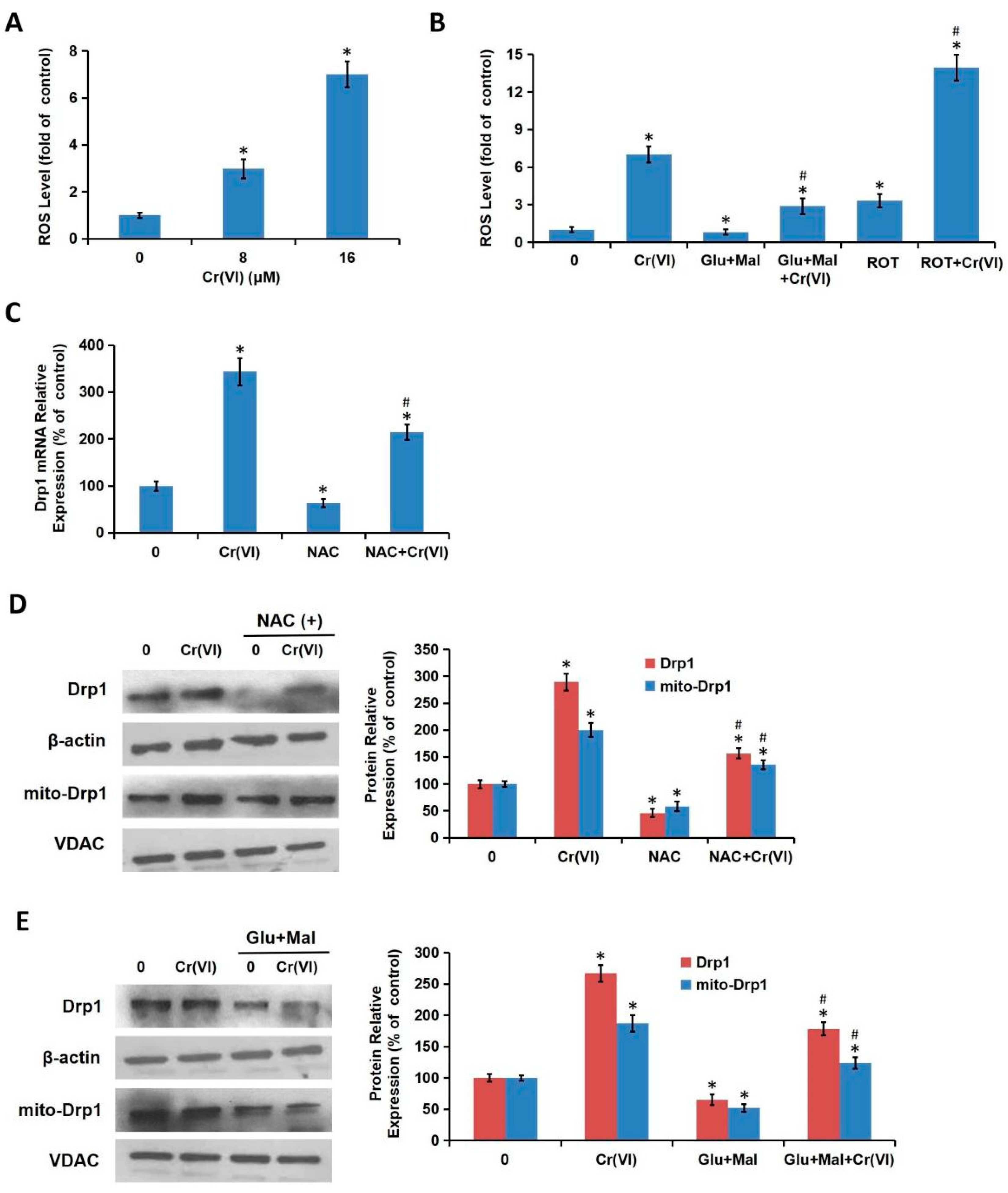
We demonstrated in the present study that MRCC I inhibition led to cytotoxicity in L02 hepatocytes, which was characterized by the increase of ALT/AST leakage, the activation of caspase-3, the inhibition of cell proliferation, and the induction of apoptosis. We then confirmed that Cr(VI) targeted MRCC I to induce ROS accumulation and then triggered mitochondrial-related cytotoxicity. Cr(VI) caused a marked increase in mPTP opening rate and decrease in MMP, declined the mtDNA copy number, mitochondrial mass, and ATP level, increased the mitochondrial release of apoptosis-inducing factors, and eventually triggered apoptosis. The pretreatment of Glu/Mal significantly alleviated Cr(VI)-induced cytotoxicity, while the cotreatment of ROT obviously aggravated Cr(VI)-induced cytotoxicity.
In theory, all four complexes (I–IV) of the ETC could generate ROS; evidence suggests that MRCC I and III are the main sites of ROS formation [
13,
14]. However, the ability of MRCC III to produce ROS has also been questioned, while MRCC I is considered to be the relevant major site of superoxide formation. It is reported that the formation of ROS can not be detected until MRCC III was inhibited by up to 71 ± 4%; in contrast, the small-extent deactivation of MRCC I (16 ± 2%) could lead to a significant increase of ROS generation [
15]. It is also demonstrated that the production rate of endogenous H
2O
2 was much lower when Glu/Mal, the substrates of MRCC I were used as respiratory fuel; the application of ROT, the inhibitor of MRCC I, led to a significantly increasing H
2O
2 release [
16]. ROT can interrupt the transfer of electrons to CoQ and enhance ROS production. The amount of ROS produced by various stimulation determines whether ROS play profitable or detrimental roles. In the physiological state, it was believed that ROS appear to be important second messengers that mediate different cell signaling pathways, while the overburdened ROS are exclusively harmful to the cells [
17]. The burst generation of ROS leads to irreversible damage to mitochondria, DNA damage, lipid peroxidation, ATP exhaustion, and, eventually, cell death [
18]. Although the precise mechanisms involved in ROS burst-generation and ROS-related cytotoxicity are still not clear, the application of specific ROS inhibitors such as NAC and Trolox to reduce the over-production of ROS under pathological conditions has been shown to ameliorate various diseases mediated by oxidative stress [
19].
The mtDNA copy number in each mitochondrion is constant, thus, the total number of mtDNA copies reflects the total number of mitochondria in cells [
20]. mtDNA is located near the ETC and its repair mechanism is incomplete, thus, mtDNA is more susceptible to damage when exposed to oxidative stress compared with the nuclear DNA [
21]. The opening of mPTP represents the abrupt change in permeability of the mitochondrial inner membrane, allowing not only protons but also various ions and solutes of up to 1.5 kDa in size to freely pass through the membrane [
22]. Apoptosis is initiated by the change of mitochondrial membrane permeability transition (MPT), and the opening of the non-specific mega-channel mPTP enables the membrane permeability suddenly increase, resulting in the release of the apoptosis executors including apoptosis-inducing factor (AIF), cytochrome c (Cyt c), and endonuclease G from the mitochondrial matrix to cytoplasm. The increase of mPTP opening may lead to the flow back of protons from mitochondrial membrane space to matrix, thus inhibiting MMP and ATP production, resulting in metabolic abnormalities and cytotoxicity [
23]. Moreover, Cr(VI) exposure triggered over-opening of mPTP, dropped MMP, decreased mtDNA number, declined ATP synthesis, indicating that Cr(VI) can directly disrupt the structure and function of mitochondria and induce cytotoxicity. As one of the early events of apoptosis induced by various stimuli, the increase of intracellular ROS can trigger Cyt c first detaches from cardiolipin and then being released into the cytoplasm. Cr(VI) exposure disrupted mitochondrial redox homeostasis and accumulated excessive ROS, then mitochondrial function such as MMP and mPTP was severely affected, thus, L02 hepatocytes undergo mitochondria-mediated apoptosis.
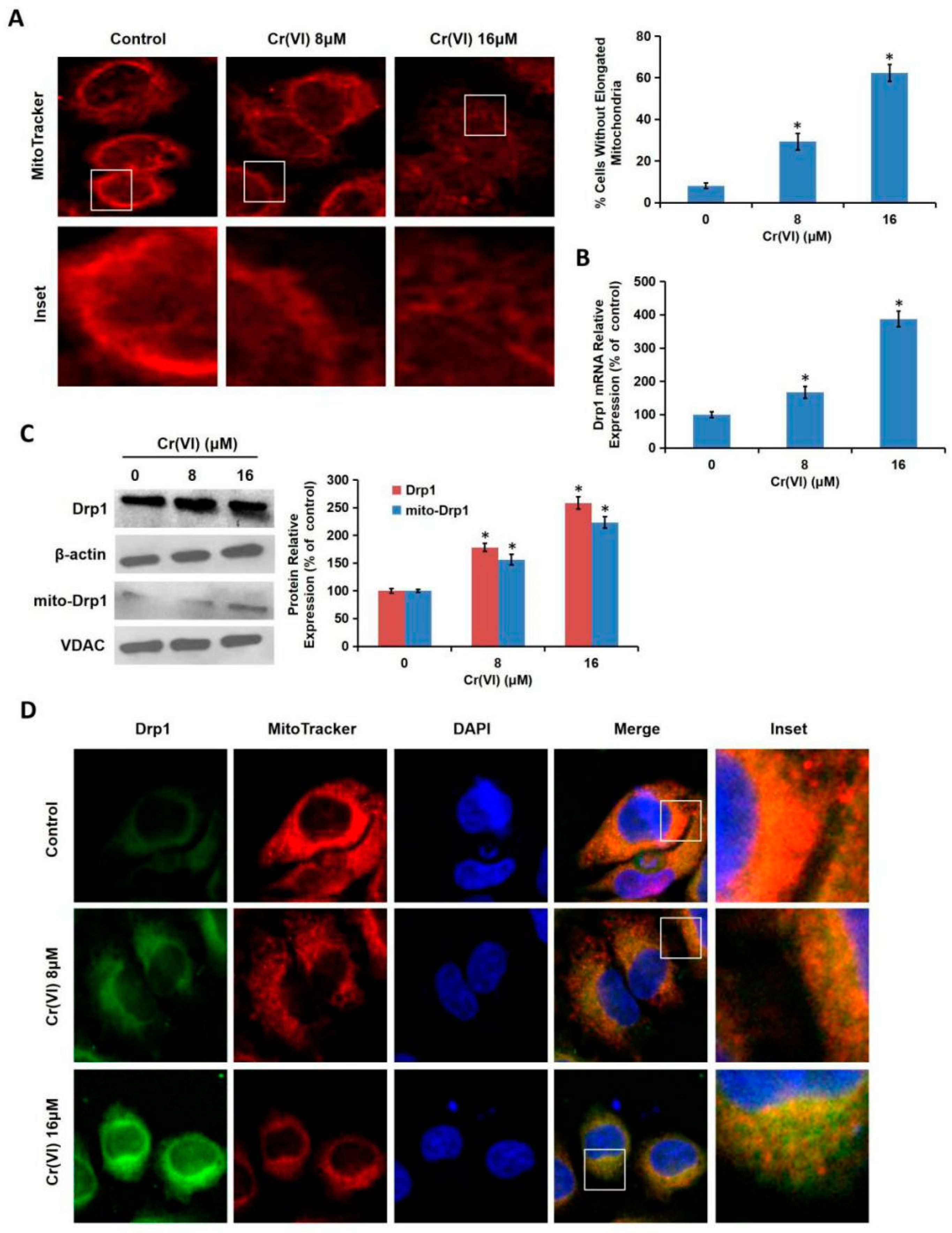
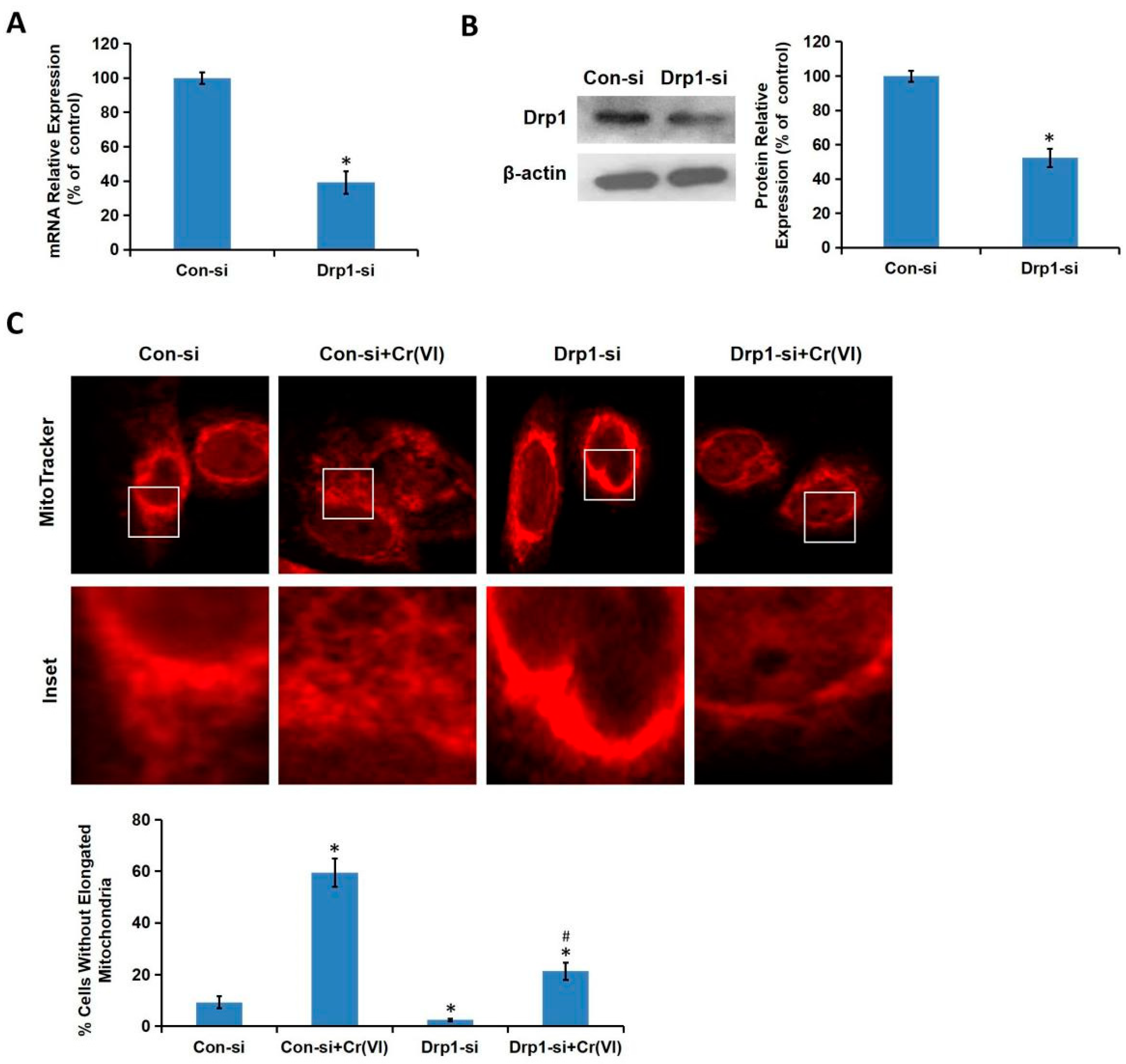
Cr(VI) induced excessive mitochondrial fragmentation and mitochondrial Drp1 translocation, the application of Drp1-siRNA alleviated Cr(VI)-induced ALT/AST leakage, caspase-3 activation, and apoptosis induction. The cytotoxicity in Drp1-si plus Cr(VI) treatment group was alleviated by the application of Glu/Mal, and aggravated by the application of ROT. Drp1 siRNA can further promote the inhibition of Glu/Mal on Cr(VI)-induced cytotoxicity, and also can further alleviate the aggravation of ROT on Cr(VI)-induced cytotoxicity. The dynamic network of mitochondria can meet the energy and metabolic needs of cells. The intracellular mitochondrial morphology network represents a perfect balance between the fusion/fission events [
24]. Mitochondrial dynamics is mainly regulated by Drp1, mitofusin (Mfn) 1 and Mfn2, and other mitochondrial fusion and fission-related proteins [
25]. Once the damage progressed to the irreversible stage, mitochondria will reveal excessive fission and fragmentation, mass decrease, as well as membrane integrity loss. The application of Drp1 siRNA or Mdivi-1 (a specific Drp1 inhibitor) can inhibit the conversion to punctate mitochondrial phenotype, weaken the insertion and oligomerization of pro-apoptotic Bax protein, thus attenuating cell apoptosis, reducing transient focal ischemia-induced infarct and neurological deficits. Drp1 activity could be altered under all kinds of stimulation, leading to mitochondrial dynamics abnormality and cell damage [
26]. It is reported that oxidative stress can enhance the activity of Drp1 and promote mitochondrial fragmentation and dysfunction mediated by Drp1, while the application of antioxidants can restore mitochondrial morphology [
27,
28]. Evidence suggested that the upstream transcription activator of Drp1 is p53, which could promote Drp1 transcription by binding to its promoter [
29]. In the present study, we confirmed that Cr(VI)-induced Drp1 modulation was dependent on MRCC I inhibition-mediated ROS production. Taken together, this study demonstrated that Drp1-mediated mitochondrial fragmentation contributes to Cr(VI)-induced MRCC I-dependent cytotoxicity.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







