Synthetic Lethality in Lung Cancer—From the Perspective of Cancer Genomics


Abstract
:1. Introduction
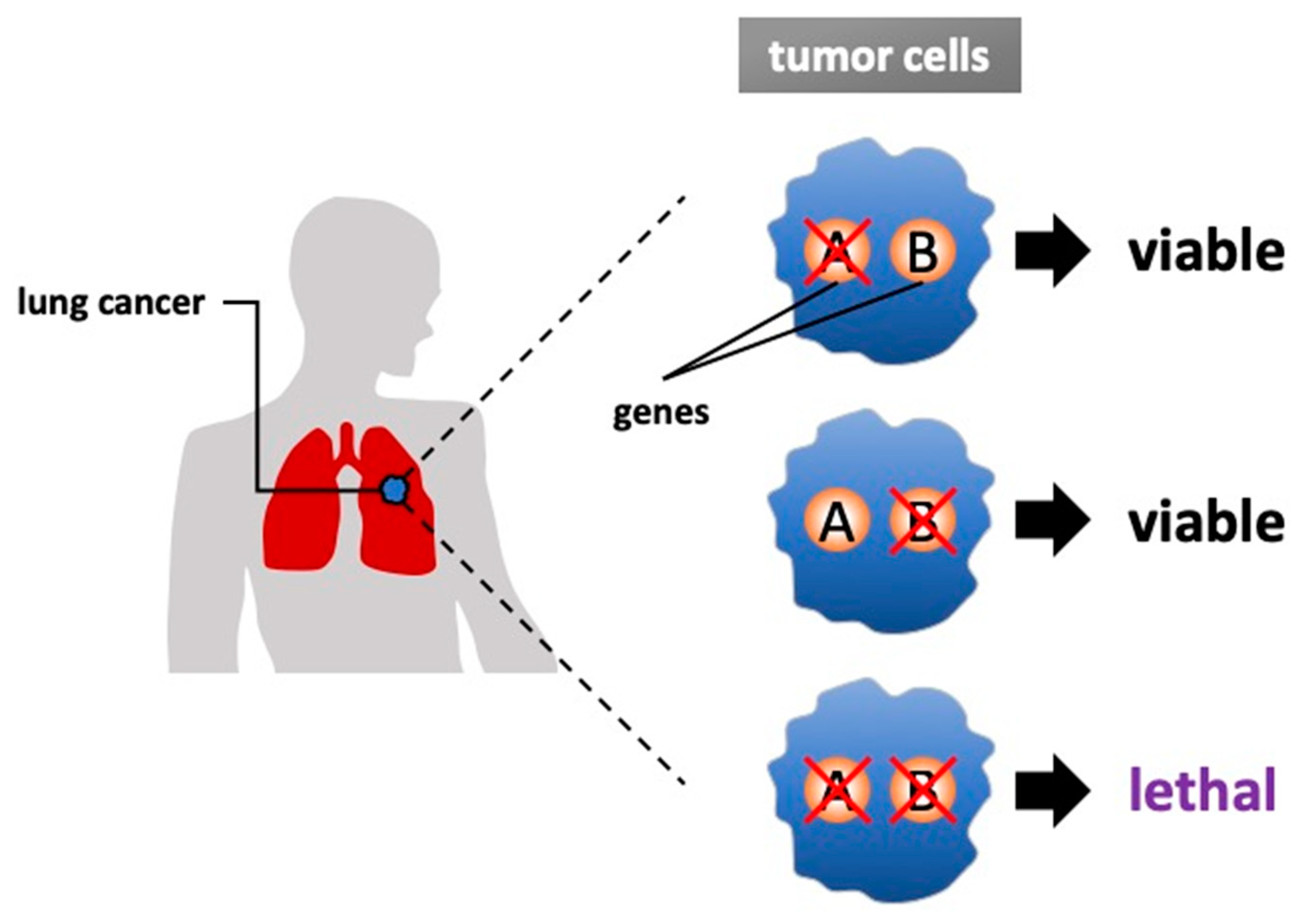
2. From Cancer Genome to a Synthetic Lethal Approach
3. Therapeutic Advances in Lung Cancer
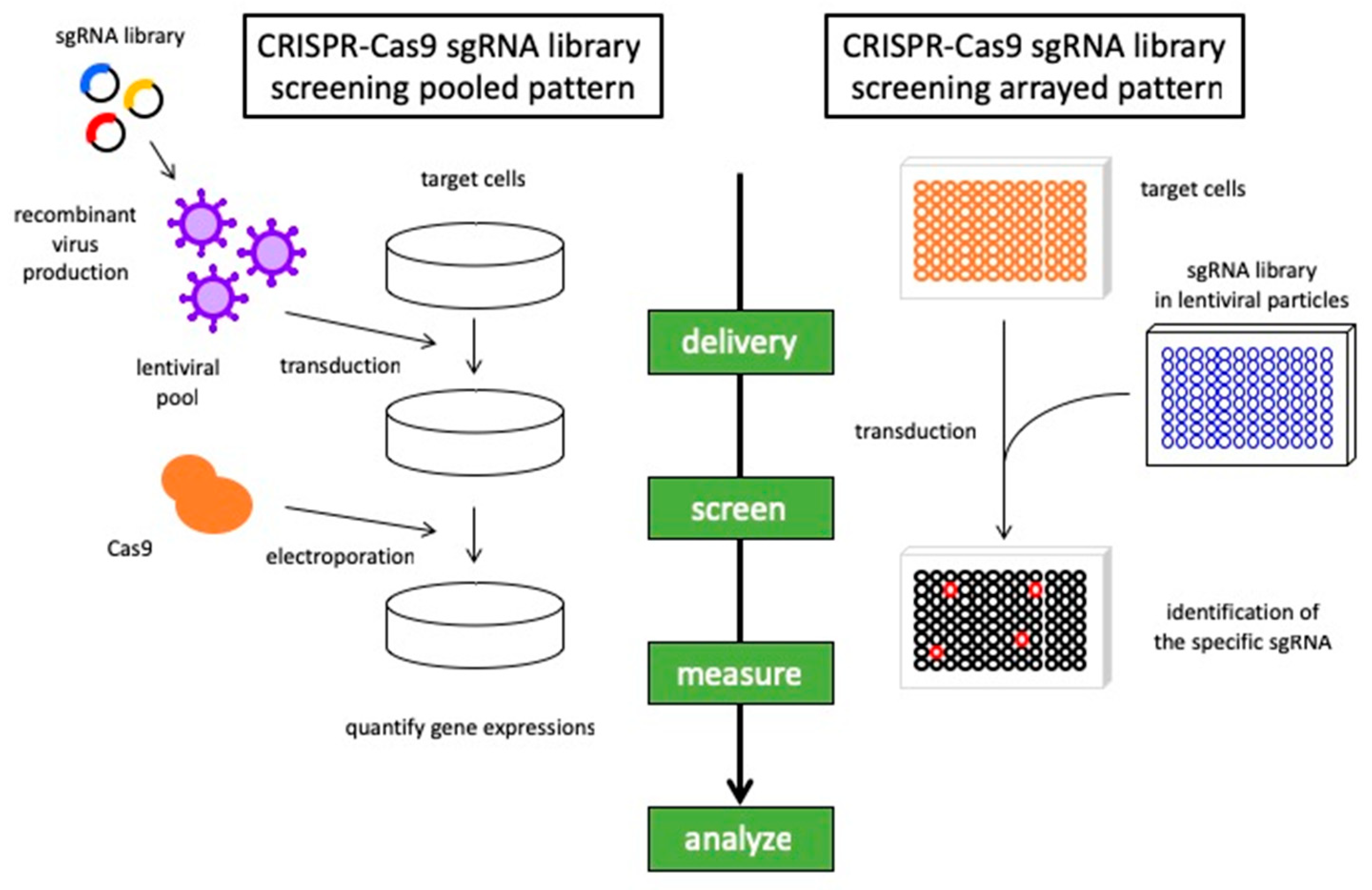
4. Discovering Synthetic Lethal Interaction in Lung Cancer
4.1. Synthetic Lethality in NSCLC
4.2. Synthetic Lethality in SCLC
5. Future Prospective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef] [PubMed]
- McLornan, D.P.; List, A.; Mufti, G.J. Applying synthetic lethality for the selective targeting of cancer. N. Engl. J. Med. 2014, 371, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, A.; Lord, C.J.; Reis-Filho, J.S. Genetic interactions in cancer progression and treatment. Cell 2011, 145, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M. Synthetic lethality: General principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011, 585, 1–6. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Szankasi, P.; Roberts, C.J.; Murray, A.W.; Friend, S.H. Integrating genetic approaches into the discovery of anticancer drugs. Science (New York N. Y.) 1997, 278, 1064–1068. [Google Scholar] [CrossRef]
- Ashworth, A.; Lord, C.J. Synthetic lethal therapies for cancer: What’s next after PARP inhibitors? Nat. Reviews. Clin. Oncol. 2018, 15, 564–576. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of brca2-deficient tumours with inhibitors of poly(adp-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Downward, J. Ras synthetic lethal screens revisited: Still seeking the elusive prize? Clin. Cancer Res. 2015, 21, 1802–1809. [Google Scholar] [CrossRef]
- Campaner, S.; Doni, M.; Hydbring, P.; Verrecchia, A.; Bianchi, L.; Sardella, D.; Schleker, T.; Perna, D.; Tronnersjo, S.; Murga, M.; et al. Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nat. Cell Biol. 2010, 12, 54–59, sup pp. 51–14. [Google Scholar] [CrossRef]
- Ogiwara, H.; Sasaki, M.; Mitachi, T.; Oike, T.; Higuchi, S.; Tominaga, Y.; Kohno, T. Targeting p300 addiction in cbp-deficient cancers causes synthetic lethality by apoptotic cell death due to abrogation of myc expression. Cancer Discov. 2016, 6, 430–445. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Emanuele, M.J.; Li, D.; Creighton, C.J.; Schlabach, M.R.; Westbrook, T.F.; Wong, K.K.; Elledge, S.J. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009, 137, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Solimini, N.L.; Luo, J.; Elledge, S.J. Non-oncogene addiction and the stress phenotype of cancer cells. Cell 2007, 130, 986–988. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.; Friend, S.H. Cancer. Potential of the synthetic lethality principle. Science (New York N. Y.) 2013, 342, 809–811. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [PubMed]
- McDonald, E.R., 3rd; de Weck, A.; Schlabach, M.R.; Billy, E.; Mavrakis, K.J.; Hoffman, G.R.; Belur, D.; Castelletti, D.; Frias, E.; Gampa, K.; et al. Project drive: A compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale, deep RNAi screening. Cell 2017, 170, 577–592.e510. [Google Scholar] [CrossRef]
- Horlbeck, M.A.; Xu, A.; Wang, M.; Bennett, N.K.; Park, C.Y.; Bogdanoff, D.; Adamson, B.; Chow, E.D.; Kampmann, M.; Peterson, T.R.; et al. Mapping the genetic landscape of human cells. Cell 2018, 174, 953–967.e922. [Google Scholar] [CrossRef]
- Najm, F.J.; Strand, C.; Donovan, K.F.; Hegde, M.; Sanson, K.R.; Vaimberg, E.W.; Sullender, M.E.; Hartenian, E.; Kalani, Z.; Fusi, N.; et al. Orthologous crispr-cas9 enzymes for combinatorial genetic screens. Nat. Biotechnol. 2018, 36, 179–189. [Google Scholar] [CrossRef]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Science (New York N. Y.) 2015, 350, 1096–1101. [Google Scholar] [CrossRef] [Green Version]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C. DNA damage and repair. Nature 2003, 421, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Boveri, T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J. Cell Sci. 2008, 121 (Suppl. 1), 1–84. [Google Scholar] [CrossRef]
- Watson, J.D.; Crick, F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Rowley, J.D. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and giemsa staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Shih, C.; Padhy, L.C.; Murray, M.; Weinberg, R.A. Transforming genes of carcinomas and neuroblastomas introduced into mouse fibroblasts. Nature 1981, 290, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Macconaill, L.E.; Garraway, L.A. Clinical implications of the cancer genome. J. Clin. Oncol. 2010, 28, 5219–5228. [Google Scholar] [CrossRef]
- Weinstein, I.B. Cancer. Addiction to oncogenes—The achilles heal of cancer. Science (New York N. Y.) 2002, 297, 63–64. [Google Scholar] [CrossRef]
- Garraway, L.A.; Lander, E.S. Lessons from the cancer genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef]
- Dulbecco, R. A turning point in cancer research: Sequencing the human genome. Science (New York N. Y.) 1986, 231, 1055–1056. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science (New York N. Y.) 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S. Initial impact of the sequencing of the human genome. Nature 2011, 470, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [CrossRef]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nature reviews. Cancer 2004, 4, 177–183. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Costanzo, M.; VanderSluis, B.; Koch, E.N.; Baryshnikova, A.; Pons, C.; Tan, G.; Wang, W.; Usaj, M.; Hanchard, J.; Lee, S.D.; et al. A global genetic interaction network maps a wiring diagram of cellular function. Science (New York N. Y.) 2016, 353, aaf1420. [Google Scholar] [CrossRef]
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef]
- Lugo, T.G.; Pendergast, A.M.; Muller, A.J.; Witte, O.N. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science (New York N. Y.) 1990, 247, 1079–1082. [Google Scholar] [CrossRef]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. Egfr mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science (New York N. Y.) 2004, 304, 1497–1500. [Google Scholar] [CrossRef]
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.; Dezube, B.J.; Janne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Weiner, G.J. Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 2015, 15, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoos, A. Development of immuno-oncology drugs—From ctla4 to pd1 to the next generations. Nat. Rev. Drug Discov. 2016, 15, 235–247. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martin-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 2017, 171, 934–949.e916. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or platinum-pemetrexed in EGFR t790m-positive lung cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Kalemkerian, G.P.; Akerley, W.; Bogner, P.; Borghaei, H.; Chow, L.Q.; Downey, R.J.; Gandhi, L.; Ganti, A.K.; Govindan, R.; Grecula, J.C.; et al. Small cell lung cancer. J. Natl. Compr. Cancer Netw. JNCCN 2013, 11, 78–98. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar]
- Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [CrossRef] [Green Version]
- Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [CrossRef] [PubMed] [Green Version]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretic, L.; Kong, G.; Leenders, F.; Lu, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Wu, S.; Liu, J.; Fang, B. Identification of a small molecule with synthetic lethality for k-ras and protein kinase c iota. Cancer Res. 2008, 68, 7403–7408. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, D.B.; Abt, E.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; et al. Lkb1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 2013, 23, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Whitehurst, A.W.; Bodemann, B.O.; Cardenas, J.; Ferguson, D.; Girard, L.; Peyton, M.; Minna, J.D.; Michnoff, C.; Hao, W.; Roth, M.G.; et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 2007, 446, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.S.; Hancock, D.C.; Molina-Arcas, M.; Steckel, M.; East, P.; Diefenbacher, M.; Armenteros-Monterroso, E.; Lassailly, F.; Matthews, N.; Nye, E.; et al. The GATA2 transcriptional network is requisite for ras oncogene-driven non-small cell lung cancer. Cell 2012, 149, 642–655. [Google Scholar] [CrossRef]
- Scholl, C.; Frohling, S.; Dunn, I.F.; Schinzel, A.C.; Barbie, D.A.; Kim, S.Y.; Silver, S.J.; Tamayo, P.; Wadlow, R.C.; Ramaswamy, S.; et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 2009, 137, 821–834. [Google Scholar] [CrossRef]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef] [Green Version]
- Vincent, S.; Chen, R.; Sayles, L.C.; Lin, C.; Walker, R.G.; Gillespie, A.K.; Subramanian, A.; Hinkle, G.; Yang, X.; Saif, S.; et al. Wilms tumor 1 (wt1) regulates KRAS-driven oncogenesis and senescence in mouse and human models. J. Clin. Investig. 2010, 120, 3940–3952. [Google Scholar] [CrossRef]
- Puyol, M.; Martin, A.; Dubus, P.; Mulero, F.; Pizcueta, P.; Khan, G.; Guerra, C.; Santamaria, D.; Barbacid, M. A synthetic lethal interaction between K-Ras oncogenes and CDK4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 2010, 18, 63–73. [Google Scholar] [CrossRef]
- Oike, T.; Ogiwara, H.; Tominaga, Y.; Ito, K.; Ando, O.; Tsuta, K.; Mizukami, T.; Shimada, Y.; Isomura, H.; Komachi, M.; et al. A synthetic lethality-based strategy to treat cancers harboring a genetic deficiency in the chromatin remodeling factor brg1. Cancer Res. 2013, 73, 5508–5518. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Yu, H.; Hughes, N.W.; Liu, B.; Kendirli, A.; Klein, K.; Chen, W.W.; Lander, E.S.; Sabatini, D.M. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic Ras. Cell 2017, 168, 890–903.e815. [Google Scholar] [CrossRef]
- Chakraborty, A.A.; Nakamura, E.; Qi, J.; Creech, A.; Jaffe, J.D.; Paulk, J.; Novak, J.S.; Nagulapalli, K.; McBrayer, S.K.; Cowley, G.S.; et al. HIF activation causes synthetic lethality between the vhl tumor suppressor and the ezh1 histone methyltransferase. Sci. Transl. Med. 2017, 9, eaal5272. [Google Scholar] [CrossRef] [PubMed]
- Unni, A.M.; Lockwood, W.W.; Zejnullahu, K.; Lee-Lin, S.Q.; Varmus, H. Evidence that synthetic lethality underlies the mutual exclusivity of oncogenic KRAS and EGFR mutations in lung adenocarcinoma. Elife 2015, 4, e06907. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Patel, M.; Ng, N.; Hsieh, M.H.; Orth, A.P.; Walker, J.R.; Batalov, S.; Harris, J.L.; Liu, J. Identification of synthetic lethality of PRKDC in MYC-dependent human cancers by pooled shRNA screening. BMC Cancer 2014, 14, 944. [Google Scholar] [CrossRef] [PubMed]
- Romero, O.A.; Torres-Diz, M.; Pros, E.; Savola, S.; Gomez, A.; Moran, S.; Saez, C.; Iwakawa, R.; Villanueva, A.; Montuenga, L.M.; et al. Max inactivation in small cell lung cancer disrupts MYC-SWI/SNF programs and is synthetic lethal with brg1. Cancer Discov. 2014, 4, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Song, M.; Kim, S.; Seo, Y.; Kim, Y.; Yoon, S. Differential regulation and synthetic lethality of exclusive RB1 and CDKN2A mutations in lung cancer. Int. J. Oncol. 2016, 48, 367–375. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Liu, S.; Adeegbe, D.O.; Christensen, C.L.; Quinn, M.M.; Dries, R.; Han, S.; Buczkowski, K.; Wang, X.; et al. NK cells mediate synergistic antitumor effects of combined inhibition of HDAC6 and BET in a SCLC preclinical model. Cancer Res. 2018, 78, 3709–3717. [Google Scholar] [CrossRef]
- Gong, X.; Du, J.; Parsons, S.H.; Merzoug, F.F.; Webster, Y.; Iversen, P.W.; Chio, L.C.; Van Horn, R.D.; Lin, X.; Blosser, W.; et al. Aurora-a kinase inhibition is synthetic lethal with loss of the RB1 tumor suppressor gene. Cancer Discov. 2019, 9, 248–263. [Google Scholar] [CrossRef]
- Oser, M.G.; Fonseca, R.; Chakraborty, A.A.; Brough, R.; Spektor, A.; Jennings, R.B.; Flaifel, A.; Novak, J.S.; Gulati, A.; Buss, E.; et al. Cells lacking the RB1 tumor suppressor gene are hyperdependent on aurora b kinase for survival. Cancer Discov. 2019, 9, 230–247. [Google Scholar] [CrossRef]
- Van Meerbeeck, J.P.; Fennell, D.A.; De Ruysscher, D.K. Small-cell lung cancer. Lancet (London England) 2011, 378, 1741–1755. [Google Scholar] [CrossRef]
- Brenner, B.; Tang, L.H.; Klimstra, D.S.; Kelsen, D.P. Small-cell carcinomas of the gastrointestinal tract: A review. J. Clin. Oncol. 2004, 22, 2730–2739. [Google Scholar] [CrossRef] [PubMed]
- Bunn, P.A., Jr.; Minna, J.D.; Augustyn, A.; Gazdar, A.F.; Ouadah, Y.; Krasnow, M.A.; Berns, A.; Brambilla, E.; Rekhtman, N.; Massion, P.P.; et al. Small cell lung cancer: Can recent advances in biology and molecular biology be translated into improved outcomes? J. Thorac. Oncol. 2016, 11, 453–474. [Google Scholar] [CrossRef]
- Pietanza, M.C.; Byers, L.A.; Minna, J.D.; Rudin, C.M. Small cell lung cancer: Will recent progress lead to improved outcomes? Clin. Cancer Res. 2015, 21, 2244–2255. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Nau, M.M.; Chiba, I.; Birrer, M.J.; Rosenberg, R.K.; Vinocour, M.; Levitt, M.; Pass, H.; Gazdar, A.F.; Minna, J.D. P53: A frequent target for genetic abnormalities in lung cancer. Science (New York N. Y.) 1989, 246, 491–494. [Google Scholar] [CrossRef]
- Peifer, M.; Fernandez-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanson, K.R.; Hanna, R.E.; Hegde, M.; Donovan, K.F.; Strand, C.; Sullender, M.E.; Vaimberg, E.W.; Goodale, A.; Root, D.E.; Piccioni, F.; et al. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun. 2018, 9, 5416. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Category | Genomic Alteration (Indicator 1) | Synthetic Target (Indicator 2) | Types of Lung Cancer | Approaching Method | References |
|---|---|---|---|---|---|
| oncogene | KRAS | protein kinase C iota (PKCi) | NSCLC | chemical library screen | [53] |
| oncogene | KRAS | PLK1 | NSCLC | RNA interference screen | [12] |
| oncogene | KRAS | STK33 | NSCLC | RNA interference screen | [57] |
| oncogene | KRAS | TBK1 | NSCLC | RNA interference screen | [58] |
| oncogene | KRAS | WT1 | NSCLC | RNA interference screen | [59] |
| oncogene | KRAS | CDK4 | NSCLC | RNA interference screen | [60] |
| oncogene | KRAS | GATA2 | NSCLC | RNA interference screen | [56] |
| other signaling | LKB1 | phenformin | NSCLC | chemical library screen | [54] |
| chromatin remodeling | BRG1 | SMARCA2 | NSCLC | RNA interference screen | [61] |
| oncogene | KRAS | EGFR | NSCLC | computational analysis | [64] |
| transcription factor | MYC | PRKDC | SCLC | RNA interference screen | [65] |
| transcription factor | MAX | BRG1 | SCLC | computational analysis | [66] |
| tumor suppressor gene | RB1 | CDKN2A | SCLC | computational analysis | [67] |
| transcription factor | BET | HDAC6 | SCLC | RNA interference screen | [68] |
| tumor suppressor gene | RB1 | Aurora A | SCLC | CRISPR screen | [69] |
| tumor suppressor gene | RB1 | Aurora B | SCLC | CRISPR screen | [70] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shimomura, I.; Yamamoto, Y.; Ochiya, T. Synthetic Lethality in Lung Cancer—From the Perspective of Cancer Genomics. Medicines 2019, 6, 38. https://doi.org/10.3390/medicines6010038
Shimomura I, Yamamoto Y, Ochiya T. Synthetic Lethality in Lung Cancer—From the Perspective of Cancer Genomics. Medicines. 2019; 6(1):38. https://doi.org/10.3390/medicines6010038
Chicago/Turabian StyleShimomura, Iwao, Yusuke Yamamoto, and Takahiro Ochiya. 2019. "Synthetic Lethality in Lung Cancer—From the Perspective of Cancer Genomics" Medicines 6, no. 1: 38. https://doi.org/10.3390/medicines6010038
APA StyleShimomura, I., Yamamoto, Y., & Ochiya, T. (2019). Synthetic Lethality in Lung Cancer—From the Perspective of Cancer Genomics. Medicines, 6(1), 38. https://doi.org/10.3390/medicines6010038