
Metagenomic Analysis for Evaluating Change in Bacterial Diversity in TPH-Contaminated Soil after Soil Remediation

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Soil Sampling and Sample Description
2.2. Soil Chemical Properties and Total Petroleum Hydrocarbon (TPH) Analysis
2.3. Metagenomic Analysis
2.3.1. DNA Extraction, Polymerase Chain Reaction (PCR) Amplification, and 16S rRNA Gene Amplicon Sequencing
2.3.2. Bioinformatic Analysis
2.4. Data Analysis
3. Results and Discussion
3.1. Soil Chemical Properties and TPH Concentration
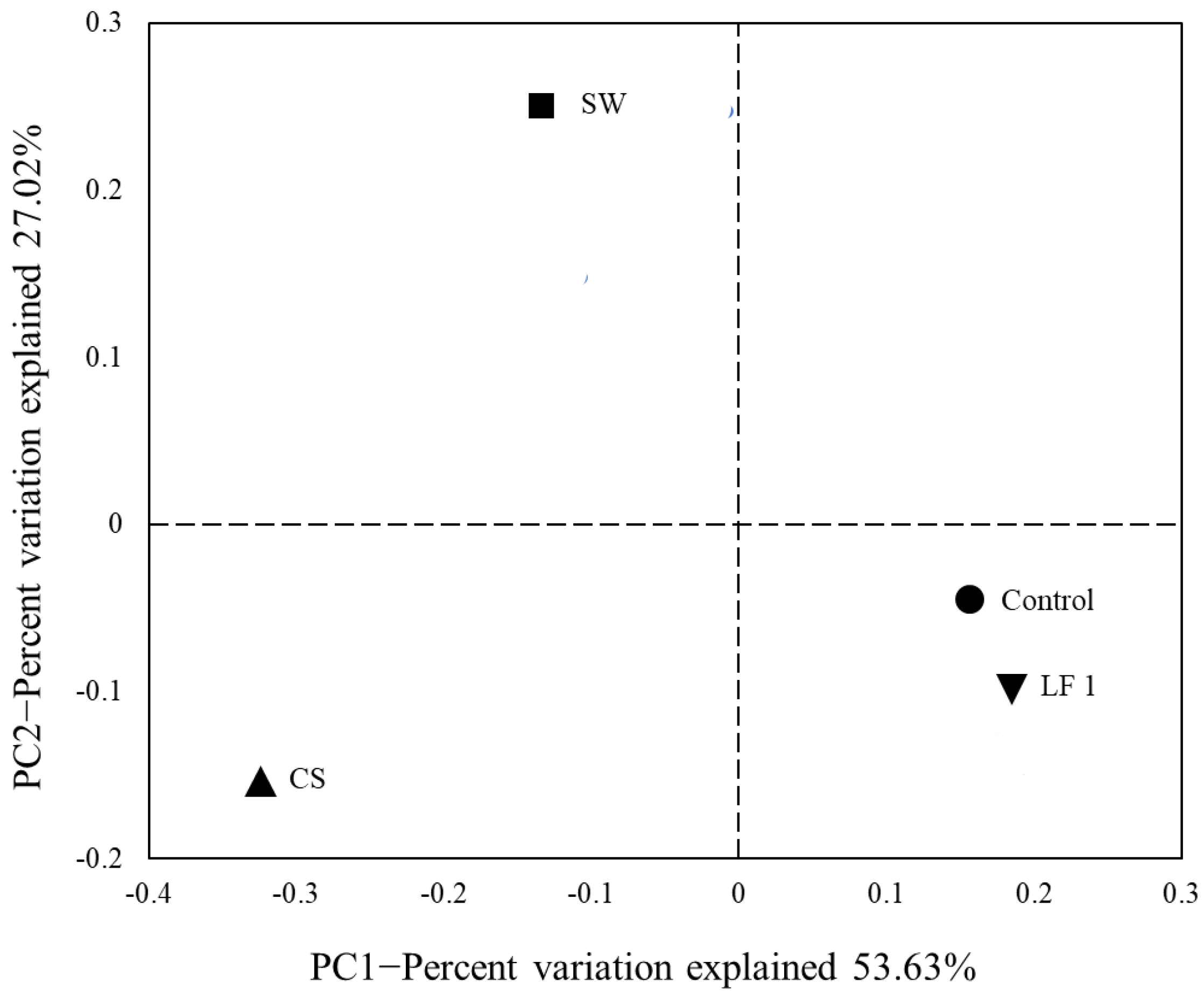
3.2. Soil Bacterial Diversity Analysis
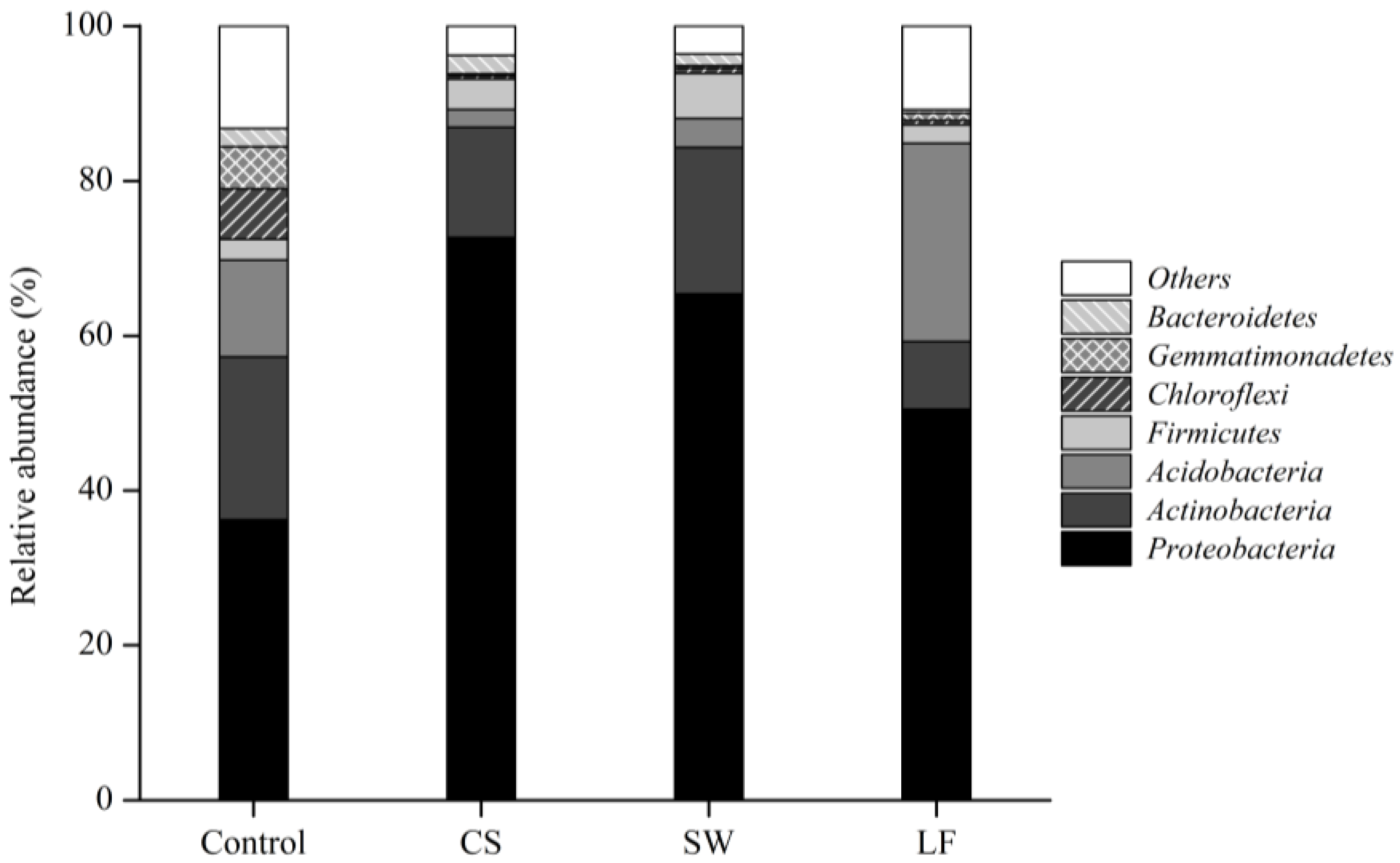
3.3. Varied Bacterial Distribution in Soil
3.4. Effect of TPH-Degrading Bacteria in Contaminated and Remediated Soil
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banks, M.K.; Mallede, H.; Rathbone, K. Rhizosphere microbial characterization in petroleum-contaminated soil. Soil Sediment Contam. 2003, 12, 371–385. [Google Scholar] [CrossRef]
- Jung, S. Environmental impact assessment schemes considering fate and transport of soil contaminants. Korea Env. Inst. 2003, 1, 8–28. [Google Scholar]
- Wang, J.; Zhang, Z.; Su, Y.; He, W.; He, F.; Song, H. Phytoremediation of petroleum polluted soil. Pet. Sci. 2008, 5, 167–171. [Google Scholar] [CrossRef] [Green Version]
- Kim, J. Policy of Soil Environmental Conservation-Current Issues and the Future. Environ. Preserv. 2005, 1, 6–9. [Google Scholar]
- Mulligan, C.; Yong, R.; Gibbs, B. Remediation technologies for metal-contaminated soils and groundwater: An evaluation. Eng. Geol. 2001, 60, 193–207. [Google Scholar] [CrossRef]
- Mann, M.J. Full-scale and pilot-scale soil washing. J. Hazard. Mater. 1999, 66, 119–136. [Google Scholar] [CrossRef]
- Besalatpour, A.; Hajabbasi, M.; Khoshgoftarmanesh, A.; Dorostkar, V. Landfarming process effects on biochemical properties of petroleum-contaminated soils. Soil Sediment Contam. 2011, 20, 234–248. [Google Scholar] [CrossRef]
- Kuo, Y.C.; Wang, S.Y.; Kao, C.M.; Chen, C.W.; Sung, W.P. Using enhanced landfarming system to remediate diesel oil-contaminated soils. Appl. Mech. Mater. 2021, 121–126, 554–558. [Google Scholar] [CrossRef]
- Wang, S.-Y.; Kuo, Y.-C.; Hong, A.; Chang, Y.-M.; Kao, C.-M. Bioremediation of diesel and lubricant oil-contaminated soils using enhanced landfarming system. Chemosphere 2016, 164, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Chaiyaraksa, C.; Sriwiriyanuphap, N. Batch washing of cadmium from soil and sludge by a mixture of Na2S2O5 and Na2EDTA. Chemosphere 2004, 56, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, K.; Walker, L.; Vigoren, L.; Bartel, J. Remediation of spilled petroleum hydrocarbons by in situ landfarming at an arctic site. Cold Reg. Sci. Technol. 2004, 40, 31–39. [Google Scholar] [CrossRef]
- Silva-Castro, G.; Uad, I.; Gónzalez-López, J.; Fandino, C.; Toledo, F.; Calvo, C. Application of selected microbial consortia combined with inorganic and oleophilic fertilizers to recuperate oil-polluted soil using land farming technology. Clean Technol. Environ. Policy 2012, 14, 719–726. [Google Scholar] [CrossRef]
- Liao, X.; Wu, Z.; Li, Y.; Cao, H.; Su, C. Effect of various chemical oxidation reagents on soil indigenous microbial diversity in remediation of soil contaminated by PAHs. Chemosphere 2019, 226, 483–491. [Google Scholar] [CrossRef]
- Hirsch, P.R.; Mauchline, T.H. The importance of the microbial N cycle in soil for crop plant nutrition. Adv. Appl. Microbiol. 2015, 93, 45–71. [Google Scholar] [PubMed]
- Lladó, S.; López-Mondéjar, R.; Baldrian, P. Forest soil bacteria: Diversity, involvement in ecosystem processes, and response to global change. Microbiol. Mol. Biol. Rev. 2017, 81, e00063-16. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, A.C.; Smith, K. Soil microbial diversity and the sustainability of agricultural soils. Plant Soil 1995, 170, 75–86. [Google Scholar] [CrossRef]
- Amann, R.I.; Ludwig, W.; Schleifer, K.-H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 1995, 59, 143–169. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, M.; Suzuki, K.; Onodera, M.; Shinano, T.; Osaki, M. Analysis of bacterial communities in soil by PCR–DGGE targeting protease genes. Soil Biol. Biochem. 2007, 39, 2777–2784. [Google Scholar] [CrossRef]
- Lukow, T.; Dunfield, P.F.; Liesack, W. Use of the T-RFLP technique to assess spatial and temporal changes in the bacterial community structure within an agricultural soil planted with transgenic and non-transgenic potato plants. FEMS Microbiol. Ecol. 2000, 32, 241–247. [Google Scholar] [CrossRef]
- Eickhorst, T.; Tippkötter, R. Improved detection of soil microorganisms using fluorescence in situ hybridization (FISH) and catalyzed reporter deposition (CARD-FISH). Soil Biol. Biochem. 2008, 40, 1883–1891. [Google Scholar] [CrossRef]
- Kuppusamy, S.; Thavamani, P.; Megharaj, M.; Venkateswarlu, K.; Lee, Y.B.; Naidu, R. Pyrosequencing analysis of bacterial diversity in soils contaminated long-term with PAHs and heavy metals: Implications to bioremediation. J. Hazard. Mater. 2016, 317, 169–179. [Google Scholar] [CrossRef]
- Jovel, J.; Patterson, J.; Wang, W.; Hotte, N.; O’Keefe, S.; Mitchel, T.; Perry, T.; Kao, D.; Mason, A.L.; Madsen, K.L. Characterization of the gut microbiome using 16S or shotgun metagenomics. Front. Microbiol. 2016, 7, 459. [Google Scholar] [CrossRef] [Green Version]
- Huaidong, H.; Waichin, L.; Riqing, Y.; Zhihong, Y. Illumina-based analysis of bulk and rhizosphere soil bacterial communities in paddy fields under mixed heavy metal contamination. Pedosphere 2017, 27, 569–578. [Google Scholar]
- Walkley, A.; Black, I.A. An examination of the Degtjareff method for determining soil organic matter, and a proposed modification of the chromic acid titration method. Soil Sci. 1934, 37, 29–38. [Google Scholar] [CrossRef]
- Bray, R.H.; Kurtz, L. Determination of total, organic, and available forms of phosphorus in soils. Soil Sci. 1945, 59, 39–46. [Google Scholar] [CrossRef]
- John, M.K. Colorimetric determination of phosphorus in soil and plant materials with ascorbic acid. Soil Sci. 1970, 109, 214–220. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Good, I.J. The population frequencies of species and the estimation of population parameters. Biometrika 1953, 40, 237–264. [Google Scholar] [CrossRef]
- Huang, Y.; Pan, H.; Wang, Q.; Ge, Y.; Liu, W.; Christie, P. Enrichment of the soil microbial community in the bioremediation of a petroleum-contaminated soil amended with rice straw or sawdust. Chemosphere 2019, 224, 265–271. [Google Scholar] [CrossRef]
- Sutton, N.B.; Maphosa, F.; Morillo, J.A.; Al-Soud, W.A.; Langenhoff, A.A.; Grotenhuis, T.; Rijnaarts, H.H.; Smidt, H. Impact of long-term diesel contamination on soil microbial community structure. Appl. Environ. Microbiol. 2013, 79, 619–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Im, J.; Yang, K.; Jho, E.H.; Nam, K. Effect of different soil washing solutions on bioavailability of residual arsenic in soils and soil properties. Chemosphere 2015, 138, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, H.; Wang, H.; Li, Q.; Li, Y. Effect of soil washing on heavy metal removal and soil quality: A two-sided coin. Ecotoxicol. Environ. Saf. 2020, 203, 110981. [Google Scholar] [CrossRef] [PubMed]
- Hejazi, R.F.; Husain, T.; Khan, F.I. Landfarming operation of oily sludge in arid region—human health risk assessment. J. Hazard. Mater. 2003, 99, 287–302. [Google Scholar] [CrossRef]
- Paudyn, K.; Rutter, A.; Rowe, R.K.; Poland, J.S. Remediation of hydrocarbon contaminated soils in the Canadian Arctic by landfarming. Cold Reg. Sci. Technol. 2008, 53, 102–114. [Google Scholar] [CrossRef]
- Marin, J.; Hernandez, T.; Garcia, C. Bioremediation of oil refinery sludge by landfarming in semiarid conditions: Influence on soil microbial activity. Environ. Res. 2005, 98, 185–195. [Google Scholar] [CrossRef]
- Cerqueira, V.S.; Hollenbach, E.B.; Maboni, F.; Vainstein, M.H.; Camargo, F.A.; Maria do Carmo, R.P.; Bento, F.M. Biodegradation potential of oily sludge by pure and mixed bacterial cultures. Bioresour. Technol. 2011, 102, 11003–11010. [Google Scholar] [CrossRef] [Green Version]
- Chikere, C.B.; Tekere, M.; Adeleke, R. Microbial communities in field-scale oil-polluted soil remediation using 16S rRNA amplicon sequencing. Int. J. Environ. Stud. 2020, 78, 410–426. [Google Scholar] [CrossRef]
- Jiang, X.-T.; Peng, X.; Deng, G.-H.; Sheng, H.-F.; Wang, Y.; Zhou, H.-W.; Tam, N.F.-Y. Illumina sequencing of 16S rRNA tag revealed spatial variations of bacterial communities in a mangrove wetland. Microb. Ecol. 2013, 66, 96–104. [Google Scholar] [CrossRef]
- Kersters, K.; De Vos, P.; Gillis, M.; Swings, J.; Vandamme, P.; Stackebrandt, E. Introduction to the Proteobacteria. In The Prokaryotes: A Handbook on the Biology of Bacteria; Springer: Berlin/Heidelberg, Germany, 2006; Volume 5, pp. 3–37. [Google Scholar]
- Kostka, J.E.; Prakash, O.; Overholt, W.A.; Green, S.J.; Freyer, G.; Canion, A.; Delgardio, J.; Norton, N.; Hazen, T.C.; Huettel, M. Hydrocarbon-degrading bacteria and the bacterial community response in Gulf of Mexico beach sands impacted by the Deepwater Horizon oil spill. Appl. Environ. Microbiol. 2011, 77, 7962–7974. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Wu, J.; Zhang, X.; Ye, X. Effect of bioaugmentation and biostimulation on hydrocarbon degradation and microbial community composition in petroleum-contaminated loessal soil. Chemosphere 2019, 237, 124456. [Google Scholar] [CrossRef]
- Abbasian, F.; Palanisami, T.; Megharaj, M.; Naidu, R.; Lockington, R.; Ramadass, K. Microbial diversity and hydrocarbon degrading gene capacity of a crude oil field soil as determined by metagenomics analysis. Biotechnol. Prog. 2016, 32, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Shahi, A.; Aydin, S.; Ince, B.; Ince, O. Reconstruction of bacterial community structure and variation for enhanced petroleum hydrocarbons degradation through biostimulation of oil contaminated soil. Chem. Eng. J. 2016, 306, 60–66. [Google Scholar] [CrossRef]
- Smith, E.; Thavamani, P.; Ramadass, K.; Naidu, R.; Srivastava, P.; Megharaj, M. Remediation trials for hydrocarbon-contaminated soils in arid environments: Evaluation of bioslurry and biopiling techniques. Int. Biodeterior. Biodegrad. 2015, 101, 56–65. [Google Scholar] [CrossRef]
- Spain, A.M.; Krumholz, L.R.; Elshahed, M.S. Abundance, composition, diversity and novelty of soil Proteobacteria. ISME J. 2009, 3, 992–1000. [Google Scholar] [CrossRef]
- Labbé, D.; Margesin, R.; Schinner, F.; Whyte, L.G.; Greer, C.W. Comparative phylogenetic analysis of microbial communities in pristine and hydrocarbon-contaminated Alpine soils. FEMS Microbiol. Ecol. 2007, 59, 466–475. [Google Scholar] [CrossRef]
- Dos Santos, H.F.; Cury, J.C.; Do Carmo, F.L.; Dos Santos, A.L.; Tiedje, J.; Van Elsas, J.D.; Rosado, A.S.; Peixoto, R.S. Mangrove bacterial diversity and the impact of oil contamination revealed by pyrosequencing: Bacterial proxies for oil pollution. PLoS ONE 2011, 6, e16943. [Google Scholar] [CrossRef] [Green Version]
- Gertler, C.; Näther, D.J.; Cappello, S.; Gerdts, G.; Quilliam, R.S.; Yakimov, M.M.; Golyshin, P.N. Composition and dynamics of biostimulated indigenous oil-degrading microbial consortia from the Irish, North and Mediterranean Seas: A mesocosm study. FEMS Microbiol. Ecol. 2012, 81, 520–536. [Google Scholar] [CrossRef]
- Ostermann, A.; Gao, J.; Welp, G.; Siemens, J.; Roelcke, M.; Heimann, L.; Nieder, R.; Xue, Q.; Lin, X.; Sandhage-Hofmann, A.; et al. Identification of soil contamination hotspots with veterinary antibiotics using heavy metal concentrations and leaching data--a field study in China. Environ. Monit. Assess. 2014, 186, 7693–7707. [Google Scholar] [CrossRef]
- Dong, Y.; Lang, Z.; Kong, X.; Lu, D.; Liu, Z. Kinetic and multidimensional profiling of accelerated degradation of oil sludge by biostimulation. Environ. Sci. Process. Impacts 2015, 17, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Ramadass, K.; Smith, E.; Palanisami, T.; Mathieson, G.; Srivastava, P.; Megharaj, M.; Naidu, R. Evaluation of constraints in bioremediation of weathered hydrocarbon-contaminated arid soils through microcosm biopile study. Int. J. Environ. Sci. Technol. 2015, 12, 3597–3612. [Google Scholar] [CrossRef] [Green Version]
- Militon, C.; Boucher, D.; Vachelard, C.; Perchet, G.; Barra, V.; Troquet, J.; Peyretaillade, E.; Peyret, P. Bacterial community changes during bioremediation of aliphatic hydrocarbon-contaminated soil. FEMS Microbiol. Ecol. 2010, 74, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Chikere, C.B.; Okoye, A.U.; Okpokwasili, G.C. Microbial community profiling of active oleophilic bacteria involved in bioreactor-based crude-oil polluted sediment treatment. J. Appl. Environ. Microbiol. 2016, 4, 1–20. [Google Scholar]
- Siles, J.A.; Margesin, R. Insights into microbial communities mediating the bioremediation of hydrocarbon-contaminated soil from an Alpine former military site. Appl. Microbiol. Biotechnol. 2018, 102, 4409–4421. [Google Scholar] [CrossRef] [Green Version]
- Zrafi-Nouira, I.; Saidane-Mosbahi, D.; Abdelghani, S.; Bakhrouf, A.; Rouabhia, M. Crude Oil Metagenomics for Better Bioremediation of Contaminated Environments. In Introduction to Enhanced Oil Recovery (EOR) Processes and Bioremediation of Oil-Contaminated Sites; Intech: Rijeka, Croatia, 2012; p. 261. [Google Scholar]
- Benedek, T.; Vajna, B.; Táncsics, A.; Márialigeti, K.; Lányi, S.; Máthé, I. Remarkable impact of PAHs and TPHs on the richness and diversity of bacterial species in surface soils exposed to long-term hydrocarbon pollution. World J. Microbiol. Biotechnol. 2013, 29, 1989–2002. [Google Scholar] [CrossRef]
- Lu, L.; Huggins, T.; Jin, S.; Zuo, Y.; Ren, Z.J. Microbial metabolism and community structure in response to bioelectrochemically enhanced remediation of petroleum hydrocarbon-contaminated soil. Environ. Sci. Technol. 2014, 48, 4021–4029. [Google Scholar] [CrossRef]
- Wu, M.; Ye, X.; Chen, K.; Li, W.; Yuan, J.; Jiang, X. Bacterial community shift and hydrocarbon transformation during bioremediation of short-term petroleum-contaminated soil. Environ. Pollut. 2017, 223, 657–664. [Google Scholar] [CrossRef]
- Roy, A.; Dutta, A.; Pal, S.; Gupta, A.; Sarkar, J.; Chatterjee, A.; Saha, A.; Sarkar, P.; Sar, P.; Kazy, S.K. Biostimulation and bioaugmentation of native microbial community accelerated bioremediation of oil refinery sludge. Bioresour. Technol. 2018, 253, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Fowler, S.J.; Laban, N.A.; Dong, X.; Sensen, C.W.; Foght, J.; Gieg, L.M. Comparative analysis of metagenomes from three methanogenic hydrocarbon-degrading enrichment cultures with 41 environmental samples. ISME J. 2015, 9, 2028–2045. [Google Scholar] [CrossRef] [Green Version]
- Margesin, R.; Labbe, D.; Schinner, F.; Greer, C.; Whyte, L. Characterization of hydrocarbon-degrading microbial populations in contaminated and pristine alpine soils. Appl. Environ. Microbiol. 2003, 69, 3085–3092. [Google Scholar] [CrossRef] [Green Version]
- Prince, R.C.; Amande, T.J.; McGenity, T.J. Prokaryotic hydrocarbon degraders. In Taxonomy, Genomics and Ecophysiology of Hydrocarbon-Degrading Microbes; Springer: New York, NY, USA, 2019; pp. 1–39. [Google Scholar]
- Kaplan, C.W.; Kitts, C.L. Bacterial succession in a petroleum land treatment unit. Appl. Environ. Microbiol. 2004, 70, 1777–1786. [Google Scholar] [CrossRef] [Green Version]
- Whyte, L.; Schultz, A.; Van Beilen, J.; Luz, A.; Pellizari, V.; Labbé, D.; Greer, C. Prevalence of alkane monooxygenase genes in Arctic and Antarctic hydrocarbon-contaminated and pristine soils. FEMS Microbiol. Ecol. 2002, 41, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Khudur, L.S.; Gleeson, D.B.; Ryan, M.H.; Shahsavari, E.; Haleyur, N.; Nugegoda, D.; Ball, A.S. Implications of co-contamination with aged heavy metals and total petroleum hydrocarbons on natural attenuation and ecotoxicity in Australian soils. Environ. Pollut. 2018, 243, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Frick, C.; Germida, J.; Farrell, R. Assessment of Phytoremediation as an In-Situ Technique for Cleaning Oil-Contaminated Sites; Petroleum Technology Alliance of Canada (PTAC): Calgary, AB, Canada, 1998; pp. 105a–124a. [Google Scholar]
- Fuentes, S.; Méndez, V.; Aguila, P.; Seeger, M. Bioremediation of petroleum hydrocarbons: Catabolic genes, microbial communities, and applications. Appl. Microbiol. Biotechnol. 2014, 98, 4781–4794. [Google Scholar] [CrossRef]
- Head, I.M.; Jones, D.M.; Röling, W.F. Marine microorganisms make a meal of oil. Nat. Rev. Microbiol. 2006, 4, 173–182. [Google Scholar] [CrossRef] [PubMed]
- de la Cueva, S.C.; Rodríguez, C.H.; Cruz, N.O.S.; Contreras, J.A.R.; Miranda, J.L. Changes in bacterial populations during bioremediation of soil contaminated with petroleum hydrocarbons. Water Air Soil Pollut. 2016, 227, 91. [Google Scholar] [CrossRef]
- Khudur, L.S.; Shahsavari, E.; Webster, G.T.; Nugegoda, D.; Ball, A.S. The impact of lead co-contamination on ecotoxicity and the bacterial community during the bioremediation of total petroleum hydrocarbon-contaminated soils. Environ. Pollut. 2019, 253, 939–948. [Google Scholar] [CrossRef]
- Wu, M.; Dick, W.A.; Li, W.; Wang, X.; Yang, Q.; Wang, T.; Xu, L.; Zhang, M.; Chen, L. Bioaugmentation and biostimulation of hydrocarbon degradation and the microbial community in a petroleum-contaminated soil. Int. Biodeterior. Biodegrad. 2016, 107, 158–164. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Treatment | pH | Electric Conductivity (EC) | Soil Organic Matter (SOM) | Available Phosphorus (Av.P) | Cation Exchange Capacity (CEC) | Total Nitrogen (TN) | TPH |
|---|---|---|---|---|---|---|---|
| 1:5 | dS m−1 | % | mg kg−1 | cmolc kg−1 | % | mg kg−1 | |
| Control | 6.23 ± 0.58 b | 0.38 ± 0.06 a | 1.54 ± 0.04 a | 24.6 ± 11.2 a | 14.9 ± 4.00 b | 0.90 ± 0.36 a | 77 ± 17 b |
| CS | 6.22 ± 0.69 b | 0.29 ± 0.10 a | 0.90 ± 0.22 b | 21.7 ± 12.1 a | 8.91 ± 2.90 c | 0.09 ± 0.09 b | 2,690 ± 680 a |
| SW | 7.23 ± 0.11 a | 0.11 ± 0.02 b | 0.81 ± 0.02 b | 2.86 ± 1.57 b | 11.7 ± 2.40 bc | 0.13 ± 0.16 b | 111 ± 39 b |
| LF | 6.90 ± 0.06 ab | 0.28 ± 0.10 a | 1.06 ± 0.01 b | 18.7 ± 0.10 a | 37.4 ± 0.80 a | 0.93 ± 0.38 a | 249 ± 39 b |
| Sample Group | Reads | Good’s Coverage (%) | OTUs | Chao1 | Shannon |
|---|---|---|---|---|---|
| Control | 14,907 ± 2188 d | 97.99 ± 0.12 | 1265 ± 308 a | 1475 ± 340 a | 8.58 ± 0.62 a |
| CS | 35,782 ± 12,110 a | 99.95 ± 0.03 | 296 ± 13 c | 308 ± 24 c | 4.76 ± 1.30 c |
| SW | 29,979 ± 5923 b | 99.94 ± 0.08 | 305 ± 47 c | 316 ± 63 c | 5.94 ± 0.57 b |
| LF | 21,345 ± 3826 c | 98.99 ± 0.16 | 815 ± 111 b | 1000 ± 101 b | 7.06 ± 0.29 b |
| Phylum | Genus | Relative Abundance (%) | |||
|---|---|---|---|---|---|
| Class | Control | CS | SW | LF | |
| Actinobacteria | |||||
| Actinobacteria | Corynebacterium | 0.01 | 0.85 | 1.20 | - |
| Dietzia | - | 0.08 | - | - | |
| Kocuria | 0.06 | 0.10 | - | - | |
| Microbacterium | 0.02 | 0.31 | 0.03 | 0.01 | |
| Micrococcus | - | 0.33 | 0.05 | - | |
| Nocardioides | 0.90 | 1.41 | 0.82 | 0.24 | |
| Rhodococcus | - | - | 0.01 | 0.05 | |
| Streptomyces | 1.49 | 0.04 | 0.08 | 0.11 | |
| Bacteroidetes | |||||
| Flavobacteriia | Chryseobacterium | - | 0.01 | 0.08 | - |
| Flavobacterium | 0.03 | 0.04 | 0.09 | - | |
| Firmicutes | |||||
| Bacilli | Bacillus | 0.26 | 0.29 | 0.32 | 0.93 |
| Paenibacillus | 0.02 | 0.06 | - | 0.04 | |
| Staphylococcus | 0.03 | 1.37 | 3.39 | - | |
| Streptococcus | - | 0.52 | 0.92 | - | |
| Proteobacteria | |||||
| Alphaproteobacteria | Azospirillum | 0.05 | 0.06 | 0.13 | 0.43 |
| Methylobacterium | 0.17 | 0.19 | 0.13 | 0.01 | |
| Paracoccus | - | 0.77 | 0.03 | - | |
| Rhizobium | 0.24 | 0.09 | 0.52 | 0.02 | |
| Betaproteobacteria | Acidovorax | 0.07 | 0.60 | 0.99 | 0.02 |
| Burkholderia | 0.25 | - | - | 0.01 | |
| Gammaproteobacteria | Acinetobacter | 0.03 | 2.92 | 5.54 | - |
| Alkanindiges | - | 0.07 | 0.01 | - | |
| Immundisolibacter | 0.02 | 1.82 | 3.26 | 8.63 | |
| Pseudomonas | 0.15 | 1.72 | 3.38 | 0.05 | |
| Pseudoxanthomonas | 0.10 | 2.85 | 0.96 | - | |
| Rhodanobacter | 0.01 | 0.64 | 0.21 | 1.70 | |
| Stenotrophomonas | 0.04 | 0.03 | 0.10 | - | |
| Thermomonas | 0.10 | 3.44 | 1.25 | 0.04 | |
| Total | 4.05 | 20.61 | 23.49 | 12.29 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-W.; Hong, Y.-K.; Kim, H.-S.; Oh, E.-J.; Park, Y.-H.; Kim, S.-C. Metagenomic Analysis for Evaluating Change in Bacterial Diversity in TPH-Contaminated Soil after Soil Remediation. Toxics 2021, 9, 319. https://doi.org/10.3390/toxics9120319
Kim J-W, Hong Y-K, Kim H-S, Oh E-J, Park Y-H, Kim S-C. Metagenomic Analysis for Evaluating Change in Bacterial Diversity in TPH-Contaminated Soil after Soil Remediation. Toxics. 2021; 9(12):319. https://doi.org/10.3390/toxics9120319
Chicago/Turabian StyleKim, Jin-Wook, Young-Kyu Hong, Hyuck-Soo Kim, Eun-Ji Oh, Yong-Ha Park, and Sung-Chul Kim. 2021. "Metagenomic Analysis for Evaluating Change in Bacterial Diversity in TPH-Contaminated Soil after Soil Remediation" Toxics 9, no. 12: 319. https://doi.org/10.3390/toxics9120319
APA StyleKim, J. -W., Hong, Y. -K., Kim, H. -S., Oh, E. -J., Park, Y. -H., & Kim, S. -C. (2021). Metagenomic Analysis for Evaluating Change in Bacterial Diversity in TPH-Contaminated Soil after Soil Remediation. Toxics, 9(12), 319. https://doi.org/10.3390/toxics9120319