Esophageal Cancer: Genomic and Molecular Characterization, Stem Cell Compartment and Clonal Evolution



Abstract
:1. Introduction
2. Genetic Alterations of Esophageal Cancer
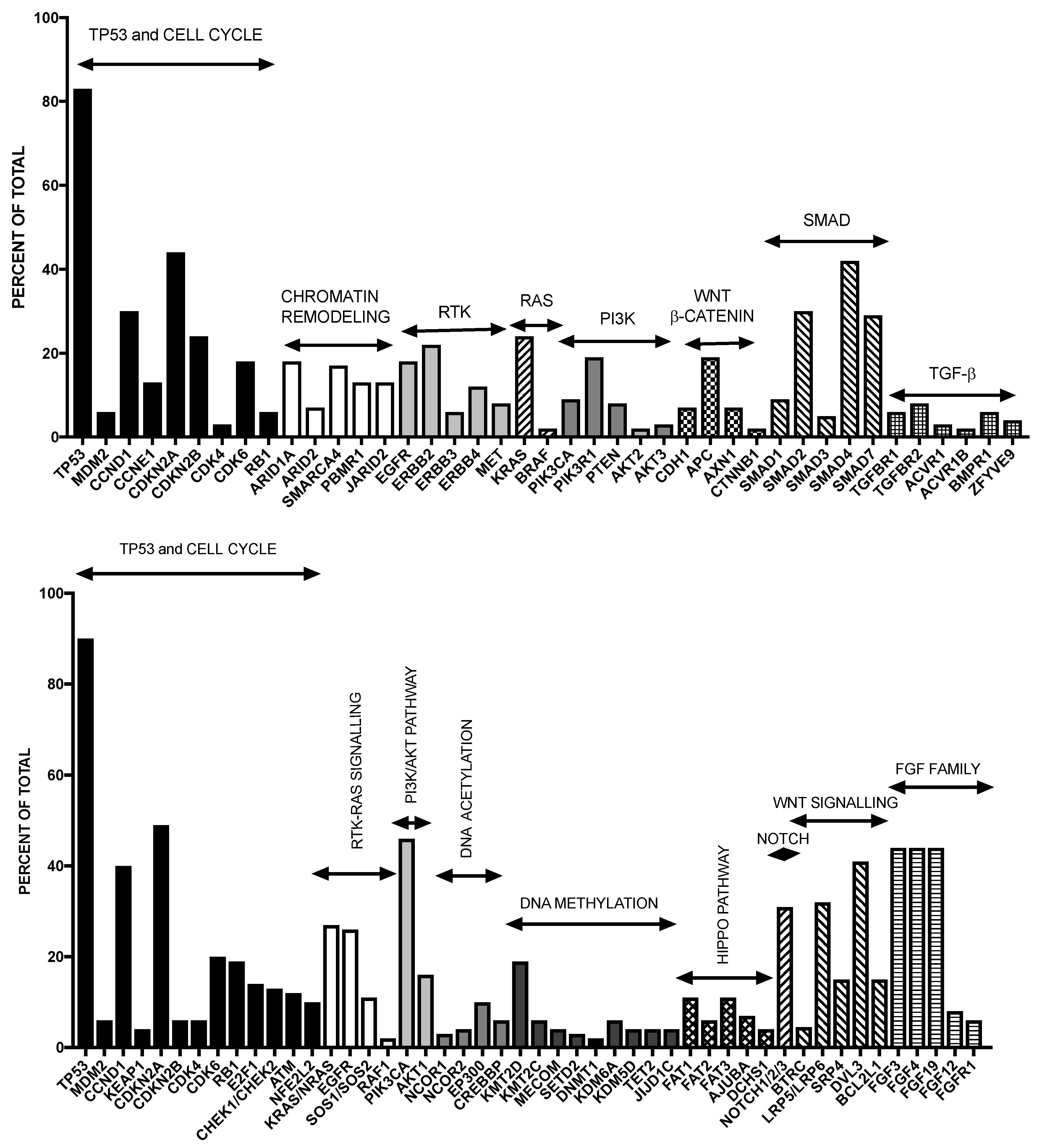
2.1. Molecular Abnormalities of EAC
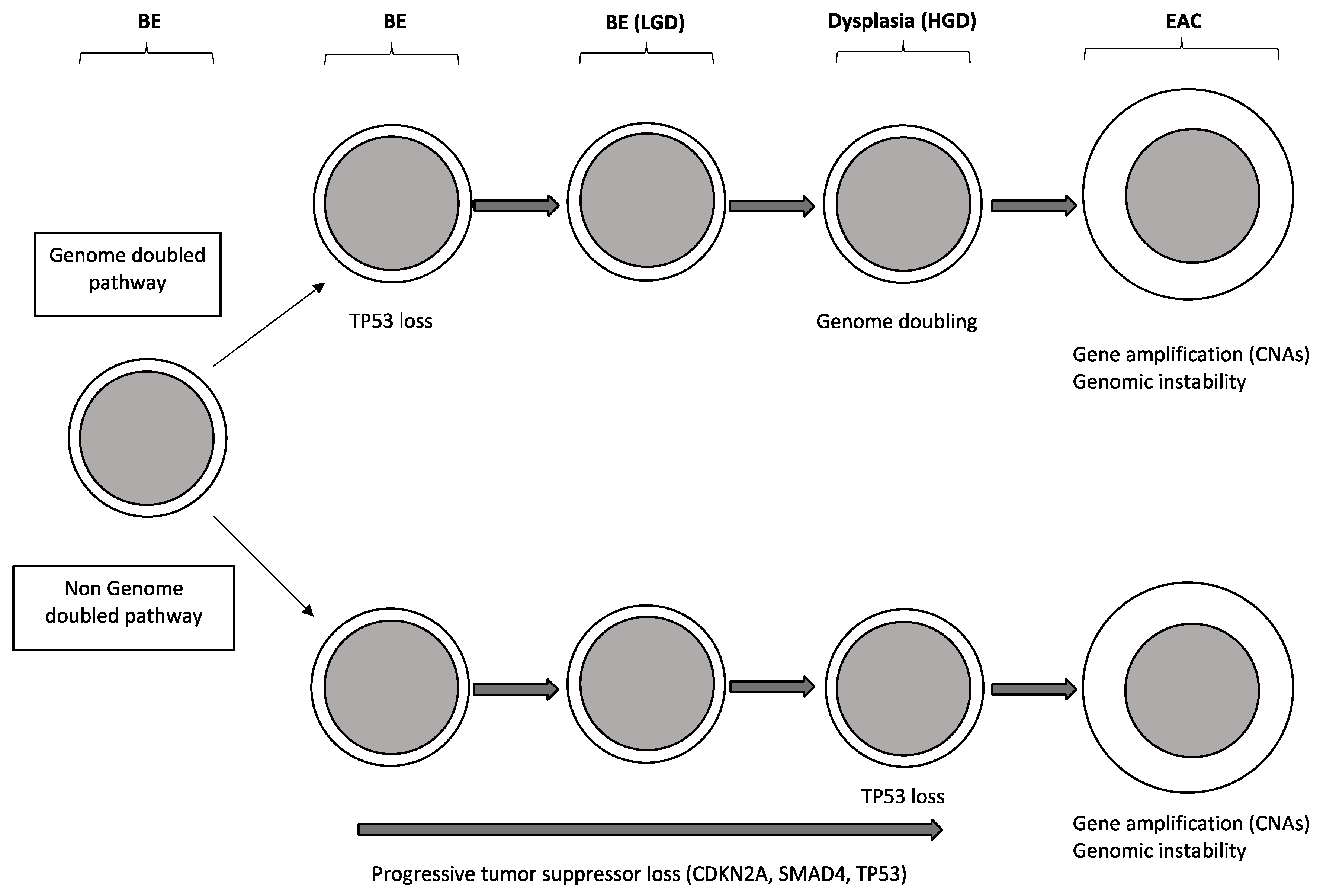
2.2. Molecular Abnormalities of Barrett’s Esophagus
2.3. Molecular Abnormalities of ESCC
2.4. Comparison between EAC and ESCC
2.5. Gene Expression Studies
3. Normal Esophageal Stem Cells
4. Cellular Origin of Barrett’s Esophagus
5. Esophageal Cancer Stem Cells
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Holmes, R.S.; Vaughan, T.L. Epidemiology and pathogenesis of esophageal cancer. Semin. Radiat. Oncol. 2007, 17, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Lagergren, J.; Smyth, E.; Cunningham, D.; Lagergren, P. Oesophageal cancer. Lancet 2017, in press. [Google Scholar] [CrossRef]
- Smyth, E.C.; Lagergren, J.; Fitzgerald, R.C.; Lordick, F.; Shah, M.A.; Lagergren, P.; Cunningham, D. Oesophageal cancer. Nat. Rev. Dis. Primers 2017, 3, 17048. [Google Scholar] [CrossRef] [PubMed]
- Contino, G.; Vaughan, T.L.; Whiteman, D.; Fitzgerald, R.C. The evolving genomic landscape of Barrett’s esophagus and esophageal adenocarcinoma. Gastroenterology 2017, 153, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Lordick, E.; Kang, J.K.; Chung, H.C.; Salman, P.; Oh, S.C.; Bodoky, G.; Kurteva, G.; Volovat, C.; Moiseyenko, V.M.; Gorbunova, V.; et al. Capecitabine and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): A randomized, open-label phase 3 trial. Lancet Oncol. 2013, 14, 490–499. [Google Scholar] [CrossRef]
- Waddell, T.; Chau, I.; Cunningham, D.; Gonzalez, D.; Okines, A.F.; Okines, C.; Wotherspoon, A.; Saffery, C.; Middleton, G.; Wadsley, J.; et al. Epirubicin, oxaliplatin, and capecitabine withg or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): A randomized, open-label phase 3 trial. Lancet Oncol. 2013, 14, 481–489. [Google Scholar] [CrossRef]
- Dulak, A.M.; Stojanov, P.; Peng, S.; Lawrence, M.S.; Fox, C.; Stewart, C.; Bandla, S.; Imamura, Y.; Schumacher, S.E.; Shefler, E.; et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat. Genet. 2013, 45, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Nones, K.; Waddel, N.; Wayte, N.; Patch, A.M.; Bailey, P.; Newell, F.; Holmes, O.; Fink, J.L.; Quinn, M.C.J.; Tang, Y.H.; et al. Genomic catastrophes frequently arise in esophageal adenocarcinoma and drive tumorigenesis. Nat. Commun. 2014, 5, 5224. [Google Scholar] [CrossRef] [PubMed]
- Secrier, M.; Li, X.; de Silva, N.; Eldridge, M.D.; Contino, G.; Bornschein, J.; MacRae, S.; Grehan, N.; O’Donovan, M.; Miremadi, A.; et al. Mutational signatures in esophageal adenocarcinoma define etiologically distinct subgroups with therapeutic relevance. Nat. Genet. 2016, 48, 1131–1140. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.C.; Hao, J.J.; Nagata, Y.; Xu, L.; Shang, L.; Meng, X.; Sato, Y.; Okuno, Y.; Varela, A.M.; Ding, L.W.; et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Li, L.; Ou, Y.; Gao, Z.; Li, E.; Li, X.; Zhang, W.; Wang, J.; Xu, L.; Zhou, Y.; et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014, 509, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.B.; Chen, Z.L.; Li, J.G.; Hu, X.D.; Shi, X.J.; Sun, Z.M.; Zhang, F.; Zhao, Z.R.; Li, Z.T.; Liu, Z.Y.; et al. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Zhou, Y.; Li, H.; Xiong, T.; Li, S.; Bi, Y.; Kong, P.; Wang, F.; Cui, H.; Li, Y.; et al. Whole-genomic sequencing reveals diverse models of structural variations in esophageal squamous cell carcinoma. Am. J. Hum. Genet. 2016, 98, 256–274. [Google Scholar] [CrossRef] [PubMed]
- Findlay, J.M.; Castro-Giner, F.; Makino, S.; Rayaner, E.; Kartsonaki, C.; Cross, W.; Kovac, M.; Ulahannan, D.; Palles, C.; Gillies, R.S.; et al. Differential clonal evolution in oesophageal cancers in response to neo-adjuvant chemotherapy. Nat. Commun. 2016, 7, 11111. [Google Scholar] [CrossRef] [PubMed]
- Murugaesu, N.; Wilson, G.A.; Birkbak, N.; Watkins, T.B.; McGranahan, N.; Kumar, S.; Abbassi-Ghadi, N.; Salm, M.; Mitter, R.; Horswell, S.; et al. Tracking the genomic evolution of easophageal adenocarcinoma through neoadjuvant chemotherapy. Cancer Discov. 2015, 5, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Noorani, A.; Bornschein, J.; Lynch, A.G.; Secrier, M.; Achilleos, A.; Elridge, M.; Bower, L.; Weaver, J.; Crawte, J.; Ong, C.A.; et al. A comparative analysis of whole genome sequencing of esophageal adenocarcinoma pre- and post-chemotherapy. Genome Res. 2017, 27, 902–912. [Google Scholar] [CrossRef] [PubMed]
- Arber, N.; Shapira, I.; Ratan, J.; Stern, B.; Hibshoosh, H.; Moshkowitz, M.; Gammon, M.; Fabian, I.; Halpern, Z. Activation of c-K-Ras mutations in human gastrointestinal tumors. Gastroenterology 2000, 118, 1045–1050. [Google Scholar] [CrossRef]
- Su, Z.; Gay, L.J.; Strange, A.; Palles, C.; Band, G.; Whiteman, D.C.; Lescai, F.; Langford, C.; Nanji, M.; Edkins, S.; et al. The Esophageal Adenocarcinoma Genetic Consortium and the Welcome Trust Case Control Consortium. Common variants at the MHC locus at chromosome 16q24.1 predispose to Barrett’s esophagus. Nat. Genet. 2012, 44, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Levine, D.M.; EK, W.E.; Zhang, R.; Liu, X.; Onstad, L.; Sather, C.; Lao-Sirieix, P.; Gammon, M.D.; Corley, D.A.; Shaheen, N.J.; et al. A genome-wide association study identifies new susceptibility loci for esophageal adenocarcinoma and Barrett’s esophagus. Nat. Genet. 2013, 45, 1487–1493. [Google Scholar] [CrossRef] [PubMed]
- Gharahkhani, P.; Fritzgerald, R.C.; Vaughan, T.L.; Palles, C.; Gockel, I.; Tomlinson, I.; Buas, M.F.; May, A.; Gerges, C.; Anders, M.; et al. Genome-wide association studies in oesophageal adenocarcinoma and Barrett’s oesophagus: A large-scale meta-analysis. Lancet Oncol. 2016, 17, 1363–1373. [Google Scholar] [CrossRef]
- Gregsobn, E.; Bornschein, J.; Fitzgerald, R.C. Genetic progression of Barrett’s esophagus to esophageal adenocarcinoma. Br. J. Cancer 2016, 115, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Maley, C.C. Multistage carcinogenesis in Barrett’s esophagus. Cancer Lett. 2007, 245, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Leedham, S.J. Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett’s esophagus. Gut 2008, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Jiao, Y.; Bettegowda, C.; Hutfless, S.M.; Wang, Y.; David, S.; Cheng, Y.; Twaddell, W.S.; Latt, N.L.; Shin, E.J.; et al. Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov. 2012, 2, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Weaver, J.M.J.; Ross-Ines, C.S.; Shannon, N.; Lynch, A.G.; Forshew, T.; Barbera, M.; Murtaza, M.; Ong, C.A.; Lao-Sirieix, P.; Dunning, M.J. Ordering of mutations in preinvasive disease stages of esophageal carcinogenesis. Nat. Genet. 2014, 46, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Ross-Innes, C.S.; Becq, J.; Warren, A.; Cheetham, R.K.; Northen, H.; O’Donovan, M.; Malhotra, S.; di Pietro, M.; Ivakhno, S.; He, M.; et al. Whole-genome sequencing provides new insights into the clonal architecture of Barrett’s esophagus and esophageal adenocarcinoma. Nat. Genet. 2015, 47, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Stachler, M.D.; Taylor-Weiner, A.; Peng, S.; McKenna, A.; Agoston, A.T.; Odze, R.D.; Davison, J.M.; Nason, K.S.; Loda, M.; Leshchiner, I.; et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat. Genet. 2015, 47, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Shigati, H.; Baba, Y.; Watanabe, M.; Murata, A.; Ishimoto, T.; Iwatsuki, M.; Iwagami, S.; Nosho, S.; Baba, H. PI3KCA mutation is associated with a favorable prognosis among patients with curatively resected esophageal squamous cell carcinoma. Clin. Cancer Res. 2013, 19, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Galipeau, P.; Paulson, T.; Sanchez, C.A.; Arnaudo, J.; Liu, K.; Sather, C.L.; Kostadinov, R.L.; Odze, R.D.; Kuhner, M.K.; et al. Temporal and spatial evolution of somatic chromosomal alterations: A case-cohort study of Barrett’s esophagus. Cancer Prev. Res. 2014, 7, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.; Timmer, M.; Lau, C.; Calpe, S.; Sancho-Serra, M.D.C.; Straub, D.; Baker, A.M.; Meijer, S.L.; ten Kate, F.J.W.; Mallant-Hent, R.C.; et al. Dynamic clonal equilibrium and predetermined cancer risk in Barrett’s esophagus. Nat. Commun. 2016, 7, 12158. [Google Scholar] [CrossRef] [PubMed]
- Kaz, A.; Grady, W.; Stachler, M.; Bass, A.J. Genetic and epigenetic alterations in Barrett’s esophagus and esophageal adenocarcinoma. Gastroenterol. Clin. N. Am. 2015, 44, 473–489. [Google Scholar] [CrossRef] [PubMed]
- Kalatskaya, I. Overview of the major molecular alterations during progression from Barrett’s esophagus to esophageal adenocarcinoma. Ann. N. Y. Acad. Sci. 2016, 1381, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Cui, H.; Zhang, L.; Jia, Z.; Song, B.; Wang, F.; Li, Y.; Liu, J.; Kong, N.P.; Shi, R.; et al. Genomic analyses reveal FAM84B and the NOTCH pathway are associated with the progression of esophageal squamous cell carcinoma. Giga Sci. 2016, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, S.; Natsuizaka, M.; Yashira-Ohtani, Y.; Kalman, R.A.; Nakagawa, M.; Wu, L.; Klein-Szanto, A.J.; Herlyn, M.; Diehl, J.A.; Katz, J.P.; et al. NOTCH1 and NOTCH3 coordinate esophageal squamous differentiation through a CSL-dependent transcriptional network. Gastroenterology 2010, 139, 2113–2123. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Fuji, T.; Hawachi, H.; Miki, Y.; Omura, K.; Morita, K.; Kayamori, K.; Katsube, K.; Yamaguchi, A. Reduction of NOPTCH1 expression pertains to maturation abnormalities in keratinocytes in squamous neoplasms. Lab. Investig. 2012, 92, 688–702. [Google Scholar] [CrossRef] [PubMed]
- Onwuegbusi, B.A.; Atchison, A.; Chin, S.F.; Kranjac, T.; Mills, I.; Huang, Y.; Lao-Sirieix, P.; Caldas, C.; Fitzgerald, R.C. Impaired transforming growth factor beta signaling in Barrett’s carcinogenesis due to frequent SMAD4 inactivation. Gut 2006, 55, 764–774. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, J.; Song, S.; Li, Y.; Maru, D.M.; Mishra, B.; Davila, M.; Hofstetter, W.L.; Mishra, L. Dysfunctional transforming growth factor-β signaling with constitutively active Notch signaling in Barrett’s esophageal adenocarcinoma. Cancer 2011, 117, 3691–3702. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Maru, D.P.; Ajani, J.A.; Chan, C.H.; Honjo, S.; Lin, H.K.; Correa, A.; Hofstetter, W.L.; Davila, M.; Stroehlein, J.; et al. Loss of TGF-β adaptor β2SP activates NOTCH signaling and SOX9 expression in esophageal adenocarcinoma. Cancer Res. 2013, 73, 2159–2169. [Google Scholar] [CrossRef] [PubMed]
- Harrawa, M.; Suzuki, S.; Dobashi, Y.; Yamane, T.; Kono, K.; Enomoto, N.; Ooi, A. EGFR protein overexpression and gene amplification in squamous cell carcinomas of esophagus. Int. J. Cancer 2006, 118, 1173–1180. [Google Scholar]
- Okawa, T.; Michayllra, C.Z.; Kalabis, J.; Douglas, B.; Stairs, D.B.; Hiroshi Nakagawa, H.; Andl, C.; Johnstone, C.M.; Klein-Szanto, A.J.; Deiry, W.S.; et al. The functional interplay between EGFR overexpression, hTERT activation, p53 mutation in esophageal epithelial cells with activation of stromal fibroblasts induces tumor development, invasion and differentiation. Genes Dev. 2007, 21, 2788–2803. [Google Scholar] [CrossRef] [PubMed]
- Bettstetter, M.; Berezowska, S.; Keller, G.; Walch, A.; Feuchtinger, A.; Slotta-Huspenina, J.; Feith, M.; Drecoll, E.; Holfer, H. Epidermal growth factor receptor, phosphatidylinositol-3-kinase catalytic subunit/PTEN and KRAS/NRAS/BRAF in primary resected esophageal adenocarcinomas: Loss of PTEN is associated with worse clinical outcome. Hum. Pathol. 2013, 44, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhu, H.; Zhang, W.; Xiao Liu, X.; Zhang, X.; He, J.; Sun, K.; Wang, L.; Xu, N. Expression of epidermal growth factor receptor is an independent prognostic factor for esophageal squamous carcinoma. World J. Surg. Oncol. 2013, 11, 278. [Google Scholar] [CrossRef] [PubMed]
- Xing, E.P.; Yang, G.Y.; Wang, L.D.; Shi, S.T.; Yang, C.S. Loss of heterozigosity of the gene correlates with pRb protein expression and associates with p53 alteration in human esophageal cancer. Clin. Cancer Res. 1999, 5, 1231–1240. [Google Scholar] [PubMed]
- Taghavi, N.; Birajimasi, F.; Sotoudeh, M.; Khademi, H.; Malekzadeh, R.; Moaven, O.; Memar, B.; A’rabi, A.; Abbaszadegan, M.R. p16INK4a hypermethylation and p53, p16 and MDM2 protein expression in esophageal squamous cell carcinoma. BMC Cancer 2010, 13, 138. [Google Scholar]
- Kuwabara, T.; Hiyama, T.; Tanaka, S.; Yoshihara, M.; Arihiro, K.; Chayama, K. Genetic pathways of multiple squamous cell carcinomas. Oncol. Rep. 2011, 25, 435–439. [Google Scholar]
- Wang, M.T.; Chen, G.; An, S.J.; Chen, Z.H.; Huang, Z.M.; Xiao, P.; Ben, X.S.; Xie, Z.; Chen, S.L.; Luo, D.L.; et al. Prognostic significance of cyclin D1 amplification and the co-alteration of cyclinD1/pRb/ppRb in patients with esophageal squamous cell carcinoma. Dis. Esophagus 2012, 25, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Phillips, W.A.; Russell, S.E.; Ciavarella, M.L.; Choong, D.Y.; Montgomery, K.G.; Smith, K.; Pearson, R.B.; Thomas, R.J.; Campbell, I.G. Mutation analysis of PI3KCA and PI3KCB in esophageal cancer and Barrett’s esophagus. Int. J. Cancer 2006, 118, 2644–2646. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Wang, T.Y.; Li, H.C.; Wei, J.C.; Song, J.X. Prognostic significance of PTEN expression is esophageal squamous cell carcinoma from Linzhou city, a high incidence area of Northern China. Dis. Esophagus 2007, 20, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Boonbe, J.; Tedn Kate, F.J.; Offerhaus, G.J.; van Diest, P.J.; Rinkes, I.H.; van Hillegersberg, R. mTOR in squamous cell carcinoma of the oesophagus: A potential target for molecular therapy? J. Clin. Pathol. 2008, 61, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Prins, M.J.; Verhage, R.; Ruurda, J.; ten Kate, F.J.; van Hillegererberg, R. Over-expression of phosphorylated mammalian target of rapamycin is associated with poor survival in oesophageal adenocarcinoma: A tissue microarray study. J. Clin. Pathol. 2013, 66, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Gen, Y.; Yasui, K.; Zen, Y.; Zen, K.; Dohi, O.; Endo, M.; Tsuji, K.; Wakabayashi, N.; Itoh, Y.; Naito, Y.; et al. SOX2 identified as a target gene for the amplification at 3q26 is frequently detected in esophageal squamous cell carcinoma. Cancer Genet. Cytogenet. 2010, 202, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Bass, A.; Watanabe, H.; Mermel, C.; Yu, S.; Perner, S.; Verhaak, R.G.; Kim, S.Y.; Wardwell, L.; Tamayo, P.; Gat-Viks, I.; et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat. Genet. 2009, 41, 1238–1242. [Google Scholar] [CrossRef] [PubMed]
- Gen, Y.; Yasui, K.; Nishikawa, T.; Yoshikawa, T. SOX2 promotes tumor growth of esophageal squamous cell carcinoma through the AKT7mammalian target of rapamycin complex 1 signaling pathway. Cancer Sci. 2013, 104, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, M.; Kwok, S.; Montgomery, K.; Lowe, A.W.; Pai, R.K. Immunohistochemical panel for distinguishing esophageal adenocarcinoma from squamous cell carcinoma: A combination of p63, cytokeratin 5/6, MUC5AC, and anterior gradient homolog 2 allows optimal subtyping. Hum. Pathol. 2012, 43, 1799–1807. [Google Scholar] [CrossRef] [PubMed]
- Sawada, G.; Niida, A.; Uchi, R.; Hirata, H.; Shimamura, T.; Suzuki, Y.; Shiraishi, Y.; Chiba, K.; Imoto, S.; Takahashi, Y.; et al. Genomic landscape of esophageal squamous cell carcinoma in a Japanese population. Gastroenterology 2016, 150, 1171–1182. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Snell, J.; Jeck, W.; Hoadley, K.A.; Wilkerson, M.D.; Parker, J.S.; Patel, N.; Mlombe, J.B.; Mulima, G.; George Liomba, N.; et al. Subtyping sub-Saharan esophageal squamous cell carcinoma by comprehensive molecular analysis. J. Clin. Investig. Insight 2016, 1, e88755. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Tan, W.; Ling, Z.; Xi, R.; Shao, M.; Chen, M.; Luo, Y.; Zhao, Y.; Liu, Y.; Huang, X.; et al. Genomic analysis of oesophageal squamous-cell carcinoma identifies alcohol drinking-related mutation signature and genomic alterations. Nat. Commun. 2017, 8, 15290. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Ko, J.; Choi, S.; Yu, Z.; Ning, L.; Zheng, H.; Gopalan, V.; Chan, K.T.; Lee, N.; Chan, K.W.; et al. Whole-exome sequencing reveals critical genes underlying metastasis in oesophageal squamous cell carcinoma. J. Pathol. 2017, 242, 500–510. [Google Scholar] [CrossRef] [PubMed]
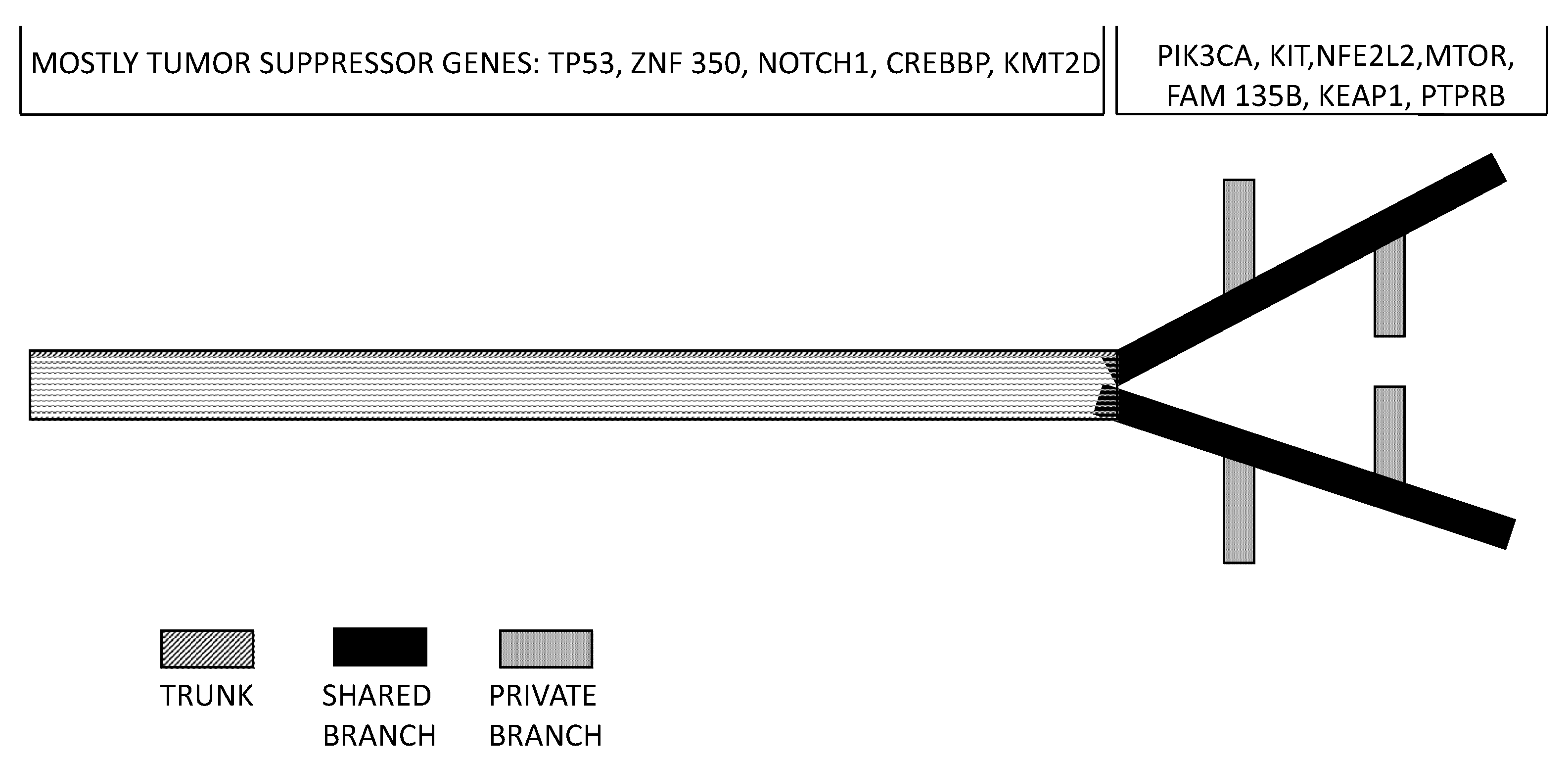
- Cao, W.; Wu, W.; Yan, M.; Tian, F.; Ma, C.; Zhang, Q.; Li, X.; Han, P.; Liu, X.; Gu, J.; et al. Multiple region whole-exome sequencing reveals dramatically evolving intratumor genomic heterogeneity in esophageal squamous cell carcinoma. Oncogenesis 2015, 4, e175. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.J.; Lin, D.C.; Dinh, H.; Mayakonda, A.; Jiang, Y.Y.; Chang, C.; Jiang, Y.; Lu, C.C.; Shi, Z.Z.; Xu, X.; et al. Spatial intratumoral heterogeneity and temporal clonal evolution in esophageal squamous cell carcinoma. Nat. Genet. 2016, 48, 1500–1507. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wang, Z.; Song, X.; Feng, X.S.; Abnet, C.C.; He, J.; Hu, N.; Zuo, X.B.; Tan, W.; Zhan, Q.; et al. Joint analysis of three genome-wide association studies of esophageal squamous cell carcinoma in Chinese population. Nat. Genet. 2014, 46, 1001–1006. [Google Scholar] [CrossRef] [PubMed]
- Dulak, A.M.; Schumacher, S.E.; Leishout, J.; Imamura, Y.; Fox, C.; Shim, B.; Ramos, A.; Saksena, G.; Baca, S.; Baselga, J. Gastrointestinal adenocarcinomas of the esophagus, stomach, and colon exhibit distinct patterns of genome instability and oncogenesis. Cancer Res. 2012, 72, 4383–4393. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Johnson, A.; Ali, S.; Klempner, S.J.; Bekaii-Saab, T.; Vacirca, J.L.; Khaira, D.; Yelensky, R.; Chmielecki, J.; Elvin, J.A.; et al. Comprehensive genomic profiling of advanced esophageal squamous cell carcinomas and esophageal adenocarcinomas reveals similarities and differences. Oncologist 2015, 20, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of esophageal carcinoma. Nature 2017, 541, 169–178. [Google Scholar]
- Visser, E.; Franken, I.A.; Brosens, L.; Ruurda, J.P.; van Hillegersberg, R. Prognostic gene expression profiling in esophageal cancer: A systematic review. Oncotarget 2017, 17, 5566–5577. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.J.; Rees, J.R.; Hardwick, R.H.; Hardwick, J.S.; Vowler, S.L.; Ong, C.A.; Zhang, C.; Save, V.; O’Donovan, M.; Rassl, D.; et al. A 4-gene signature predicts survival of patients with resected adenocarcinoma of the esophagus, junction, and gastric cardia. Gastroenterology 2010, 139, 1995–2004. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Park, Y.Y.; Park, E.S.; Cho, J.Y.; Izzo, J.G.; Zhang, D.; Kim, S.B.; Lee, J.H.; Bhutani, M.S.; Swisher, S.G.; et al. Prognostic biomarkers for esophageal adenocarcinoma identified by analysis of tumor transcriptome. PLoS ONE 2010, 5, e15074. [Google Scholar] [CrossRef] [PubMed]
- Goh, X.Y.; Rees, J.R.; Paterson, A.L.; Chin, S.F.; Marioni, J.C.; Save, V.; O’Donovan, M.; Eijk, P.P.; Alderson, D.; Ylstra, B.; et al. Integrative analysis of array-comparative genomic hybridization and matched gene expression profiling data reveals novel genes with prognostic significance in oesophageal adenocarcinoma. Gut 2011, 60, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Qiao, J.; He, W.; Wang, J.; Jia, Y.; Sun, Y.; Tang, S.; Fu, L.; Qin, Y. genome-wide gene expression profile analyses identify CTTN as a potential prognostic marker in oesophageal cancer. PLoS ONE 2014, 9, e88918. [Google Scholar]
- Rao, S.; Welsh, L.; Cunningham, D.; te-Poele, R.H.; Benson, M.; Norman, A.; Saffery, C.; Giddings, I.; Workman, P.; Clarke, P.A. Correlation of overall survival with gene expression profiles in a prospective study of resectable esophageal cancer. Clin. Colorectal Cancer 2011, 10, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Motoori, M.; Takemasa, I.; Yamasaki, M.; Komori, T.; Takeno, A.; Miyata, H.; Takiguchi, S.; Fujiwara, Y.; Yasuda, T.; Yano, M.; et al. Prediction of response to chemotherapy in advanced esophageal cancer by gene expression profiling of biopsy samples. Int. J. Oncol. 2010, 37, 1113–1120. [Google Scholar] [PubMed]
- Maher, S.G.; Gillham, C.M.; Duggan, S.P.; Smyth, P.C.; Miller, N.; Muldoon, C.; O’Byrne, K.J.; Sheils, O.M.; Hollywood, D.; Reynolds, J.V. Gene expression analysis of diagnostic biopsies predicts pathological response to neoadjuvant chemo-radiotherapy of esophageal cancer. Ann. Surg. 2009, 250, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Tamoto, E.; Tada, M.; Murakawa, K.; Takada, M.; Shindo, G.; Teramoto, K.; Matsunaga, A.; Komuro, K.; Kanai, M.; Kawakami, A.; et al. Gene-expression profile changes correlated with tumor progression and lymph node metastasis in esophageal cancer. Clin. Cancer. Res. 2004, 10, 3629–3638. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Hu, Z.; He, Z.; Jia, W.; Wang, F.; Zhou, Y.; Liu, Z.; Zhou, Q.; Liu, Y.; Yu, D.; et al. Genome-wide association study identifies three new susceptibility loci for esophageal squamous-cell carcinoma in Chinese populations. Nat. Genet. 2011, 43, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Kraft, P.; Zhai, K.; Chang, J.; Wang, Z.; Li, Y.; Hu, Z.; He, Z.; Jia, W.; Abuet, C.C.; et al. Genome-wide association analyses of esophageal squamous cell carcinoma in Chinese identify multiple susceptibility loci and gene-environment interactions. Nat. Genet. 2012, 44, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Xiong, T.; Wang, M.; Zhao, J.; Liu, Q.; Yang, C.; Luo, W.; Li, X.; Yang, H.; Kristiansen, K.; Roy, B.; et al. An esophageal squamous cell carcinoma classification system that reveals potential targets for therapy. Oncotarget 2017, 8, 49851–49860. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.W.; Karakasheva, T.A.; Lee, D.J.; Lee, J.S.; Long, Q.; Bass, A.J.; Wong, K.K.; Rustgi, A.K. Comparative transcriptomics of adenocarcinomas and squamous cell carcinomas reveal molecular similarities that span classical anatomic boundaries. PLoS Genet. 2017, 13, e1006938. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiang, M.; Kim, E.; Lin, S.; Liu, K.; Lan, X.; Que, J. Development of stem cells of the esophagus. Sem. Cell Dev. Biol. 2016, 66, S1084–S9521. [Google Scholar] [CrossRef] [PubMed]
- Kalabis, J.; Oyama, K.; Okawa, T.; Nakagawa, H.; Michaylira, C.Z.; Stairs, D.B.; Figueiredo, J.L.; Mahmood, U.; Diehl, J.A.; Herlyn, M.; et al. A subpopulation of mouse esophageal basal cells has properties of stem cells with the capacity for self-renewal and lineage specification. J. Clin. Investig. 2008, 118, 3860–3870. [Google Scholar] [CrossRef] [PubMed]
- Doupé, D.; Alcolea, M.; Roshan, A.; Zhang, G.; Klein, A.M.; Simons, B.D.; Jones, P.H. A single progenitor population switches behavior to maintain and repair esophageal epithelium. Science 2012, 337, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Giroux, V.; Lento, A.A.; Islam, M.; Pitarresi, J.R.; Kharbanda, A.; Hamilton, K.E.; Whelon, K.A.; Lang, A.; Rhoades, B.; Tang, Q.; et al. Long-lived keratin 15+ esophageal progenitor cells contribute to homeostasis and regeneration. J. Clin. Investig. 2017, 127, 2378–2391. [Google Scholar] [CrossRef] [PubMed]
- Seery, J.P.; Watt, F.M. Asymmetric stem-cell divisions define the architecture of human esophageal epithelium. Curr. Biol. 2000, 10, 1447–1450. [Google Scholar] [CrossRef]
- DeWard, A.; Cramer, J.; Lagasse, E. Cellular heterogeneity in the mouse esophagus implicates the presence of a non-quiescent epithelium stem cell population. Cell Rep. 2014, 9, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okumura, T.; Shimada, Y.; Imamura, M.; Yasumoto, S. Neutrophin receptor p75NTR characterizes human esophageal keratinocyte stem cells in vitro. Oncogene 2003, 22, 4017–4026. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Nicholson, A.M.; Barr, H.; Harrison, L.A.; Wilson, G.D.; Burkert, J.; Jeffery, R.; Alison, M.R.; Looijenga, L.; Lin, W.R.; et al. Identification of lineage-uncommitted, long-lived, label-retaining cells in healthy human esophagus and stomach, and in metaplastic esophagus. Gastroenterology 2013, 144, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Barbera, M.; Di Pietro, M.; Walker, E.; Brierley, C.; MacRae, S.; Simons, B.D.; Jones, P.H.; Stingl, J.; Fitzgerald, R.C. The human squamous esophagus has widespread capacity for clonal expansion from cells at diverse stages of differentiation. Gut 2015, 64, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Alcolea, M.P.; Greulich, P.; Wabik, A.; Frede, J.; Simons, B.D.; Jones, P.H. Differentiation imbalance in single oesophageal progenitor cells causes clonal immortalization and field change. Nat. Cell Biol. 2014, 16, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Hvid-Jensen, F.; Pedersen, L.; Drewes, A.M.; Sørensen, H.T.; Funch-Jensen, P. Incidence of adenocarcinoma among patients with Barrett’s esophagus. N. Engl. J. Med. 2011, 365, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.; Coleman, H.G.; Yousef, F.; Johnston, B.T.; McManus, D.T.; Gavin, A.T.; Murray, L.J. Risk of malignant progression in Barrett’s esophagus patients: Results from a large-population based study. J. Natl. Cancer Inst. 2011, 103, 1049–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Galipeau, P.; Paulson, T.G. Temporal and spatial evolution of somatic chromosomal alterations: A case-cohort study of Barrett’s esophagus. Cancer Prev. Res. 2013, 7, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Bhagat, G.; Abrams, J.; Marache, F.; Good, P.; Lee, M.D.; Lee, Y.; Friedman, R.; Asfaha, S.; Dubeykovskaya, Z.; et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell 2012, 21, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Schellnegger, R.; Quante, A.; Rospleszcz, S.; Schernhammer, M.; Hohl, B.; Tobiasch, M.; Pastula, A.; Brandtner, A.; Abrams, J.A.; Staruch, K.; et al. Goblet cell ratio in combination with differentiation and stem cell markers in Barrett esophagus allow distinction of patients with and without esophageal adenocarcinoma. Cancer Prev. Res. 2017, 10, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Kostandinov, R.L.; Kuhner, M.K.; Li, X.; Sanchez, C.A.; Galipeau, P.C.; Paulson, T.G.; Sather, C.L.; Srivastava, A.; Odze, R.D.; Blount, P.L.; et al. NSAIDs modulate clonal evolution in Barrett’s esophagus. PLoS Genet. 2013, 9, e1003553. [Google Scholar]
- Wang, X.; Ouyang, H.; Yamamoto, Y.; Kumar, P.A.; Wei, T.S.; Dagher, R.; Vincent, M.; Lu, X.; Bellizzi, A.M.; Ho, K.Y.; et al. Residual embryonic cells as precursors of a Barrett’s-like metaplasia. Cell 2011, 145, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Leedham, S.J.; Preston, S.L.; Mc Doualt, S.A.C.; Elia, G.; Bhandari, P.; Poller, D.; Harrison, R.; Novelli, M.R.; Jankowski, J.A.; Wright, N.A. Individual crypt heterogeneity and the origin of metaplastic glandular epithelium in human Barrett’s esophagus. Gut 2008, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Paulson, T.G.; Xu, L.; Sanchez, C.; Blount, P.L.; Ayub, K.; Odze, R.D.; Reid, B.J. Neosquamous epithelium does not typically arise from Barrett’s epithelium. Clin. Cancer Res. 2006, 12, 1701–1706. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Fang, Y.; Tavebaugh, W.; Orlando, R.C.; Shaheen, N.J.; Chen, X. Molecular mechanisms of Barrett’s esophagus tissue. Dig. Dis. Sci. 2011, 56, 3405–3420. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, A.; Graham, T.; Simpson, A.; Humphries, A.; Burch, N.; Rodriguez-Justo, M.; Novelli, M.; Harrison, R.; Wright, N.A.; McDonald, S.A.; et al. Barrett’s metaplasia glands are clonal, contain multiple stem cells and share a common squamous progenitor. Gut 2012, 61, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.H.; Clemons, N.J.; Miyashita, T.; Dupuy, A.J.; Zhang, W.; Szczepny, A.; Corcoran-Schwartz, I.M.; Wilburn, D.L.; Montgomery, E.A.; Wang, J.S.; et al. Aberrant epithelial-mesenchymal Hedgehog signaling characterizes Barrett’s metaplasia. Gastroenterology 2010, 138, 1810–1822. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.H.; Tiwari, A.; Kim, M.; Clemons, N.J.; Regmi, N.L.; Hodges, W.A.; Berman, D.M.; Montgomery, E.A.; Watkins, D.N.; Zhang, X.; et al. Hedgehog signaling regulates FOXA2 in esophageal embryogenesis and Barrett’s metaplasia. J. Clin. Investig. 2014, 124, 3767–3780. [Google Scholar] [CrossRef] [PubMed]
- Mori, L.; Milano, F.; Parikh, K.; Straub, D.; Everts, V.; Hoeben, K.K.; Fockens, P.; Buttar, N.S.; Krishnadath, K.K. A pSMAD/CDX2 complex is essential for the internalization of epithelial metaplasia. Cell Rep. 2014, 7, 1197–1210. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yamamoto, Y.; Wilson, L.H.; Zhang, T.; Howitt, B.E.; Farrow, M.A.; Kern, F.; Ning, G.; Hong, Y.; Khor, C.C.; et al. Cloning and variation of ground state intestinal stem cells. Nature 2015, 522, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Wang, X.; Bertrand, D.; Kern, F.; Zhang, T.; Duleba, M.; Srivastava, S.; Khor, C.C.; Hu, Y.; Wilson, L.H.; et al. Mutational spectrum of Barrett’s stem cells suggests paths to initiation of a precancerous lesion. Nat. Commun. 2016, 7, 10380. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.S.; Li, W.J.; Ge, D.; Zhang, P.J.; Li, J.J.; Lu, C.L.; Ji, X.D.; Guan, D.X.; Gao, H.; Xu, L.Y.; et al. Tumor initiating cells in esophageal squamous cell carcinoma express high levels of CD44. PLoS ONE 2011, 6, e21419. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.D.; Yuan, Y.; Liu, X.H.; Gong, D.J.; Bai, C.G.; Wang, F.; Luo, J.H.; Xu, Z.Y. Self-renewal and chemotherapy resistance of p75NTR positive cells in esophageal squamous cell carcinomas. BMC Cancer 2009, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Okumura, T.; Tsunoda, S.; Mori, Y.; Ito, T.; Kikuchi, K.; Wang, T.C.; Yasumoto, S.; Shimada, Y. The biological role of the low-affinity p75 neurotrophin receptor in esophageal squamous cell carcinoma. Clin. Cancer. Res. 2006, 12, 5096–5103. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.T.; Wang, P.J.; Liou, N.J.; Lin, P.S.; Chen, C.H.; Chang, W.C. ICAM1 is a potential cancer stem cell marker of esophageal squamous carcinoma. PLoS ONE 2015, 10, e0142834. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.H.; Dai, Y.D.; Tong, M.; Chan, Y.P.; Kwan, P.S.; Fu, L.; Qin, Y.R.; Tsao, S.W.; Lung, H.L.; Lung, M.L.; et al. A CD90+ tumor-initiating cell population with an aggressive signature and metastatic capacity in esophageal cancer. Cancer Res. 2013, 73, 2322–2332. [Google Scholar] [CrossRef] [PubMed]
- Ming, X.Y.; Fu, F.; Zhang, L.Y.; Qin, Y.R.; Cao, T.T.; Chan, K.W.; Ma, S.; Xie, D.; Guan, X.Y. Integrin α7 is a functional cancer stem cell surface marker in esophageal squamous cell carcinoma. Nat. Commun. 2006, 7, 13568. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Lu, Q.; Han, Y.; Li, Z.; Zhang, Z.; Li, X. ABCG2/V-ATPase was associated with the drug resistance and tumor metastasis of esophageal squamous cancer cells. Diagn. Pathol. 2012, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mathur, M.; Zhang, Y.; Xi, S.; Atay, S.; Hong, J.A.; Datrice, N.; Upham, T.; Kemp, C.D.; Ripley, R.T.; et al. Mitharmycin represses basal and cigarette smoke-induced expression of ABCG2 and inhibits stem cell signaling in lung and esophageal cancer cells. Cancer Res. 2012, 72, 4178–4192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Ma, L.; Xie, Y.; Miao, X.B.; Jin, C. Esophageal cancer tumor-spheres involve cancer stem-like populations with elevated aldehyde dehydrogenase enzymatic activity. Mol. Med. Rep. 2012, 6, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, D.; Kato, K.; Nohara, S.; Iwanuma, Y.; Kajiyama, Y. The usefulness of three-dimensional cell culture in induction of cancer stem cells from esophageal squamous cell carcinoma cell lines. Biochem. Biophys. Res. Commun. 2013, 434, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ren, Y.; Yu, X.; Qian, F.; Bian, B.S.; Xiao, H.L.; Wang, W.G.; Xu, S.L.; Yang, J.; Cui, W.; et al. ALDHA1 defines invasive cancer stem-like cells and predicts poor prognosis in patients with esophageal squamous cell carcinoma. Mod. Pathol. 2014, 27, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Ajani, J.A.; Wang, X.; Song, S.; Suzuki, A.; Taketa, T.; Sudo, K.; Wadhwa, R.; Hofstetter, W.L.; Komaki, R.; Maru, D.M.; et al. ALDH-1 expression levels predicts response or resistance to preoperative chemoradiation in resectable esophageal cancer patients. Mol. Oncol. 2014, 8, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Honjo, S.; Ajani, J.A.; Scott, A.M.; Chen, Q.; Skinner, H.D.; Stroehlein, J.; Johnson, R.L.; Song, S. Metformin sensitizes to chemotherapy by targeting cancer stem cells and the mTOR pathway in esophageal cancer. Int. J. Oncol. 2014, 45, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, T.; Lun, X.; Zhang, J.; Zheng, S.; Yang, W.; Li, W.; Xiang, A.P.; Chen, Z. Contribution of nestin-positive esophageal squamous cancer cells on malignant proliferation, apoptosis and poor prognosis. Cancer Cell Int. 2014, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jiang, T.; Mao, A.; Xu, J. Esophageal cancer stem cells express PLGF to increase cancer invasion through MMP9 activation. Tumor Biol. 2014, 35, 12749–12755. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Komaki, R.; Wang, L.; Fang, B.; Chang, J.Y. Treatment of radioresistant stem-like esophageal cancer cells by an apoptotic gene armed, telomerase-specific oncolytic adenovirus. Clin. Cancer Res. 2008, 14, 2813–2823. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L.; Yu, J.P.; Sun, Z.Q.; Sun, S.P. Radiobiological characteristics of cancer stem cells from esophageal cancer cell lines. World J. Gastroenterol. 2014, 20, 18296–18305. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Da Silva, T.; Jin, K.; Han, X.; Ranganathan, P.; Zhu, X.; Sanchez-Mejias, A.; Bai, F.; Li, B.; Fei, D.L.; et al. Notch signaling drives stemness and tumorigenicity of esophageal adenocarcinoma. Cancer Res. 2014, 74, 63646374. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ying, Z.; Lin, X.; Lin, H.; Wu, J.; Li, M.; Song, L. Acylglycerol kinase augments JAK2/STAT3 signaling in esophageal squamous cells. J. Clin. Investig. 2013, 123, 2576–2589. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Jiang, M.; Lu, Y.; Chen, H.; Sun, J.; Wu, S.; Ku, W.Y.; Nakagawa, H.; Kita, Y.; Natsugoe, S.; et al. Sox2 cooperates with inflammation-mediated Stat3 activation in the malignant transformation of foregut basal progenitor cells. Cell Stem Cell 2013, 12, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Ajani, J.; Honjo, S.; Maru, D.M.; Chen, Q.; Scott, A.W.; Heallen, T.D.; Xiao, L.; Hofstetter, W.L.; Weston, B.; et al. Hippo coactivator YAP1 upregulates SOX9 and endows stem-like properties to esophageal cancer cells. Cancer Res. 2014, 74, 4170–4182. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Honjo, S.; Jin, J.; Chang, S.S.; Scott, A.W.; Chen, Q.; Kalhor, N.; Correa, A.M.; Hofstetter, W.L.; Albarracin, C.T.; et al. Hippo coactivator YAP1 mediates EGFR overexpression and confers chemo-resistance in esophageal cancer. Clin. Cancer Res. 2015, 21, 2580–2590. [Google Scholar] [CrossRef] [PubMed]
- Sato, F.; Kubota, Y.; Natsuizaka, M.; Maehara, O.; Hatanaka, Y.; Marukawa, K.; Terashita, K.; Suda, G.; Ohnishi, S.; Shimizu, Y.; et al. EGFR inhibitors prevent induction of cancer stem-like cells in esophageal squamous cell carcinoma by suppressing epithelial-mesenchymal transition. Cancer Biol. Ther. 2015, 16, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yuan, G.; Fang, Y.; Qui, M.; Lin, J.; Sun, J.; Yang, D. Increased WNT6 expression in tumor cells predicts unfavorable survival in esophageal squamous cell carcinoma patients. Int. J. Clin. Exp. Pathol. 2015, 8, 11421–11427. [Google Scholar] [PubMed]
- Ge, C.; Wu, S.; Wang, W.; Liu, W.; Zhang, J.; Wang, Z.; Li, R.; Zhang, Z.; Li, Z.; Dong, S.; et al. miR-942 promotes cancer stem cell-like traits in esophageal squanmous cell carcinoma through activation of Wnt7beta-catenin signaling pathway. Oncotarget 2015, 6, 10964–10977. [Google Scholar] [CrossRef] [PubMed]
- Long, A.; Giroux, V.; Whelan, K.A.; Hamilton, K.E.; Tétreault, M.P.; Tanaka, K.; Lee, J.S.; Klein-Szanto, A.J.; Nakagawa, H.; Rustgi, A.K. WNT10A promotes an invasive and self-renewing phenotype in esophageal squamous cell carcinoma. Carcinogenesis 2015, 36, 598–606. [Google Scholar] [CrossRef] [PubMed]
- De Both, N.J.; Wijnhoven, B.P.; Sleddens, H.F.; Tilanus, H.W.; Dinjens, W.N.M. Establishment of cell lines from adenocarcinomas of the esophagus and gastric cardia growing in vivo and in vitro. Virchows Arch. 2001, 438, 451–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Rifai, W.; Harper, J.C.; Cummings, O.W.; Hyytinen, E.R.; Frierson, H.F.; Knuutila, J.S.; Powell, S.M. Consistent genetic alterations in xenografts of proximal stomach and gastro-esophageal junction adenocarcinomas. Cancer Res. 1998, 58, 34–37. [Google Scholar] [PubMed]
- Kitamura, M.; Suda, M.; Nishihira, T.; Watanabe, T.; Kasai, M. Heterotransplantation of human esophageal carcinoma to nude mice. Tohoku J. Exp. Med. 1981, 135, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Dodbiba, L.; Teichman, J.; Fleet, A.; Thai, H.; Sun, B.; Panchal, D.; Patel, D.; Tse, A.; Chen, Z.; Faluyi, O.O.; et al. Primary esophageal and gastro-esophageal junction cancer xenograft models: Clinicopathological features and engraftment. Lab. Investig. 2013, 93, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Dodbiba, L.; Teichman, J.; Fleet, A.; Thai, H.; Starmans, M.H.W.; Navab, R.; Chen, Z.; Girgis, H.; Eng, L.; Espin-Garcia, O.; et al. Appropriateness of using patient-derived xenograft models for pharmacologic evaluation of novel therapies for esophageal/gastro-esophageal junction cancers. PLoS ONE 2015, 10, e121872. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, D.; Li, X.; Lv, J.; Xie, L.; Zheng, L.; Gavine, P.R.; Hu, Q.; Shi, Y.; Tan, L.; et al. Establishment and characterization of esophageal squamous cell carcinoma patient-derived xenograft mouse models for preclinical drug discovery. Lab. Investig. 2014, 94, 917–926. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker | Function | Biological properties | Assays | Subpopulations | Clinical Significance |
|---|---|---|---|---|---|
| ABCG2 (ATP-Binding Cassette sub-family G member 2) | This membrane protein functions as a xenobiotic transporter and may have a role in multi drug resistance. | Tumor spheres isolated fromEC cell lines are enriched in ABCG2+ cells. Cigarette smoke condensate induces ABCG2 expression and potentiates CSC properties. | ABCG2+ cells isolated from EC cell lines and EC biopsies form tumor-spheres in vitro and tumors in vivo in nude mice. | ABCG2+/ALDH1+ cells are tumorigenic. | ABCG2 is overexpressed in ESCC, in association with tumor grade, stage and metastasis. 5-FU treatment increases ABCG2+ cells; 5-FU+COX2 inhibitor decreases ABCG2+ cells. |
| CD44 Also, known as HCAM (Homing Cell Adhesion Molecule) | CD44 is a cell surface glycoprotein involved in cell-cell interactions, cell adhesion and migration. | CD44+/CD24- cells isolated from EC cell lines and EC biopsies have phenotypic and functional properties of CSCs. | CD44+/CD24- cells isolated from EC cell lines and EC biopsies form tumor-spheres in vitro and tumors in vivo in nude mice. | CD44+/CD24- cells have high sphere-forming potential, are resistant to irradiation, reside in hypoxic niches, formed tumors in xenotransplantation assays and their number correlated with tumor grade. CD44+/ICAM1+, CD44+/ALDH1+ and CD44+CD133+ have high CSC potential. | The number of CD44+/CD24- cells correlated with response of residual EAC to chemo/radiation. CD44 expression increases during the progression of Barrett’s metaplasia to EAC. |
| CD90 Also, known as Thy-1 | CD90 is a heavily glycosylated, glyco- Psphatidylinositol anchored cell surface protein. CD90 is used as a marker for a variety of stem cell populations. | CD90+ cells isolated from cancer cell lines have CSC properties. CD90 expression is increased in esophageal tumor tissues, compared to normal ones. | CD90+ cells isolated from KYSE140 or KYSE520 cell lines form tumor-spheres in vitro and tumors in vivo in nude mice. | Only a minority of CD90+ cells co-express CD271 or CD44. | CD90 expression is upregulated in primary ESCC cells. 2–10% CD90+ cells in ESCCs. CD90+ cells are radio-resistant. |
| CD133 Also, known as Promin-1 | CD133 is a membrane glycoprotein, encoded by the PROM1 gene, member of the pentasman transmembrane glycoproteins. | CD133+ or CD133+/CXCR4+ cells isolated from EC cell lines and EC biopsies have phenotypic and functional properties of CSCs. | CD133+/CXCR4+ cells isolated from TE-1 cell line have a high colony-forming capacity. | CD133+/CXCR4+ cells have CSC properties with high proliferative potential. | CD133 expression associated with lymph node metastasis, clinical stage and histo- pathological grade. 21% of ESCCs have high CD133+/CXCR4 expression, associated with negative prognosis. CD133 expression increases during the progression of Barrett’s metaplasia to EAC. |
| CD271 Also, known as p75 Neutrophin Receptor (p75NTR) | CD271 is the low-affinity nerve growth factor receptor, one of the two receptor types for neutrophins. | CD271+ cells isolated from cancer cell lines (KYSE70) have CSC properties. | CD271+ cells isolated from EC cell lines and EC biopsies form tumor-spheres in vitro and tumors in vivo in nude mice. | CD271+/CD90+, CD271+/CD44+ cells have the same CSC function, compared to CD271+/CD90- , CD271+/CD44- cells. Quiescent CD271+ cells express ABCG2 and are chemoresistant. | CD271 expression is higher in ESCC tissue than in normal esophageal epithelium. High CD271 expression is observed in 34% of ESCCs. |
| ITGA7 (Integrin alpha 7) | ITGA7 is a member of the integrin family of cell surface proteins that mediate cellular interactions with the extracellular matrix. Together with a beta subunit, it forms the functional receptor that binds laminin. | ITGA7+ cells isolated from cancer cell lines have CSC properties. ITGA7 overexpression pro- Motes CSC properties, metastasis and EMT. ITGA7 knockdown decreases CSC properties. | in vitro: tumor-sphere In vivo: xenotranspanta- tion in nude mice. | ITGA7 and CD90 are co-expressed. ITGA7+/CD90+ cells have a high clono- genic potential. | ITGA7 expression is associated with poor prognosis in ESCC, poor differentiation and lymph node metastasis. Chemotherapy enriches ITGA7+ cells. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Testa, U.; Castelli, G.; Pelosi, E. Esophageal Cancer: Genomic and Molecular Characterization, Stem Cell Compartment and Clonal Evolution. Medicines 2017, 4, 67. https://doi.org/10.3390/medicines4030067
Testa U, Castelli G, Pelosi E. Esophageal Cancer: Genomic and Molecular Characterization, Stem Cell Compartment and Clonal Evolution. Medicines. 2017; 4(3):67. https://doi.org/10.3390/medicines4030067
Chicago/Turabian StyleTesta, Ugo, Germana Castelli, and Elvira Pelosi. 2017. "Esophageal Cancer: Genomic and Molecular Characterization, Stem Cell Compartment and Clonal Evolution" Medicines 4, no. 3: 67. https://doi.org/10.3390/medicines4030067
APA StyleTesta, U., Castelli, G., & Pelosi, E. (2017). Esophageal Cancer: Genomic and Molecular Characterization, Stem Cell Compartment and Clonal Evolution. Medicines, 4(3), 67. https://doi.org/10.3390/medicines4030067