A Visualization Tool for Cryo-EM Protein Validation with an Unsupervised Machine Learning Model in Chimera Platform



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
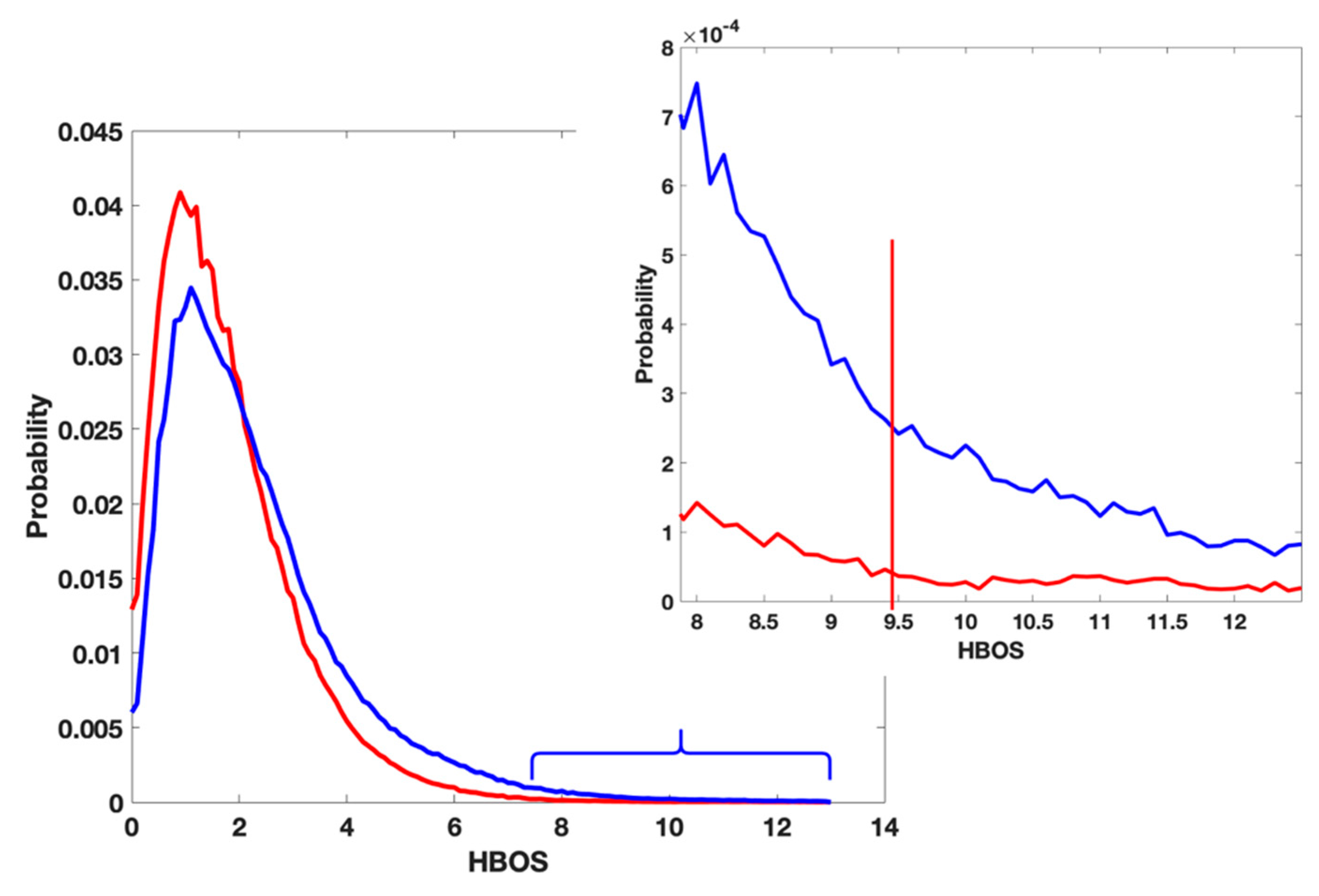
3.1. Cons in the Current Validation Tool
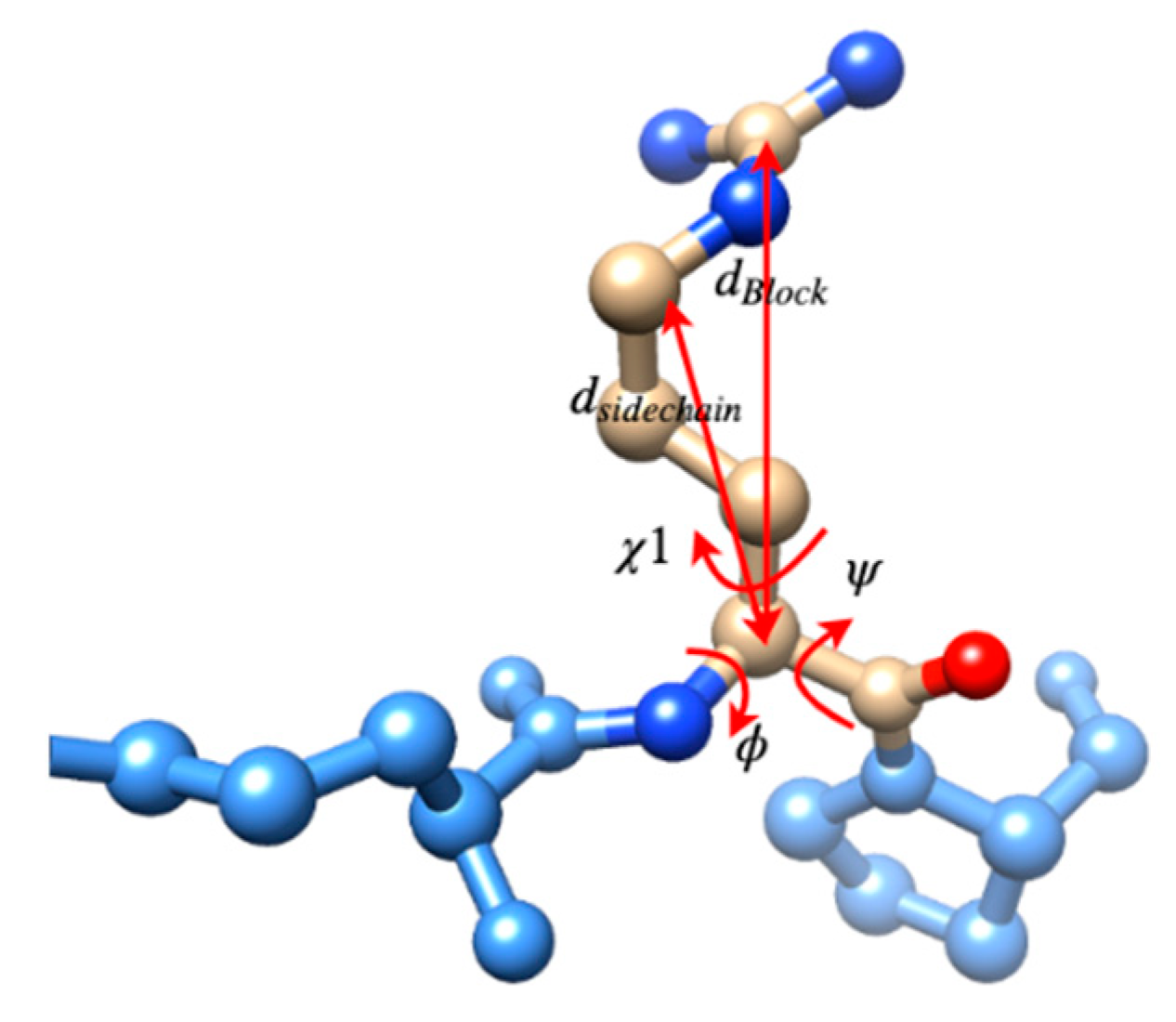
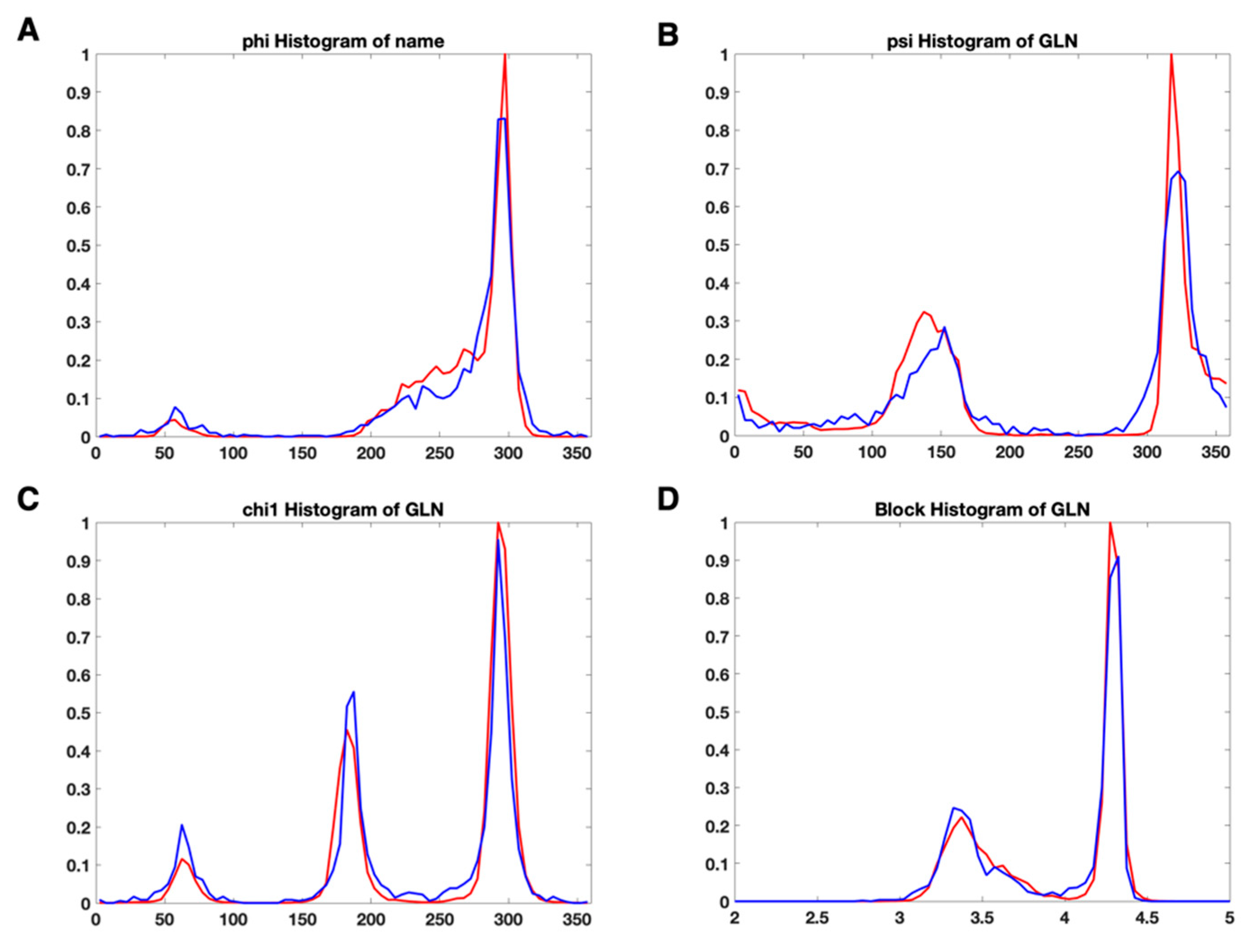
3.2. Use Combined Multi-Features
3.3. A Complement of the Current PDB Validation Tool
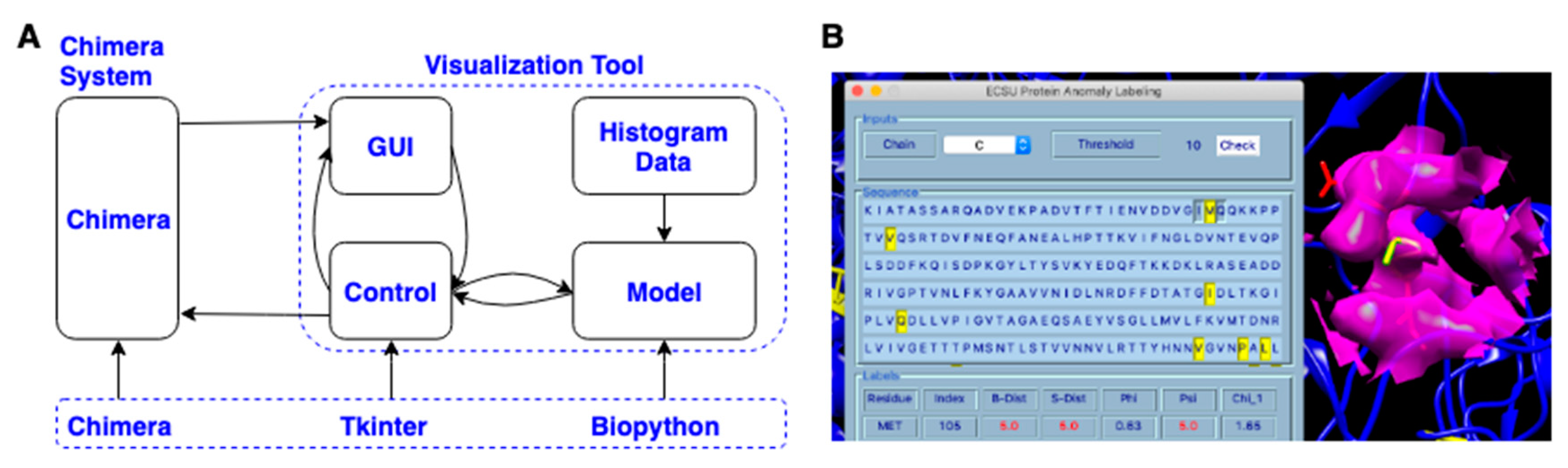
3.4. Visualization Chimera Tool
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chung, J.-H.; Kim, H.M. The Nobel Prize in Chemistry 2017: High-Resolution Cryo-Electron Microscopy. Appl. Microsc. 2017, 47, 218–222. [Google Scholar] [CrossRef] [Green Version]
- Baker, T.S.; Henderson, R. Electron cryomicroscopy. In International Tables for Crystallography Volume F: Crystallography Ofbiological Macromolecules; Rossmann, M.G., Arnold, E., Eds.; Springer: Dordrecht, The Netherlands, 2001; pp. 451–463. [Google Scholar]
- Grünewald, K.; Desai, P.; Winkler, D.C.; Heymann, J.B.; Belnap, D.M.; Baumeister, W.; Steven, A.C. Three-Dimensional Structure of Herpes Simplex Virus from Cryo-Electron Tomography. Science 2003, 302, 1396–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medalia, O.; Weber, I.; Frangakis, A.S.; Nicastro, D.; Gerisch, G.; Baumeister, W. Macromolecular Architecture in Eukaryotic Cells Visualized by Cryoelectron Tomography. Science 2002, 298, 1209–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinothkumar, K.R.; Henderson, R. Single particle electron cryomicroscopy: Trends, issues and future perspective. Q. Rev. Biophys. 2016, 49, e13. [Google Scholar] [CrossRef] [PubMed]
- Deptuch, G.; Besson, A.; Rehak, P.; Szelezniak, M.; Wall, J.; Winter, M.; Zhu, Y. Direct electron imaging in electron microscopy with monolithic active pixel sensors. Ultramicroscopy 2007, 107, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Milazzo, A.-C.; Leblanc, P.; Duttweiler, F.; Jin, L.; Bouwer, J.C.; Peltier, S.; Ellisman, M.; Bieser, F.; Matis, H.S.; Wieman, H.; et al. Active pixel sensor array as a detector for electron microscopy. Ultramicroscopy 2005, 104, 152–159. [Google Scholar] [CrossRef]
- Kühlbrandt, W. The Resolution Revolution. Science 2014, 343, 1443–1444. [Google Scholar] [CrossRef]
- Renaud, J.-P.; Chari, A.; Ciferri, C.; Liu, W.-T.; Rémigy, H.-W.; Stark, H.; Wiesmann, C. Cryo-EM in drug discovery: Achievements, limitations and prospects. Nat. Rev. Drug Discov. 2018, 17, 471–492. [Google Scholar] [CrossRef]
- RCSB PDB. Available online: https://www.rcsb.org/ (accessed on 3 August 2019).
- PDB Statistics: Growth of Structures from 3DEM Experiments Released per Year. Available online: https://www.rcsb.org/stats/growth/em (accessed on 28 July 2019).
- Chen, L.; He, J.; Sazzed, S.; Walker, R. An Investigation of Atomic Structures Derived from X-ray Crystallography and Cryo-Electron Microscopy Using Distal Blocks of Side-Chains. Molecules 2018, 23, 610. [Google Scholar] [CrossRef]
- Janssen, B.J.C.; Read, R.J.; Brünger, A.T.; Gros, P. Crystallographic evidence for deviating C3b structure. Nature 2007, 448, E1–E3. [Google Scholar] [CrossRef]
- Read, R.J.; Adams, P.D.; Arendall, W.B., 3rd; Brunger, A.T.; Emsley, P.; Joosten, R.P.; Kleywegt, G.J.; Krissinel, E.B.; Lütteke, T.; Otwinowski, Z.; et al. A new generation of crystallographic validation tools for the protein data bank. Structure 2011, 19, 1395–1412. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. Cryo-electron microscopy shapes up. Nature 2018, 561, 565–567. [Google Scholar] [CrossRef] [PubMed]
- Montelione, G.T.; Nilges, M.; Bax, A.; Güntert, P.; Herrmann, T.; Richardson, J.S.; Schwieters, C.D.; Vranken, W.F.; Vuister, G.W.; Wishart, D.S.; et al. Recommendations of the wwPDB NMR Validation Task Force. Structure 2013, 21, 1563–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, R.; Sali, A.; Baker, M.L.; Carragher, B.; Devkota, B.; Downing, K.H.; Egelman, E.H.; Feng, Z.; Frank, J.; Grigorieff, N.; et al. Outcome of the first electron microscopy validation task force meeting. Structure 2012, 20, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Sousa, D.; Grigorieff, N. Ab initio resolution measurement for single particle structures. J. Struct. Biol. 2007, 157, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Penczek, P.A. Resolution measures in molecular electron microscopy. Methods Enzymol. 2010, 482, 73–100. [Google Scholar] [PubMed]
- Schwede, T.; Sali, A.; Honig, B.; Levitt, M.; Berman, H.M.; Jones, D.; Brenner, S.E.; Burley, S.K.; Das, R.; Dokholyan, N.V.; et al. Outcome of a workshop on applications of protein models in biomedical research. Structure 2009, 17, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Harauz, G.; Heel, M.V. Exact filters for general geometry three dimensional reconstruction. Optik 1986, 73, 146–156. [Google Scholar]
- Wlodawer, A.; Li, M.; Dauter, Z. High-Resolution Cryo-EM Maps and Models: A Crystallographer’s Perspective. Structure 2017, 25, 1589–1597.e1. [Google Scholar] [CrossRef]
- Scheres, S.H.W.; Chen, S. Prevention of overfitting in cryo-EM structure determination. Nat. Methods 2012, 9, 853–854. [Google Scholar] [CrossRef]
- XML Metadata of Protein Validation Reports. Available online: ftp://ftp.rcsb.org/pub/pdb/validation_reports/ (accessed on 3 August 2019).
- Rawson, S.; Bisson, C.; Hurdiss, D.L.; Fazal, A.; McPhillie, M.J.; Sedelnikova, S.E.; Baker, P.J.; Rice, D.W.; Muench, S.P. Elucidating the structural basis for differing enzyme inhibitor potency by cryo-EM. Proc. Natl. Acad. Sci. USA 2018, 115, 1795–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Autzen, H.E.; Myasnikov, A.G.; Campbell, M.G.; Asarnow, D.; Julius, D.; Cheng, Y. Structure of the human TRPM4 ion channel in a lipid nanodisc. Science 2018, 359, 228–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kater, L.; Thoms, M.; Barrio-Garcia, C.; Cheng, J.; Ismail, S.; Ahmed, Y.L.; Bange, G.; Kressler, D.; Berninghausen, O.; Sinning, I.; et al. Visualizing the Assembly Pathway of Nucleolar Pre-60S Ribosomes. Cell 2017, 171, 1599–1610.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; She, J.; Zeng, W.; Chen, Q.; Bai, X.-C.; Jiang, Y. Structures of the calcium-activated, non-selective cation channel TRPM4. Nature 2017, 552, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Zwart, P.H.; Grosse-Kunstleve, R.W.; Adams, P.D. Xtriage and Fest: Automatic assessment of X-ray data and substructure structure factor estimation. CCP4 Newsl. 2005, 43, 27–35. [Google Scholar]
- Jones, T.A.; Zou, J.-Y.; Cowan, S.W.; Kjeldgaard, M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. Sect. A 1991, 47, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Hooft, R.W.W.; Vriend, G.; Sander, C.; Abola, E.E. Errors in protein structures. Nature 1996, 381, 272. [Google Scholar] [CrossRef]
- Bruno, I.J.; Cole, J.C.; Kessler, M.; Luo, J.; Motherwell, W.D.S.; Purkis, L.H.; Smith, B.R.; Taylor, R.; Cooper, R.I.; Harris, S.E.; et al. Retrieval of Crystallographically-Derived Molecular Geometry Information. J. Chem. Inf. Comput. Sci. 2004, 44, 2133–2144. [Google Scholar] [CrossRef]
- Kleywegt, G.J.; Harris, M.R.; Zou, J.-Y.; Taylor, T.C.; Wählby, A.; Jones, T.A. The Uppsala Electron-Density Server. Acta Crystallogr. Sect. D 2004, 60, 2240–2249. [Google Scholar] [CrossRef] [Green Version]
- Gore, S.; Velankar, S.; Kleywegt, G.J. Implementing an X-ray validation pipeline for the Protein Data Bank. Acta Crystallogr. Sect. D 2012, 68, 478–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gore, S.; Sanz García, E.; Hendrickx, P.M.S.; Gutmanas, A.; Westbrook, J.D.; Yang, H.; Feng, Z.; Baskaran, K.; Berrisford, J.M.; Hudson, B.P.; et al. Validation of Structures in the Protein Data Bank. Structure 2017, 25, 1916–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonine, P.V.; Klaholz, B.P.; Moriarty, N.W.; Poon, B.K.; Sobolev, O.V.; Terwilliger, T.C.; Adams, P.D.; Urzhumtsev, A. New tools for the analysis and validation of cryo-EM maps and atomic models. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 814–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Chen, L.; He, J. Using Combined Features to Analyze Atomic Structures derived from Cryo-EM Density Maps. In Proceedings of the 2018 ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, Washington, DC, USA, 29 August–1 September 2018; pp. 651–655. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Guerra Pedregal, J.; Maréchal, J.-D. PyChimera: Use UCSF Chimera modules in any Python 2.7 project. Bioinformatics 2018, 34, 1784–1785. [Google Scholar] [CrossRef] [PubMed]
- Raza, S.; Ranaghan, K.E.; van der Kamp, M.W.; Woods, C.J.; Mulholland, A.J.; Azam, S.S. Visualizing protein–ligand binding with chemical energy-wise decomposition (CHEWD): Application to ligand binding in the kallikrein-8 S1 Site. J. Comput. Aided Mol. Des. 2019, 33, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Qu, K.; Glass, B.; Doležal, M.; Schur, F.K.M.; Murciano, B.; Rein, A.; Rumlová, M.; Ruml, T.; Kräusslich, H.-G.; Briggs, J.A.G. Structure and architecture of immature and mature murine leukemia virus capsids. Proc. Natl. Acad. Sci. USA 2018, 115, E11751–E11760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garzón, J.I.; Kovacs, J.; Abagyan, R.; Chacón, P. ADP_EM: Fast exhaustive multi-resolution docking for high-throughput coverage. Bioinformatics 2006, 23, 427–433. [Google Scholar] [CrossRef]
- Sehnal, D.; Vařeková, R.S.; Pravda, L.; Ionescu, C.-M.; Geidl, S.; Horský, V.; Jaiswal, D.; Wimmerová, M.; Koča, J. ValidatorDB: Database of up-to-date validation results for ligands and non-standard residues from the Protein Data Bank. Nucleic Acids Res. 2014, 43, D369–D375. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Schmid, M.F.; Zhou, Z.H.; Rixon, F.; Chiu, W. Finding and using local symmetry in identifying lower domain movements in hexon subunits of the herpes simplex virus type 1 B capsid. J. Mol. Biol. 2001, 309, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol. 1963, 7, 95–99. [Google Scholar] [CrossRef]
- Chen, L.; He, J. A distance- and orientation-dependent energy function of amino acid key blocks. Biopolymers 2014, 101, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Biopython Download. Available online: https://biopython.org/wiki/Download (accessed on 3 August 2019).
- Chimera Download. Available online: https://www.cgl.ucsf.edu/chimera/download.html (accessed on 3 August 2019).
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Baker, B.; Santos, E.; Sheep, M.; Daftarian, D. A Visualization Tool for Cryo-EM Protein Validation with an Unsupervised Machine Learning Model in Chimera Platform. Medicines 2019, 6, 86. https://doi.org/10.3390/medicines6030086
Chen L, Baker B, Santos E, Sheep M, Daftarian D. A Visualization Tool for Cryo-EM Protein Validation with an Unsupervised Machine Learning Model in Chimera Platform. Medicines. 2019; 6(3):86. https://doi.org/10.3390/medicines6030086
Chicago/Turabian StyleChen, Lin, Brandon Baker, Eduardo Santos, Michell Sheep, and Darius Daftarian. 2019. "A Visualization Tool for Cryo-EM Protein Validation with an Unsupervised Machine Learning Model in Chimera Platform" Medicines 6, no. 3: 86. https://doi.org/10.3390/medicines6030086
APA StyleChen, L., Baker, B., Santos, E., Sheep, M., & Daftarian, D. (2019). A Visualization Tool for Cryo-EM Protein Validation with an Unsupervised Machine Learning Model in Chimera Platform. Medicines, 6(3), 86. https://doi.org/10.3390/medicines6030086