Heparin Binding Proteins as Therapeutic Target: An Historical Account and Current Trends

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Heparin Binding Growth Factors and Their Inhibitors
2.1. Semi-Synthetic and Fully Synthetic Heparin-Like Oligosaccharides
2.2. Non-Carbohydrate Small Molecules as FGF Antagonists
3. GAG Metabolic Enzymes as Target
3.1. Galactosyl Transferase Inhibitors and Decoys
3.2. Sulfotransferase Inhibitors
3.3. Heparanase Inhibitors
4. Heparin-Binding Proteins as Biological Drugs
5. Summary and Closing Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Pomin, V.H.; Mulloy, B. Current Structural Biology of the Heparin Interactome. Curr. Opin. Struct. Biol. 2015, 34, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, U.; Couchman, J.; Kimata, K.; Esko, J.D. Proteoglycans and Sulfated Glycosaminoglycans. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015. [Google Scholar]
- Peysselon, F.; Ricard-Blum, S. Heparin-Protein Interactions: From Affinity and Kinetics to Biological Roles. Application to an Interaction Network Regulating Angiogenesis. Matrix Biol. 2014, 35, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Sasisekharan, R.; Raman, R.; Prabhakar, V. Glycomics Approach to Structure-Function Relationships of Glycosaminoglycans. Annu. Rev. Biomed. Eng. 2006, 8, 181–231. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.S.; Yazzie, B.; Middaugh, C.R. Polyanions and the Proteome. Mol. Cell. Proteom. 2004, 3, 746–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyrek, E.; Dubin, P. Glycosaminoglycans As Polyelectrolytes. Adv. Colloid Interface Sci. 2010, 158, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Gigli, M.; Consonni, A.; Ghiselli, G.; Rizzo, V.; Naggi, A.; Torri, G. Heparin Binding to Human Plasma Low-Density Lipoproteins: Dependence on Heparin Sulfation Degree and Chain Length. Biochemistry 1992, 31, 5996–6003. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, A.I.S.; Pitt, S.J.; Stewart, A.J. Glycosaminoglycan Neutralization in Coagulation Control. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1258–1270. [Google Scholar] [CrossRef] [Green Version]
- Medeiros, G.F.; Mendes, A.; Castro, R.A.; Bau, E.C.; Nader, H.B.; Dietrich, C.P. Distribution of Sulfated Glycosaminoglycans in the Animal Kingdom: Widespread Occurrence of Heparin-Like Compounds in Invertebrates. Biochim. Biophys. Acta 2000, 1475, 287–294. [Google Scholar] [CrossRef]
- Nader, H.B.; Ferreira, T.M.; Toma, L.; Chavante, S.F.; Dietrich, C.P.; Casu, B.; Torri, G. Maintenance of Heparan Sulfate Structure Throughout Evolution: Chemical and Enzymic Degradation, and 13C-n.m.r.-Spectral Evidence. Carbohydr. Res. 1988, 184, 292–300. [Google Scholar] [CrossRef]
- Mulloy, B.; Hogwood, J.; Gray, E.; Lever, R.; Page, C.P. Pharmacology of Heparin and Related Drugs. Pharmacol. Rev. 2016, 68, 76–141. [Google Scholar] [CrossRef]
- Oduah, E.I.; Linhardt, R.J.; Sharfstein, S.T. Heparin: Past, Present, and Future. Pharmaceuticals 2016, 9, 38. [Google Scholar] [CrossRef]
- Cassinelli, G.; Naggi, A. Old and New Applications of Non-Anticoagulant Heparin. Int. J. Cardiol. 2016, 212, S14–S21. [Google Scholar] [CrossRef]
- Gray, E.; Hogwood, J.; Mulloy, B. The Anticoagulant and Anti-thrombotic Mechanisms of Heparin; Springer: Berlin/Heidelberg, Germany, 2012; pp. 43–61. [Google Scholar]
- Petitou, M.; Herault, J.P.; Bernat, A.; Driguez, P.A.; Duchaussoy, P.; Lormeau, J.C.; Herbert, J.M. Synthesis of Thrombin-Inhibiting Heparin Mimetics Without Side Effects. Nature 1999, 398, 417–422. [Google Scholar] [CrossRef]
- Petitou, M.; van Boeckel, C.A. A Synthetic Antithrombin III Binding Pentasaccharide Is Now a Drug! What Comes Next? Angew. Chem. Int. Ed. 2004, 43, 3118–3133. [Google Scholar] [CrossRef]
- Page, C. Heparin and Related Drugs: Beyond Anticoagulant Activity. ISRN Pharmacol. 2013, 2013, 910743. [Google Scholar] [CrossRef]
- Christianson, H.C.; Belting, M. Heparan Sulfate Proteoglycan As a Cell-Surface Endocytosis Receptor. Matrix Biol. 2014, 35, 51–55. [Google Scholar] [CrossRef]
- Parsons, B.J. Oxidation of Glycosaminoglycans by Free Radicals and Reactive Oxidative Species: A Review of Investigative Methods. Free Radic. Res. 2015, 49, 618–632. [Google Scholar] [CrossRef]
- Ori, A.; Wilkinson, M.C.; Fernig, D.G. A Systems Biology Approach for the Investigation of the Heparin/Heparan Sulfate Interactome. J. Biol. Chem. 2011, 286, 19892–19904. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Esko, J.D. Demystifying Heparan Sulfate-Protein Interactions. Annu. Rev. Biochem. 2014, 83, 129–157. [Google Scholar] [CrossRef]
- Bernfield, M.; Gotte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of Cell Surface Heparan Sulfate Proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef]
- Chiodelli, P.; Bugatti, A.; Urbinati, C.; Rusnati, M. Heparin/Heparan Sulfate Proteoglycans Glycomic Interactome in Angiogenesis: Biological Implications and Therapeutical Use. Molecules 2015, 20, 6342–6388. [Google Scholar] [CrossRef] [Green Version]
- Li, J.P.; Kusche-Gullberg, M. Heparan Sulfate: Biosynthesis, Structure, and Function. Int. Rev. Cell Mol. Biol. 2016, 325, 215–273. [Google Scholar]
- Thornton, S.C.; Mueller, S.N.; Levine, E.M. Human Endothelial Cells: Use of Heparin in Cloning and Long-Term Serial Cultivation. Science 1983, 222, 623–625. [Google Scholar] [CrossRef]
- Kan, M.; Wang, F.; Xu, J.; Crabb, J.W.; Hou, J.; McKeehan, W.L. An Essential Heparin-Binding Domain in the Fibroblast Growth Factor Receptor Kinase. Science 1993, 259, 1918–1921. [Google Scholar] [CrossRef]
- Zugmaier, G.; Lippman, M.E.; Wellstein, A. Inhibition by Pentosan Polysulfate (PPS) of Heparin-Binding Growth Factors Released From Tumor Cells and Blockage by PPS of Tumor Growth in Animals. J. Natl. Cancer Inst. 1992, 84, 1716–1724. [Google Scholar] [CrossRef]
- Miao, H.Q.; Ishai-Michaeli, R.; Peretz, T.; Vlodavsky, I. Laminarin Sulfate Mimics the Effects of Heparin on Smooth Muscle Cell Proliferation and Basic Fibroblast Growth Factor-Receptor Binding and Mitogenic Activity. J. Cell. Physiol. 1995, 164, 482–490. [Google Scholar] [CrossRef]
- Miao, H.Q.; Ornitz, D.M.; Aingorn, E.; Ben-Sasson, S.A.; Vlodavsky, I. Modulation of Fibroblast Growth Factor-2 Receptor Binding, Dimerization, Signaling, and Angiogenic Activity by a Synthetic Heparin-Mimicking Polyanionic Compound. J. Clin. Investig. 1997, 99, 1565–1575. [Google Scholar] [CrossRef]
- Lozano, R.M.; Rivas, G.; Gimenez-Gallego, G. Destabilization, Oligomerization and Inhibition of the Mitogenic Activity of Acidic Fibroblast-Growth Factor by Aurintricarboxylic Acid. Eur. J. Biochem. 1997, 248, 30–36. [Google Scholar] [CrossRef]
- Folkman, J.; Szabo, S.; Stovroff, M.; McNeil, P.; Li, W.; Shing, Y. Duodenal Ulcer. Discovery of a New Mechanism and Development of Angiogenic Therapy That Accelerates Healing. Ann. Surg. 1991, 214, 414–425. [Google Scholar] [CrossRef]
- Zhu, X.; Hsu, B.T.; Rees, D.C. Structural Studies of the Binding of the Anti-Ulcer Drug Sucrose Octasulfate to Acidic Fibroblast Growth Factor. Structure 1993, 1, 27–34. [Google Scholar] [CrossRef]
- Yeh, B.K.; Eliseenkova, A.V.; Plotnikov, A.N.; Green, D.; Pinnell, J.; Polat, T.; Gritli-Linde, A.; Linhardt, R.J.; Mohammadi, M. Structural Basis for Activation of Fibroblast Growth Factor Signaling by Sucrose Octasulfate. Mol. Cell. Biol. 2002, 22, 7184–7192. [Google Scholar] [CrossRef] [Green Version]
- Pineda-Lucena, A.; Jimenez, M.A.; Nieto, J.L.; Santoro, J.; Rico, M.; Gimenez-Gallego, G. 1H-NMR Assignment and Solution Structure of Human Acidic Fibroblast Growth Factor Activated by Inositol Hexasulfate. J. Mol. Biol. 1994, 242, 81–98. [Google Scholar] [CrossRef]
- Gagliardi, A.; Hadd, H.; Collins, D.C. Inhibition of Angiogenesis by Suramin. Cancer Res. 1992, 52, 5073–5075. [Google Scholar]
- Zaniboni, A. Suramin: The Discovery of an Old Anticancer Drug. Med. Oncol. Tumor Pharmacother. 1990, 7, 287–290. [Google Scholar]
- Stein, C.A. Suramin: A Novel Antineoplastic Agent With Multiple Potential Mechanisms of Action. Cancer Res. 1993, 53, 2239–2248. [Google Scholar]
- Pesenti, E.; Sola, F.; Mongelli, N.; Grandi, M.; Spreafico, F. Suramin Prevents Neovascularisation and Tumour Growth Through Blocking of Basic Fibroblast Growth Factor Activity. Br. J. Cancer 1992, 66, 367–372. [Google Scholar] [CrossRef]
- Zamai, M.; Caiolfa, V.R.; Pines, D.; Pines, E.; Parola, A.H. Nature of Interaction Between Basic Fibroblast Growth Factor and the Antiangiogenic Drug 7,7-(Carbonyl-Bis[Imino-N-Methyl-4, 2-Pyrrolecarbonylimino[N-Methyl-4,2-Pyrrole]-Carbonylimino])Bis-(1, 3-Naphthalene Disulfonate). Biophys. J. 1998, 75, 672–682. [Google Scholar] [CrossRef]
- Ciomei, M.; Pastori, W.; Mariani, M.; Sola, F.; Grandi, M.; Mongelli, N. New Sulfonated Distamycin A Derivatives With BFGF Complexing Activity. Biochem. Pharmacol. 1994, 47, 295–302. [Google Scholar] [CrossRef]
- Manetti, F.; Corelli, F.; Botta, M. Fibroblast Growth Factors and Their Inhibitors. Curr. Pharm. Des. 2000, 6, 1897–1924. [Google Scholar] [CrossRef]
- Xu, R.; Ori, A.; Rudd, T.R.; Uniewicz, K.A.; Ahmed, Y.A.; Guimond, S.E.; Skidmore, M.A.; Siligardi, G.; Yates, E.A.; Fernig, D.G. Diversification of the Structural Determinants of Fibroblast Growth Factor-Heparin Interactions: Implications for Binding Specificity. J. Biol. Chem. 2012, 287, 40061–40073. [Google Scholar] [CrossRef]
- Giacomini, A.; Chiodelli, P.; Matarazzo, S.; Rusnati, M.; Presta, M.; Ronca, R. Blocking the FGF/FGFR System As a “Two-Compartment” Antiangiogenic/Antitumor Approach in Cancer Therapy. Pharmacol. Res. 2016, 107, 172–185. [Google Scholar] [CrossRef]
- Mohammadi, M.; Olsen, S.K.; Goetz, R. A Protein Canyon in the FGF-FGF Receptor Dimer Selects From an a La Carte Menu of Heparan Sulfate Motifs. Curr. Opin. Struct. Biol. 2005, 15, 506–516. [Google Scholar] [CrossRef]
- Pellegrini, L. Role of Heparan Sulfate in Fibroblast Growth Factor Signalling: A Structural View. Curr. Opin. Struct. Biol. 2001, 11, 629–634. [Google Scholar] [CrossRef]
- Plotnikov, A.N.; Hubbard, S.R.; Schlessinger, J.; Mohammadi, M. Crystal Structures of Two FGF-FGFR Complexes Reveal the Determinants of Ligand-Receptor Specificity. Cell 2000, 101, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Salmivirta, M.; Lidholt, K.; Lindahl, U. Heparan Sulfate: A Piece of Information. FASEB J. 1996, 10, 1270–1279. [Google Scholar] [CrossRef]
- Kreuger, J.; Spillmann, D.; Li, J.P.; Lindahl, U. Interactions Between Heparan Sulfate and Proteins: The Concept of Specificity. J. Cell Biol. 2006, 174, 323–327. [Google Scholar] [CrossRef]
- Casu, B.; Lindahl, U. Structure and Biological Interactions of Heparin and Heparan Sulfate. Adv. Carbohydr. Chem. Biochem. 2001, 57, 159–206. [Google Scholar]
- Fu, L.; Suflita, M.; Linhardt, R.J. Bioengineered Heparins and Heparan Sulfates. Adv. Drug Deliv. Rev. 2016, 97, 237–249. [Google Scholar] [CrossRef]
- Miller, G.J.; Hansen, S.U.; Avizienyte, E.; Rushton, G.; Cole, C.; Jayson, G.C.; Gardiner, J.M. Efficient Chemical Synthesis of Heparin-like Octa-, Deca- and Dodecasaccharides and Inhibition of FGF2- and VEGF165- Mediated Endothelial Cell Functions. Chem. Sci. 2013, 4, 3218–3222. [Google Scholar] [CrossRef]
- Schlessinger, J.; Plotnikov, A.N.; Ibrahimi, O.A.; Eliseenkova, A.V.; Yeh, B.K.; Yayon, A.; Linhardt, R.J.; Mohammadi, M. Crystal Structure of a Ternary FGF-FGFR-Heparin Complex Reveals a Dual Role for Heparin in FGFR Binding and Dimerization. Mol. Cell 2000, 6, 743–750. [Google Scholar] [CrossRef]
- Jastrebova, N.; Vanwildemeersch, M.; Lindahl, U.; Spillmann, D. Heparan Sulfate Domain Organization and Sulfation Modulate FGF-Induced Cell Signaling. J. Biol. Chem. 2010, 285, 26842–26851. [Google Scholar] [CrossRef]
- Qiu, H.; Shi, S.; Yue, J.; Xin, M.; Nairn, A.V.; Lin, L.; Liu, X.; Li, G.; Rcher-Hartmann, S.A.; Dela, R.M.; et al. A Mutant-Cell Library for Systematic Analysis of Heparan Sulfate Structure-Function Relationships. Nat. Methods 2018, 15, 889–899. [Google Scholar] [CrossRef]
- Avizienyte, E.; Cole, C.L.; Rushton, G.; Miller, G.J.; Bugatti, A.; Presta, M.; Gardiner, J.M.; Jayson, G.C. Synthetic Site-Selectively Mono-6-O-Sulfated Heparan Sulfate Dodecasaccharide Shows Anti-Angiogenic Properties In Vitro and Sensitizes Tumors to Cisplatin In Vivo. PLoS ONE 2016, 11, e0159739. [Google Scholar] [CrossRef]
- Zong, C.; Venot, A.; Li, X.; Lu, W.; Xiao, W.; Wilkes, J.L.; Salanga, C.L.; Handel, T.M.; Wang, L.; Wolfert, M.A.; et al. Heparan Sulfate Microarray Reveals That Heparan Sulfate-Protein Binding Exhibits Different Ligand Requirements. J. Am. Chem. Soc. 2017, 139, 9534–9543. [Google Scholar] [CrossRef]
- Braddock, P.S.; Hu, D.E.; Fan, T.P.; Stratford, I.J.; Harris, A.L.; Bicknell, R. A Structure-Activity Analysis of Antagonism of the Growth Factor and Angiogenic Activity of Basic Fibroblast Growth Factor by Suramin and Related Polyanions. Br. J. Cancer 1994, 69, 890–898. [Google Scholar] [CrossRef]
- Lozano, R.M.; Jimenez, M.; Santoro, J.; Rico, M.; Gimenez-Gallego, G. Solution Structure of Acidic Fibroblast Growth Factor Bound to 1,3, 6-Naphthalenetrisulfonate: A Minimal Model for the Anti-Tumoral Action of Suramins and Suradistas. J. Mol. Biol. 1998, 281, 899–915. [Google Scholar] [CrossRef]
- Fernandez-Tornero, C.; Lozano, R.M.; Redondo-Horcajo, M.; Gomez, A.M.; Lopez, J.C.; Quesada, E.; Uriel, C.; Valverde, S.; Cuevas, P.; Romero, A.; et al. Leads for Development of New Naphthalenesulfonate Derivatives With Enhanced Antiangiogenic Activity: Crystal Structure of Acidic Fibroblast Growth Factor in Complex With 5-Amino-2-Naphthalene Sulfonate. J. Biol. Chem. 2003, 278, 21774–21781. [Google Scholar] [CrossRef]
- Fernandez, I.S.; Cuevas, P.; Angulo, J.; Lopez-Navajas, P.; Canales-Mayordomo, A.; Gonzalez-Corrochano, R.; Lozano, R.M.; Valverde, S.; Jimenez-Barbero, J.; Romero, A.; et al. Gentisic Acid, a Compound Associated With Plant Defense and a Metabolite of Aspirin, Heads a New Class of in Vivo Fibroblast Growth Factor Inhibitors. J. Biol. Chem. 2010, 285, 11714–11729. [Google Scholar] [CrossRef]
- Angulo, J.; Cuevas, P.; Cuevas, B.; El, Y.M.; Fernandez, A.; Martinez-Salamanca, E.; Gonzalez-Corrochano, R.; Gimenez-Gallego, G. Diacetyloxyl Derivatization of the Fibroblast Growth Factor Inhibitor Dobesilate Enhances Its Anti-Inflammatory, Anti-Angiogenic and Anti-Tumoral Activities. J. Transl. Med. 2015, 13, 48. [Google Scholar] [CrossRef]
- Taraboletti, G.; Rusnati, M.; Ragona, L.; Colombo, G. Targeting Tumor Angiogenesis With TSP-1-Based Compounds: Rational Design of Antiangiogenic Mimetics of Endogenous Inhibitors. Oncotarget 2010, 1, 662–673. [Google Scholar] [CrossRef]
- Pagano, K.; Torella, R.; Foglieni, C.; Bugatti, A.; Tomaselli, S.; Zetta, L.; Presta, M.; Rusnati, M.; Taraboletti, G.; Colombo, G.; et al. Direct and Allosteric Inhibition of the FGF2/HSPGs/FGFR1 Ternary Complex Formation by an Antiangiogenic, Thrombospondin-1-Mimic Small Molecule. PLoS ONE 2012, 7, e36990. [Google Scholar] [CrossRef]
- Colombo, G.; Margosio, B.; Ragona, L.; Neves, M.; Bonifacio, S.; Annis, D.S.; Stravalaci, M.; Tomaselli, S.; Giavazzi, R.; Rusnati, M.; et al. Non-Peptidic Thrombospondin-1 Mimics As Fibroblast Growth Factor-2 Inhibitors: An Integrated Strategy for the Development of New Antiangiogenic Compounds. J. Biol. Chem. 2010, 285, 8733–8742. [Google Scholar] [CrossRef]
- Foglieni, C.; Pagano, K.; Lessi, M.; Bugatti, A.; Moroni, E.; Pinessi, D.; Resovi, A.; Ribatti, D.; Bertini, S.; Ragona, L.; et al. Integrating Computational and Chemical Biology Tools in the Discovery of Antiangiogenic Small Molecule Ligands of FGF2 Derived From Endogenous Inhibitors. Sci. Rep. 2016, 6, 23432. [Google Scholar] [CrossRef]
- Zhang, J.; Riverst, G.; Zhu, Y.; Jacobson, A.; Peyers, J.; Grundstrom, G.; Burch, P.; Hussein, S.; Marolewski, A.; Herlihy, W.; et al. Identification of Inhibitors of Heparin-Growth Factor Interactions From Combinatorial Libraries of Four-Component Condensation Reactions. Bioorg. Med. Chem. 2001, 9, 825–836. [Google Scholar] [CrossRef]
- Cochran, S.; Li, C.P.; Bytheway, I. An Experimental and Molecular-Modeling Study of the Binding of Linked Sulfated Tetracyclitols to FGF-1 and FGF-2. ChemBioChem 2005, 6, 1882–1890. [Google Scholar] [CrossRef]
- Liu, L.; Li, C.; Cochran, S.; Feder, D.; Guddat, L.W.; Ferro, V. A Focused Sulfated Glycoconjugate Ugi Library for Probing Heparan Sulfate-Binding Angiogenic Growth Factors. Bioorg. Med. Chem. Lett. 2012, 22, 6190–6194. [Google Scholar] [CrossRef]
- Liu, L.; Li, C.; Cochran, S.; Jimmink, S.; Ferro, V. Synthesis of a Heparan Sulfate Mimetic Library Targeting FGF and VEGF Via Click Chemistry on a Monosaccharide Template. ChemMedChem 2012, 7, 1267–1275. [Google Scholar] [CrossRef]
- Mikami, T.; Kitagawa, H. Biosynthesis and Function of Chondroitin Sulfate. Biochim. Biophys. Acta 2013, 1830, 4719–4733. [Google Scholar] [CrossRef]
- Prydz, K. Determinants of Glycosaminoglycan (GAG) Structure. Biomolecules 2015, 5, 2003–2022. [Google Scholar] [CrossRef] [Green Version]
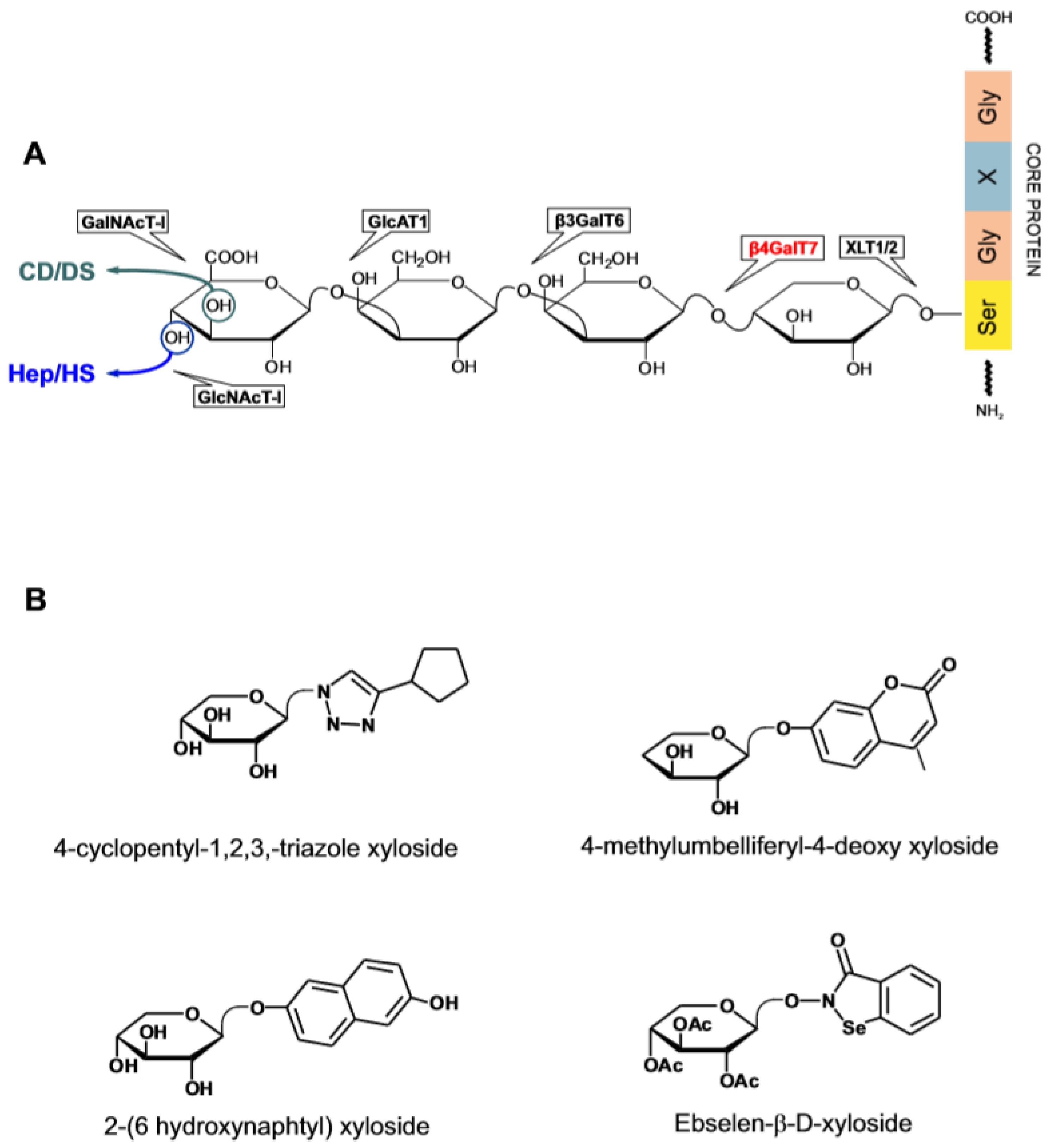
- Garcia-Garcia, J.F.; Corrales, G.; Casas, J.; Fernandez-Mayoralas, A.; Garcia-Junceda, E. Synthesis and Evaluation of Xylopyranoside Derivatives As “Decoy Acceptors” of Human Beta-1,4-Galactosyltransferase 7. Mol. Biosyst. 2011, 7, 1312–1321. [Google Scholar] [CrossRef]
- Chua, J.S.; Kuberan, B. Synthetic Xylosides: Probing the Glycosaminoglycan Biosynthetic Machinery for Biomedical Applications. Acc. Chem. Res. 2017, 50, 2693–2705. [Google Scholar] [CrossRef]
- Ghiselli, G. Drug-Mediated Regulation of Glycosaminoglycan Biosynthesis. Med. Res. Rev. 2017, 37, 1051–1094. [Google Scholar] [CrossRef]
- Persson, A.; Ellervik, U.; Mani, K. Fine-Tuning the Structure of Glycosaminoglycans in Living Cells Using Xylosides. Glycobiology 2018, 28, 499–511. [Google Scholar] [CrossRef]
- Siegbahn, A.; Manner, S.; Persson, A.; Tykesson, E.; Holmqvist, K.; Ochocinska, A.; Ronnols, J.; Sundin, A.; Mani, K.; Westergren-Thorsson, G.; et al. Rules for Priming and Inhibition of Glycosaminoglycan Biosynthesis; Probing the B4GalT7 Active Site. Chem. Sci. 2014, 5, 3501–3508. [Google Scholar] [CrossRef]
- Siegbahn, A.; Thorsheim, K.; Stahle, J.; Manner, S.; Hamark, C.; Persson, A.; Tykesson, E.; Mani, K.; Westergren-Thorsson, G.; Widmalm, G.; et al. Exploration of the Active Site of Beta4GalT7: Modifications of the Aglycon of Aromatic Xylosides. Org. Biomol. Chem. 2015, 13, 3351–3362. [Google Scholar] [CrossRef]
- Saliba, M.; Ramalanjaona, N.; Gulberti, S.; Bertin-Jung, I.; Thomas, A.; Dahbi, S.; Lopin-Bon, C.; Jacquinet, J.C.; Breton, C.; Ouzzine, M.; et al. Probing the Acceptor Active Site Organization of the Human Recombinant Beta1,4-Galactosyltransferase 7 and Design of Xyloside-Based Inhibitors. J. Biol. Chem. 2015, 290, 7658–7670. [Google Scholar] [CrossRef]
- Tsutsui, Y.; Ramakrishnan, B.; Qasba, P.K. Crystal Structures of Beta-1,4-Galactosyltransferase 7 Enzyme Reveal Conformational Changes and Substrate Binding. J. Biol. Chem. 2013, 288, 31963–31970. [Google Scholar] [CrossRef]
- Chen, Y.H.; Narimatsu, Y.; Clausen, T.M.; Gomes, C.; Karlsson, R.; Steentoft, C.; Spliid, C.B.; Gustavsson, T.; Salanti, A.; Persson, A.; et al. The GAGOme: A Cell-Based Library of Displayed Glycosaminoglycans. Nat. Methods 2018, 15, 881–888. [Google Scholar] [CrossRef]
- Martin, N.B.; Masson, P.; Sepulchre, C.; Theveniaux, J.; Millet, J.; Bellamy, F. Pharmacologic and Biochemical Profiles of New Venous Anti-thrombotic Beta-D-Xyloside Derivatives: Potential Antiathero/Thrombotic Drugs. Semin. Thromb. Hemost. 1996, 22, 247–254. [Google Scholar] [CrossRef]
- Toomey, J.R.; Abboud, M.A.; Valocik, R.E.; Koster, P.F.; Burns-Kurtis, C.L.; Pillarisetti, K.; Danoff, T.M.; Erhardt, J.A. A Comparison of the Beta-D-Xyloside, Odiparcil, to Warfarin in a Rat Model of Venous Thrombosis. J. Thromb. Haemost. 2006, 4, 1989–1996. [Google Scholar] [CrossRef]
- Chua, J.S.; Tran, V.M.; Kalita, M.; Quintero, M.V.; Antelope, O.; Muruganandam, G.; Saijoh, Y.; Kuberan, B. A Glycan-Based Approach to Therapeutic Angiogenesis. PLoS ONE 2017, 12, e0182301. [Google Scholar] [CrossRef]
- Shaukat, I.; Barre, L.; Venkatesan, N.; Li, D.; Jaquinet, J.C.; Fournel-Gigleux, S.; Ouzzine, M. Targeting of Proteoglycan Synthesis Pathway: A New Strategy to Counteract Excessive Matrix Proteoglycan Deposition and Transforming Growth Factor-Beta1-Induced Fibrotic Phenotype in Lung Fibroblasts. PLoS ONE 2016, 11, e0146499. [Google Scholar] [CrossRef]
- Mani, K.; Belting, M.; Ellervik, U.; Falk, N.; Svensson, G.; Sandgren, S.; Cheng, F.; Fransson, L.A. Tumor Attenuation by 2(6-Hydroxynaphthyl)-Beta-D-Xylopyranoside Requires Priming of Heparan Sulfate and Nuclear Targeting of the Products. Glycobiology 2004, 14, 387–397. [Google Scholar] [CrossRef]
- Persson, A.; Tykesson, E.; Westergren-Thorsson, G.; Malmstrom, A.; Ellervik, U.; Mani, K. Xyloside-Primed Chondroitin Sulfate/Dermatan Sulfate From Breast Carcinoma Cells With a Defined Disaccharide Composition Has Cytotoxic Effects in Vitro. J. Biol. Chem. 2016, 291, 14871–14882. [Google Scholar] [CrossRef]
- Tran, V.M.; Victor, X.V.; Yockman, J.W.; Kuberan, B. RGD-Xyloside Conjugates Prime Glycosaminoglycans. Glycoconj. J. 2010, 27, 625–633. [Google Scholar] [CrossRef]
- Chen, Z.; Jiang, Z.; Chen, N.; Shi, Q.; Tong, L.; Kong, F.; Cheng, X.; Chen, H.; Wang, C.; Tang, B. Target Discovery of Ebselen With a Biotinylated Probe. Chem. Commun. 2018, 54, 9506–9509. [Google Scholar] [CrossRef]
- Tang, Y.; Zhang, S.; Chang, Y.; Fan, D.; Agostini, A.; Zhang, L.; Jiang, T. Aglycone Ebselen and Beta-d-Xyloside Primed Glycosaminoglycans Co-Contribute to Ebselen Beta-d-Xyloside-Induced Cytotoxicity. J. Med. Chem. 2018, 61, 2937–2948. [Google Scholar] [CrossRef]
- Lindahl, U.; Li, J.P. Interactions Between Heparan Sulfate and Proteins-Design and Functional Implications. Int. Rev. Cell Mol. Biol. 2009, 276, 105–159. [Google Scholar]
- Gama, C.I.; Tully, S.E.; Sotogaku, N.; Clark, P.M.; Rawat, M.; Vaidehi, N.; Goddard, W.A., III; Nishi, A.; Hsieh-Wilson, L.C. Sulfation Patterns of Glycosaminoglycans Encode Molecular Recognition and Activity. Nat. Chem. Biol. 2006, 2, 467–473. [Google Scholar] [CrossRef]
- Townley, R.A.; Bulow, H.E. Deciphering Functional Glycosaminoglycan Motifs in Development. Curr. Opin. Struct. Biol. 2018, 50, 144–154. [Google Scholar] [CrossRef]
- Stancanelli, E.; Elli, S.; Hsieh, P.H.; Liu, J.; Guerrini, M. Recognition and Conformational Properties of an Alternative Antithrombin Binding Sequence Obtained by Chemoenzymatic Synthesis. ChemBioChem 2018, 19, 1178–1188. [Google Scholar] [CrossRef]
- Casu, B.; Naggi, A.; Torri, G. Re-Visiting the Structure of Heparin. Carbohydr. Res. 2015, 403, 60–68. [Google Scholar] [CrossRef]
- Meneghetti, M.C.; Hughes, A.J.; Rudd, T.R.; Nader, H.B.; Powell, A.K.; Yates, E.A.; Lima, M.A. Heparan Sulfate and Heparin Interactions With Proteins. J. R. Soc. Interface 2015, 12, 0589. [Google Scholar] [CrossRef]
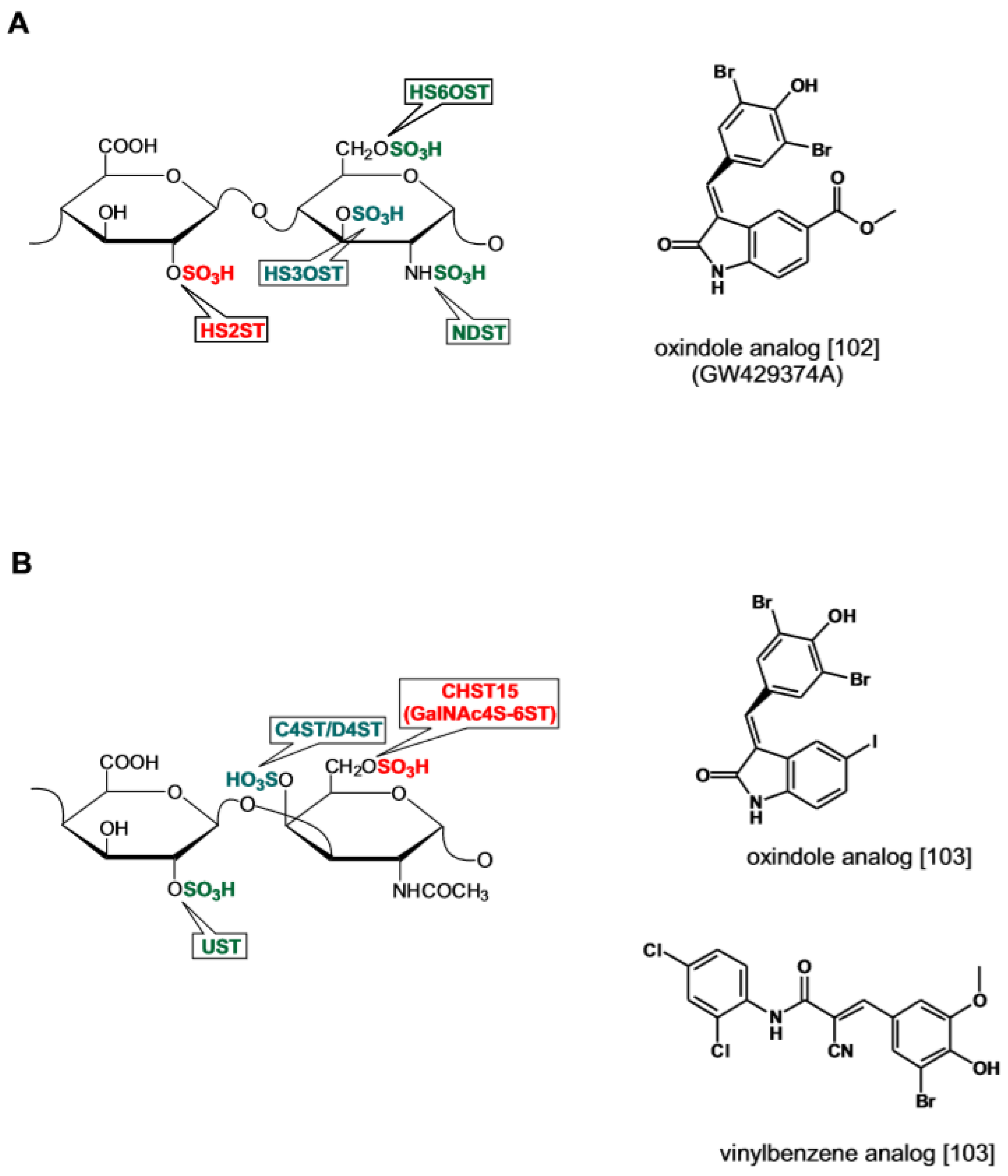
- Liu, J.; Moon, A.F.; Sheng, J.; Pedersen, L.C. Understanding the Substrate Specificity of the Heparan Sulfate Sulfotransferases by an Integrated Biosynthetic and Crystallographic Approach. Curr. Opin. Struct. Biol. 2012, 22, 550–557. [Google Scholar] [CrossRef]
- Liu, C.; Sheng, J.; Krahn, J.M.; Perera, L.; Xu, Y.; Hsieh, P.H.; Dou, W.; Liu, J.; Pedersen, L.C. Molecular Mechanism of Substrate Specificity for Heparan Sulfate 2-O-Sulfotransferase. J. Biol. Chem. 2014, 289, 13407–13418. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, J.I.; Portley, A.R.; Chang, Y.T.; Nierengarten, D.M.; Cook, B.N.; Bowman, K.G.; Bishop, A.; Gray, N.S.; Shokat, K.M.; Schultz, P.G.; et al. Discovery of Carbohydrate Sulfotransferase Inhibitors From a Kinase-Directed Library. Angew. Chem. 2000, 39, 1359–1362. [Google Scholar] [CrossRef]
- Rath, V.L.; Verdugo, D.; Hemmerich, S. Sulfotransferase Structural Biology and Inhibitor Discovery. Drug Discov. Today 2004, 9, 1003–1011. [Google Scholar] [CrossRef]
- Ghiselli, G.; Maccarana, M. Drugs Affecting Glycosaminoglycan Metabolism. Drug Discov. Today 2016, 21, 1162–1169. [Google Scholar] [CrossRef]
- Hagner-McWhirter, A.; Li, J.P.; Oscarson, S.; Lindahl, U. Irreversible Glucuronyl C5-Epimerization in the Biosynthesis of Heparan Sulfate. J. Biol. Chem. 2004, 279, 14631–14638. [Google Scholar] [CrossRef]
- Byrne, D.P.; Li, Y.; Ngamlert, P.; Ramakrishnan, K.; Eyers, C.E.; Wells, C.; Drewry, D.H.; Zuercher, W.J.; Berry, N.G.; Fernig, D.G.; et al. New Tools for Evaluating Protein Tyrosine Sulfation: Tyrosylprotein Sulfotransferases (TPSTs) Are Novel Targets for RAF Protein Kinase Inhibitors. Biochem. J. 2018, 475, 2435–2455. [Google Scholar] [CrossRef]
- Cheung, S.T.; Miller, M.S.; Pacoma, R.; Roland, J.; Liu, J.; Schumacher, A.M.; Hsieh-Wilson, L.C. Discovery of a Small-Molecule Modulator of Glycosaminoglycan Sulfation. ACS Chem. Biol. 2017, 12, 3126–3133. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.M.; Xia, J.; Zhuang, B.; Cho, K.S.; Rogers, C.J.; Gama, C.I.; Rawat, M.; Tully, S.E.; Uetani, N.; Mason, D.E.; et al. A Sulfated Carbohydrate Epitope Inhibits Axon Regeneration After Injury. Proc. Natl. Acad. Sci. USA 2012, 109, 4768–4773. [Google Scholar] [CrossRef]
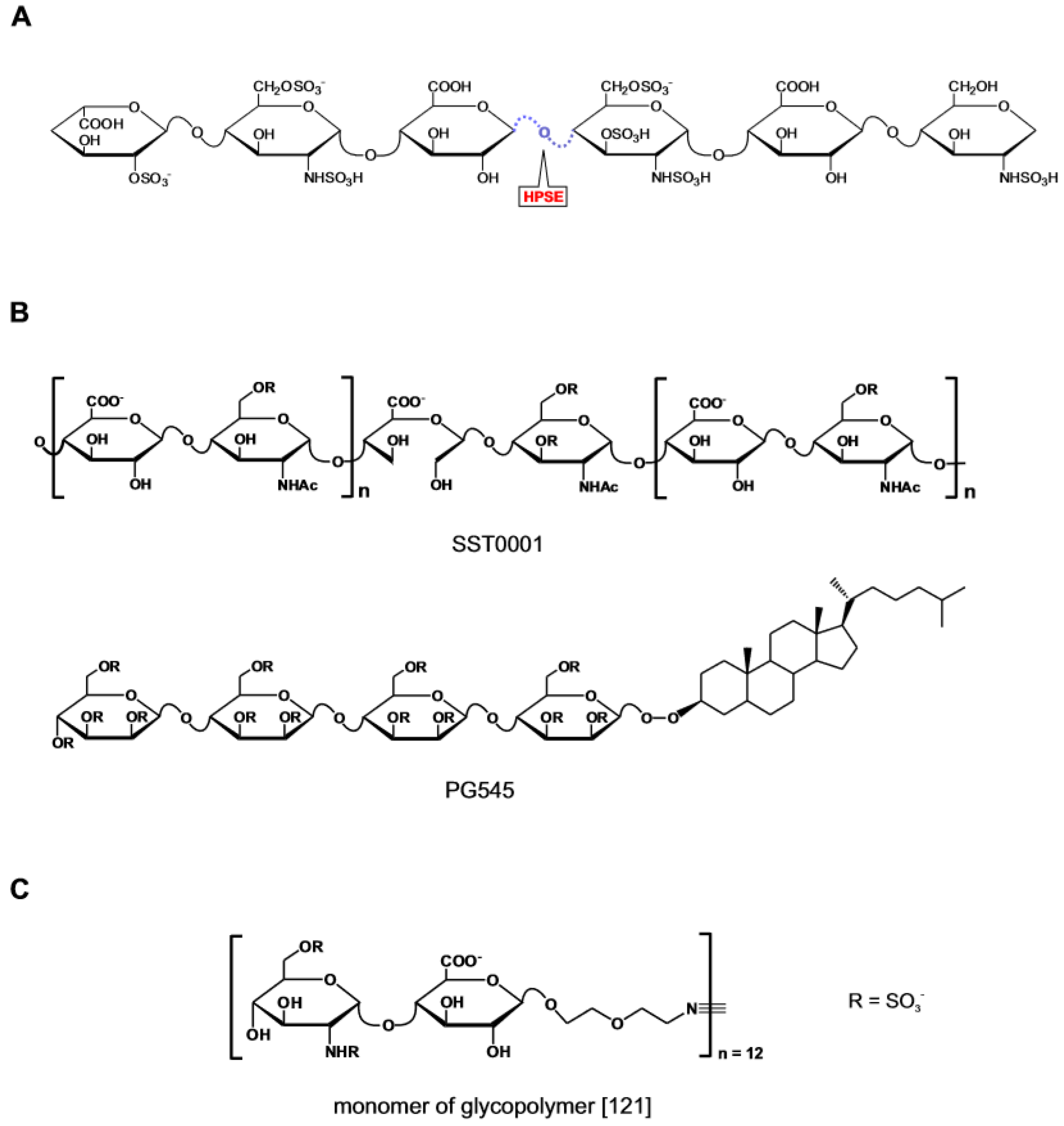
- Peterson, S.B.; Liu, J. Multi-Faceted Substrate Specificity of Heparanase. Matrix Biol. 2013, 32, 223–227. [Google Scholar] [CrossRef]
- Wu, L.; Viola, C.M.; Brzozowski, A.M.; Davies, G.J. Structural Characterization of Human Heparanase Reveals Insights into Substrate Recognition. Nat. Struct. Mol. Biol. 2015, 22, 1016–1022. [Google Scholar] [CrossRef]
- Hammond, E.; Khurana, A.; Shridhar, V.; Dredge, K. The Role of Heparanase and Sulfatases in the Modification of Heparan Sulfate Proteoglycans Within the Tumor Microenvironment and Opportunities for Novel Cancer Therapeutics. Front. Oncol. 2014, 4, 195. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Singh, P.; Boyango, I.; Gutter-Kapon, L.; Elkin, M.; Sanderson, R.D.; Ilan, N. Heparanase: From Basic Research to Therapeutic Applications in Cancer and Inflammation. Drug Resist. Updat. 2016, 29, 54–75. [Google Scholar] [CrossRef]
- Heyman, B.; Yang, Y. Mechanisms of Heparanase Inhibitors in Cancer Therapy. Exp. Hematol. 2016, 44, 1002–1012. [Google Scholar] [CrossRef]
- Ferro, V. Heparan Sulfate Inhibitors and Their Therapeutic Implications in Inflammatory Illnesses. Expert Opin. Ther. Targets 2013, 17, 965–975. [Google Scholar] [CrossRef]
- Thakkar, N.; Yadavalli, T.; Jaishankar, D.; Shukla, D. Emerging Roles of Heparanase in Viral Pathogenesis. Pathogens 2017, 6, 43. [Google Scholar] [CrossRef]
- Jin, H.; Zhou, S. The Functions of Heparanase in Human Diseases. Mini Rev. Med. Chem. 2017, 17, 541–548. [Google Scholar] [CrossRef]
- Rivara, S.; Milazzo, F.M.; Giannini, G. Heparanase: A Rainbow Pharmacological Target Associated to Multiple Pathologies Including Rare Diseases. Future Med. Chem. 2016, 8, 647–680. [Google Scholar] [CrossRef]
- Jia, L.; Ma, S. Recent Advances in the Discovery of Heparanase Inhibitors As Anti-Cancer Agents. Eur. J. Med. Chem. 2016, 121, 209–220. [Google Scholar] [CrossRef]
- Casu, B.; Naggi, A.; Torri, G. Heparin-Derived Heparan Sulfate Mimics to Modulate Heparan Sulfate-Protein Interaction in Inflammation and Cancer. Matrix Biol. 2010, 29, 442–452. [Google Scholar] [CrossRef]
- Cassinelli, G.; Favini, E.; Dal, B.L.; Tortoreto, M.; De, M.M.; Dagrada, G.; Pilotti, S.; Zunino, F.; Zaffaroni, N.; Lanzi, C. Antitumor Efficacy of the Heparan Sulfate Mimic Roneparstat (SST0001) Against Sarcoma Models Involves Multi-Target Inhibition of Receptor Tyrosine Kinases. Oncotarget 2016, 7, 47848–47863. [Google Scholar] [CrossRef]
- Brennan, T.V.; Lin, L.; Brandstadter, J.D.; Rendell, V.R.; Dredge, K.; Huang, X.; Yang, Y. Heparan Sulfate Mimetic PG545-Mediated Antilymphoma Effects Require TLR9-Dependent NK Cell Activation. J. Clin. Investig. 2016, 126, 207–219. [Google Scholar] [CrossRef]
- Loka, R.S.; Yu, F.; Sletten, E.T.; Nguyen, H.M. Design, Synthesis, and Evaluation of Heparan Sulfate Mimicking Glycopolymers for Inhibiting Heparanase Activity. Chem. Commun. 2017, 53, 9163–9166. [Google Scholar] [CrossRef]
- Pala, D.; Rivara, S.; Mor, M.; Milazzo, F.M.; Roscilli, G.; Pavoni, E.; Giannini, G. Kinetic Analysis and Molecular Modeling of the Inhibition Mechanism of Roneparstat (SST0001) on Human Heparanase. Glycobiology 2016, 26, 640–654. [Google Scholar] [CrossRef]
- Ferro, V.; Liu, L.; Johnstone, K.D.; Wimmer, N.; Karoli, T.; Handley, P.; Rowley, J.; Dredge, K.; Li, C.P.; Hammond, E.; et al. Discovery of PG545: A Highly Potent and Simultaneous Inhibitor of Angiogenesis, Tumor Growth, and Metastasis. J. Med. Chem. 2012, 55, 3804–3813. [Google Scholar] [CrossRef]
- Loka, R.S.; Sletten, E.T.; Barash, U.; Vlodavsky, I.; Nguyen, H.M. Specific Inhibition of Heparanase by Glycopolymer With Well-Defined Sulfation Pattern Prevents Breast Cancer Metastasis in Mice. ACS Appl. Mater. Interfaces 2018, 11, 244–254. [Google Scholar] [CrossRef]
- Sanderson, R.D.; Elkin, M.; Rapraeger, A.C.; Ilan, N.; Vlodavsky, I. Heparanase Regulation of Cancer, Autophagy and Inflammation: New Mechanisms and Targets for Therapy. FEBS J. 2017, 284, 42–55. [Google Scholar] [CrossRef]
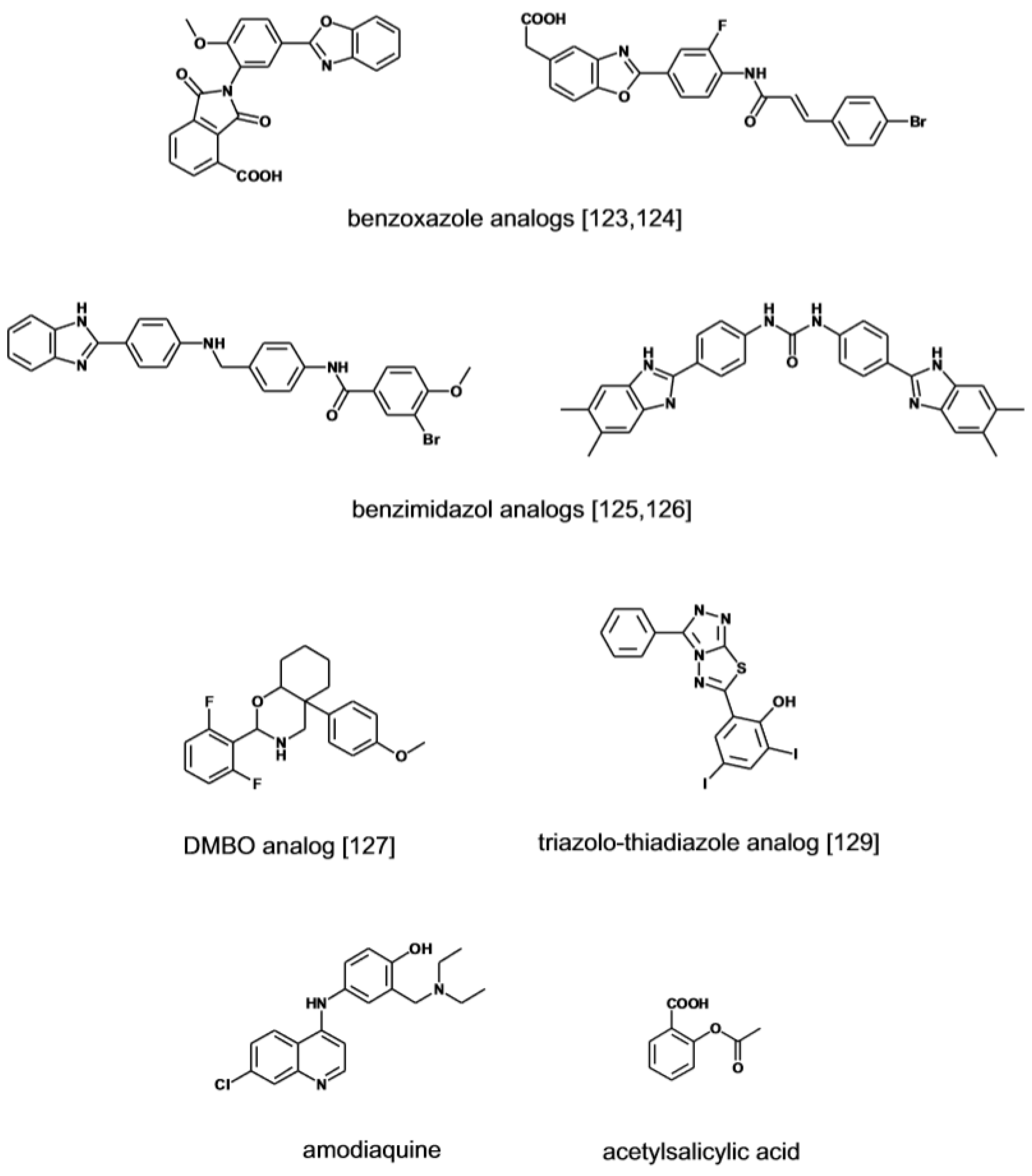
- Courtney, S.M.; Hay, P.A.; Buck, R.T.; Colville, C.S.; Porter, D.W.; Scopes, D.I.; Pollard, F.C.; Page, M.J.; Bennett, J.M.; Hircock, M.L.; et al. 2,3-Dihydro-1,3-Dioxo-1H-Isoindole-5-Carboxylic Acid Derivatives: A Novel Class of Small Molecule Heparanase Inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 3269–3273. [Google Scholar] [CrossRef]
- Courtney, S.M.; Hay, P.A.; Buck, R.T.; Colville, C.S.; Phillips, D.J.; Scopes, D.I.; Pollard, F.C.; Page, M.J.; Bennett, J.M.; Hircock, M.L.; et al. Furanyl-1,3-Thiazol-2-Yl and Benzoxazol-5-Yl Acetic Acid Derivatives: Novel Classes of Heparanase Inhibitor. Bioorg. Med. Chem. Lett. 2005, 15, 2295–2299. [Google Scholar] [CrossRef]
- Xu, Y.J.; Miao, H.Q.; Pan, W.; Navarro, E.C.; Tonra, J.R.; Mitelman, S.; Camara, M.M.; Deevi, D.S.; Kiselyov, A.S.; et al. N-(4-{[4-(1H-Benzoimidazol-2-Yl)-Arylamino]-Methyl}-Phenyl)-Benzamide Derivatives As Small Molecule Heparanase Inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 404–408. [Google Scholar] [CrossRef]
- Pan, W.; Miao, H.Q.; Xu, Y.J.; Navarro, E.C.; Tonra, J.R.; Corcoran, E.; Lahiji, A.; Kussie, P.; Kiselyov, A.S.; Wong, W.C.; et al. 1-[4-(1H-Benzoimidazol-2-Yl)-Phenyl]-3-[4-(1H-Benzoimidazol-2-Yl)-Phenyl]-Urea Derivatives As Small Molecule Heparanase Inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 409–412. [Google Scholar] [CrossRef]
- Song, Y.; Hu, B.; Qu, H.; Wang, L.; Zhang, Y.; Tao, J.; Cui, J. Novel 1, 3-N, O-Spiroheterocyclic Compounds Inhibit Heparanase Activity and Enhance Nedaplatin-Induced Cytotoxicity in Cervical Cancer Cells. Oncotarget 2016, 7, 36154–36167. [Google Scholar] [CrossRef]
- Murugan, S.; Kavitha, C.V.; Purushothaman, A.; Nevin, K.G.; Sugahara, K.; Rangappa, K.S. A Small Oxazine Compound As an Anti-Tumor Agent: A Novel Pyranoside Mimetic That Binds to VEGF, HB-EGF, and TNF-Alpha. Cancer Lett. 2010, 297, 231–243. [Google Scholar]
- Baburajeev, C.P.; Mohan, C.D.; Rangappa, S.; Mason, D.J.; Fuchs, J.E.; Bender, A.; Barash, U.; Vlodavsky, I.; Rangappa, K.S. Identification of Novel Class of Triazolo-Thiadiazoles As Potent Inhibitors of Human Heparanase and Their Anticancer Activity. BMC Cancer 2017, 17, 235. [Google Scholar] [CrossRef]
- Nakajima, M.; DeChavigny, A.; Johnson, C.E.; Hamada, J.; Stein, C.A.; Nicolson, G.L. Suramin. A Potent Inhibitor of Melanoma Heparanase and Invasion. J. Biol. Chem. 1991, 266, 9661–9666. [Google Scholar]
- Rondanin, R.; Fochi, S.; Baruchello, R.; Bernardi, T.; Oliva, P.; Semeraro, F.; Simoni, D.; Giannini, G. Arylamidonaphtalene Sulfonate Compounds As a Novel Class of Heparanase Inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 4421–4425. [Google Scholar] [CrossRef]
- Simmons, S.C.; McKenzie, E.A.; Harris, L.K.; Aplin, J.D.; Brenchley, P.E.; Velasco-Garcia, M.N.; Missailidis, S. Development of Novel Single-Stranded Nucleic Acid Aptamers Against the Pro-Angiogenic and Metastatic Enzyme Heparanase (HPSE1). PLoS ONE 2012, 7, e37938. [Google Scholar] [CrossRef]
- Simmons, S.C.; Jamsa, H.; Silva, D.; Cortez, C.M.; McKenzie, E.A.; Bitu, C.C.; Salo, S.; Nurmenniemi, S.; Nyberg, P.; Risteli, J.; et al. Anti-Heparanase Aptamers As Potential Diagnostic and Therapeutic Agents for Oral Cancer. PLoS ONE 2014, 9, e96846. [Google Scholar] [CrossRef]
- Gozalbes, R.; Mosulen, S.; Orti, L.; Rodriguez-Diaz, J.; Carbajo, R.J.; Melnyk, P.; Pineda-Lucena, A. Hit Identification of Novel Heparanase Inhibitors by Structure- and Ligand-Based Approaches. Bioorg. Med. Chem. 2013, 21, 1944–1951. [Google Scholar] [CrossRef]
- Dai, X.; Yan, J.; Fu, X.; Pan, Q.; Sun, D.; Xu, Y.; Wang, J.; Nie, L.; Tong, L.; Shen, A.; et al. Aspirin Inhibits Cancer Metastasis and Angiogenesis Via Targeting Heparanase. Clin. Cancer Res. 2017, 23, 6267–6278. [Google Scholar] [CrossRef]
- Handel, T.M.; Johnson, Z.; Crown, S.E.; Lau, E.K.; Proudfoot, A.E. Regulation of Protein Function by Glycosaminoglycans—As Exemplified by Chemokines. Annu. Rev. Biochem. 2005, 74, 385–410. [Google Scholar] [CrossRef]
- Proudfoot, A.E.I.; Johnson, Z.; Bonvin, P.; Handel, T.M. Glycosaminoglycan Interactions With Chemokines Add Complexity to a Complex System. Pharmaceuticals 2017, 10, 70. [Google Scholar] [CrossRef]
- Pomin, V.H. Sulfated Glycans in Inflammation. Eur. J. Med. Chem. 2015, 92, 353–369. [Google Scholar] [CrossRef]
- Farrugia, B.L.; Lord, M.S.; Melrose, J.; Whitelock, J.M. The Role of Heparan Sulfate in Inflammation, and the Development of Biomimetics As Anti-Inflammatory Strategies. J. Histochem. Cytochem. 2018, 66, 321–336. [Google Scholar] [CrossRef]
- Proudfoot, A.E.; Fritchley, S.; Borlat, F.; Shaw, J.P.; Vilbois, F.; Zwahlen, C.; Trkola, A.; Marchant, D.; Clapham, P.R.; Wells, T.N. The BBXB Motif of RANTES Is the Principal Site for Heparin Binding and Controls Receptor Selectivity. J. Biol. Chem. 2001, 276, 10620–10626. [Google Scholar] [CrossRef] [Green Version]
- Severin, I.C.; Soares, A.; Hantson, J.; Teixeira, M.; Sachs, D.; Valognes, D.; Scheer, A.; Schwarz, M.K.; Wells, T.N.; Proudfoot, A.E.; et al. Glycosaminoglycan Analogs As a Novel Anti-Inflammatory Strategy. Front. Immunol. 2012, 3, 293. [Google Scholar] [CrossRef]
- Nonaka, M.; Bao, X.; Matsumura, F.; Gotze, S.; Kandasamy, J.; Kononov, A.; Broide, D.H.; Nakayama, J.; Seeberger, P.H.; Fukuda, M. Synthetic Di-Sulfated Iduronic Acid Attenuates Asthmatic Response by Blocking T-Cell Recruitment to Inflammatory Sites. Proc. Natl. Acad. Sci. USA 2014, 111, 8173–8178. [Google Scholar] [CrossRef]
- Lau, E.K.; Paavola, C.D.; Johnson, Z.; Gaudry, J.P.; Geretti, E.; Borlat, F.; Kungl, A.J.; Proudfoot, A.E.; Handel, T.M. Identification of the Glycosaminoglycan Binding Site of the CC Chemokine, MCP-1: Implications for Structure and Function in Vivo. J. Biol. Chem. 2004, 279, 22294–22305. [Google Scholar] [CrossRef]
- Proudfoot, A.E.; Handel, T.M.; Johnson, Z.; Lau, E.K.; LiWang, P.; Clark-Lewis, I.; Borlat, F.; Wells, T.N.; Kosco-Vilbois, M.H. Glycosaminoglycan Binding and Oligomerization Are Essential for the in Vivo Activity of Certain Chemokines. Proc. Natl. Acad. Sci. USA 2003, 100, 1885–1890. [Google Scholar] [CrossRef]
- Ali, S.; O’Boyle, G.; Hepplewhite, P.; Tyler, J.R.; Robertson, H.; Kirby, J.A. Therapy With Nonglycosaminoglycan-Binding Mutant CCL7: A Novel Strategy to Limit Allograft Inflammation. Am. J. Transplant. 2010, 10, 47–58. [Google Scholar] [CrossRef]
- Severin, I.C.; Gaudry, J.P.; Johnson, Z.; Kungl, A.; Jansma, A.; Gesslbauer, B.; Mulloy, B.; Power, C.; Proudfoot, A.E.; Handel, T. Characterization of the Chemokine CXCL11-Heparin Interaction Suggests Two Different Affinities for Glycosaminoglycans. J. Biol. Chem. 2010, 285, 17713–17724. [Google Scholar] [CrossRef] [Green Version]
- Johnson, Z.; Kosco-Vilbois, M.H.; Herren, S.; Cirillo, R.; Muzio, V.; Zaratin, P.; Carbonatto, M.; Mack, M.; Smailbegovic, A.; Rose, M.; et al. Interference With Heparin Binding and Oligomerization Creates a Novel Anti-Inflammatory Strategy Targeting the Chemokine System. J. Immunol. 2004, 173, 5776–5785. [Google Scholar] [CrossRef]
- Bedke, J.; Nelson, P.J.; Kiss, E.; Muenchmeier, N.; Rek, A.; Behnes, C.L.; Gretz, N.; Kungl, A.J.; Grone, H.J. A Novel CXCL8 Protein-Based Antagonist in Acute Experimental Renal Allograft Damage. Mol. Immunol. 2010, 47, 1047–1057. [Google Scholar] [CrossRef]
- Adage, T.; Konya, V.; Weber, C.; Strutzmann, E.; Fuchs, T.; Zankl, C.; Gerlza, T.; Jeremic, D.; Heinemann, A.; Kungl, A.J. Targeting Glycosaminoglycans in the Lung by an Engineered CXCL8 As a Novel Therapeutic Approach to Lung Inflammation. Eur. J. Pharmacol. 2015, 748, 83–92. [Google Scholar] [CrossRef]
- Adage, T.; Del, B.F.; Fiorentini, F.; Doornbos, R.P.; Zankl, C.; Bartley, M.R.; Kungl, A.J. PA401, a Novel CXCL8-Based Biologic Therapeutic With Increased Glycosaminoglycan Binding, Reduces Bronchoalveolar Lavage Neutrophils and Systemic Inflammatory Markers in a Murine Model of LPS-Induced Lung Inflammation. Cytokine 2015, 76, 433–441. [Google Scholar] [CrossRef]
- Liehn, E.A.; Piccinini, A.M.; Koenen, R.R.; Soehnlein, O.; Adage, T.; Fatu, R.; Curaj, A.; Popescu, A.; Zernecke, A.; Kungl, A.J.; et al. A New Monocyte Chemotactic Protein-1/Chemokine CC Motif Ligand-2 Competitor Limiting Neointima Formation and Myocardial Ischemia/Reperfusion Injury in Mice. J. Am. Coll. Cardiol. 2010, 56, 1847–1857. [Google Scholar] [CrossRef] [Green Version]
- Roblek, M.; Strutzmann, E.; Zankl, C.; Adage, T.; Heikenwalder, M.; Atlic, A.; Weis, R.; Kungl, A.; Borsig, L. Targeting of CCL2-CCR2-Glycosaminoglycan Axis Using a CCL2 Decoy Protein Attenuates Metastasis Through Inhibition of Tumor Cell Seeding. Neoplasia 2016, 18, 49–59. [Google Scholar] [CrossRef]
- Gschwandtner, M.; Trinker, M.U.; Hecher, B.; Adage, T.; Ali, S.; Kungl, A.J. Glycosaminoglycan Silencing by Engineered CXCL12 Variants. FEBS Lett. 2015, 589, 2819–2824. [Google Scholar] [CrossRef]
- Cecchi, F.; Pajalunga, D.; Fowler, C.A.; Uren, A.; Rabe, D.C.; Peruzzi, B.; Macdonald, N.J.; Blackman, D.K.; Stahl, S.J.; Byrd, R.A.; et al. Targeted Disruption of Heparan Sulfate Interaction With Hepatocyte and Vascular Endothelial Growth Factors Blocks Normal and Oncogenic Signaling. Cancer Cell 2012, 22, 250–262. [Google Scholar] [CrossRef]
- Datta-Mannan, A.; Yaden, B.; Krishnan, V.; Jones, B.E.; Croy, J.E. An Engineered Human Follistatin Variant: Insights into the Pharmacokinetic and Pharmocodynamic Relationships of a Novel Molecule With Broad Therapeutic Potential. J. Pharmacol. Exp. Ther. 2013, 344, 616–623. [Google Scholar] [CrossRef]
- Guerrini, M.; Mourier, P.A.; Torri, G.; Viskov, C. Antithrombin-Binding Oligosaccharides: Structural Diversities in a Unique Function? Glycoconj. J. 2014, 31, 409–416. [Google Scholar] [CrossRef]
- Pomin, V.H. Marine Non-Glycosaminoglycan Sulfated Glycans As Potential Pharmaceuticals. Pharmaceuticals 2015, 8, 848–864. [Google Scholar] [CrossRef]
- Nahain, A.A.; Ignjatovic, V.; Monagle, P.; Tsanaktsidis, J.; Ferro, V. Heparin Mimetics With Anticoagulant Activity. Med. Res. Rev. 2018, 38, 1582–1613. [Google Scholar] [CrossRef]
- Gulati, K.; Jamsandekar, M.; Poluri, K.M. Mechanistic Insights into Molecular Evolution of Species-Specific Differential Glycosaminoglycan Binding Surfaces in Growth-Related Oncogene Chemokines. R. Soc. Open Sci. 2017, 4, 171059. [Google Scholar] [CrossRef]
- Li, Y.; Sun, C.; Yates, E.A.; Jiang, C.; Wilkinson, M.C.; Fernig, D.G. Heparin Binding Preference and Structures in the Fibroblast Growth Factor Family Parallel Their Evolutionary Diversification. Open Biol. 2016, 6, 150275. [Google Scholar] [CrossRef]
- Boittier, E.D.; Ganghi, N.S.; Ferro, V.; Coombe, D.R. Cross-Species Analysis of Glycosaminoglycan Binding Proteins Reveals Some Animals Models Are “More Equal” Than Others. Molecules 2019, 24, 924. [Google Scholar] [CrossRef]
- Rusnati, M.; Presta, M. Angiogenic Growth Factors Interactome and Drug Discovery: The Contribution of Surface Plasmon Resonance. Cytokine Growth Factor Rev. 2015, 26, 293–310. [Google Scholar] [CrossRef]
- Rusnati, M.; Chiodelli, P.; Bugatti, A.; Urbinati, C. Bridging the Past and the Future of Virology: Surface Plasmon Resonance As a Powerful Tool to Investigate Virus/Host Interactions. Crit. Rev. Microbiol. 2015, 41, 238–260. [Google Scholar] [CrossRef]
- Ori, A.; Free, P.; Courty, J.; Wilkinson, M.C.; Fernig, D.G. Identification of Heparin-Binding Sites in Proteins by Selective Labeling. Mol. Cell. Proteom. 2009, 8, 2256–2265. [Google Scholar] [CrossRef]









© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghiselli, G. Heparin Binding Proteins as Therapeutic Target: An Historical Account and Current Trends. Medicines 2019, 6, 80. https://doi.org/10.3390/medicines6030080
Ghiselli G. Heparin Binding Proteins as Therapeutic Target: An Historical Account and Current Trends. Medicines. 2019; 6(3):80. https://doi.org/10.3390/medicines6030080
Chicago/Turabian StyleGhiselli, Giancarlo. 2019. "Heparin Binding Proteins as Therapeutic Target: An Historical Account and Current Trends" Medicines 6, no. 3: 80. https://doi.org/10.3390/medicines6030080
APA StyleGhiselli, G. (2019). Heparin Binding Proteins as Therapeutic Target: An Historical Account and Current Trends. Medicines, 6(3), 80. https://doi.org/10.3390/medicines6030080